

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Overview and Introduction
Genes and Disease Risk
Celiac disease, autoimmune hepatitis (AIH), and the inflammatory bowel diseases (IBDs), Crohn's disease and ulcerative colitis (UC), are chronic inflammatory diseases of unknown etiology. They are considered complex genetic diseases because both inherited and environmental influences appear to be important in determining risk. Complex genetic diseases are more common than Mendelian diseases in the population. Prevalence ranges for AIH, IBD and celiac disease are given in Table 1. Generally, accepted average prevalences are 1 in 1,000 persons for Crohn's disease and UC, and 3 in 1,000 for celiac disease. However, the prevalence rate of Crohn's, UC and celiac disease are much lower in some populations (e.g., 1.25 in 100,000 for Crohn's disease in Hong Kong1). The average prevalence of AIH is 10-fold lower at 1 in 10,000.
Table 1
Epidemiologic and genetic data for IBD, celiac disease and AIH.
Some effort has been devoted to understanding the relative contribution of environmental and genetic factors to disease risk. One method for ascertaining the genetic component of a disease is to compute the risk to siblings divided by the population risk (λs). Mendelian genetic diseases have extremely high λs values (λs = 500 for cystic fibrosis), while diseases with weaker genetic components have lower values (λs = 2 for hypertension). Crohn's disease, UC, and celiac disease have sibling relative risks that are higher than most complex traits (shown in Table 1). The risk to any first-degree family member for Crohn's disease is 10–32 fold higher than in the general population; for UC it is 5–12 fold, and for celiac disease it is 20–53 fold.2,3 Familial risk has not been reported in AIH, perhaps due to its lower prevalence. Notably, both celiac disease and AIH more commonly affect women (Table 1), suggesting that sex steroids or other female-specific factors may play a role in the etiology of these diseases.
In this chapter, we address the etiology of each disease by first providing a summary of the known environmental influences, and then reviewing the genetic studies aimed at the identification of susceptibility loci. One of the genetic loci we will give attention to is the major histocompatibility complex (MHC). The MHC is home to over 140 genes, including the eight highly variable classical human leukocyte antigen (HLA) loci. HLA genes encode cell surface molecules that present antigenic peptides to T cells, thereby initiating acquired immune response to invading pathogens and other foreign antigens. T cells determine whether antigens are “self ” or “nonself ” based on the specific antigenic peptide sequence as well as the HLA variant that presents the peptide. Given their central role in self/nonself distinction, associations between autoimmune diseases and variation in the classical HLA genes are among the most consistent findings in human genetics.4 The relative contribution of the MHC to disease risk for celiac disease, Crohn's disease, and UC can be determined by assessing the sibling risk based on MHC haplotype sharing (MHC λs). The ratio of the MHC λs value to the λs implies the degree to which non-MHC genes are involved in the risk to disease. We see that the non-MHC component is larger for Crohn's disease and UC risk is associated more with the MHC.
Complex Trait Genetics
Genetic studies fall into two main categories: linkage studies and association tests. Linkage studies, which are most often performed on a genomewide scale, seek to identify genomic regions (linkage peaks) with increased allele sharing between affected family members. These peaks may be interpreted as large regions where causal variation may lie. Association testing aims to determine whether statistically significant correlation exists between a particular allele and a given phenotype.
Genomewide linkage studies have the benefit of assessing the entire genome in an unbiased manner. A linkage peak is considered suggestive evidence when it is expected to occur by chance about once per genome scan, and becomes significant when this expectation decreases to once per 20 scans.16 Confirmed linkage is only achieved after significant linkage in one study is replicated in a second independent study. Because replication involves one or a small number of tests, and is derived from prior evidence of linkage in a genomic scan, a modest threshold is sufficient (e.g., p < 0.05 for a single test). However, extremely large study sizes and dense coverage are required for linkage studies to obtain the statistical power necessary for identifying loci of modest effect. One approach to increase statistical power is through meta-analyses of multiple linkage studies. We will review the meta-analyses that have recently been performed for Crohn's and celiac disease later in the chapter.
Association studies are more powerful than linkage studies at assessing the role of a particular genomic region in disease. It is difficult to perform association studies in a genomewide manner due to the current costs of genotyping. However, the recent finding that the human genome is organized in “haplotype blocks” of relatively low diversity17,18 is reducing the effort required to thoroughly test large regions for association to disease. Haplotype blocks are regions in the human genome (extending from 1–2 to >100 kb) within which the underlying genetic variation exists in linkage disequilibrium and little recombination has occurred.17-19 Approximately 90% of the genetic variation observed in human chromosomes can be parsed into only 3–5 common patterns of variation, or haplotypes, per block. As a result of this genomic structure, the common haplotypes within a block can be identified with a subset of variants or haplotype tagging SNPs (htSNPs).18 Consequently, these haplotype frameworks offer a reduction in genotyping costs and an increase in statistical power of association studies because not every SNP in a particular haplotype block needs to be tested. Instead, a few haplotype-identifying SNPs can be employed to test all the variation at any given block for association to disease.
An international effort—the International HapMap Project—aims to identify the haplotype structure of the entire human genome (http://www.hapmap.org).20 The completion of this haplotype map holds great promise for future genetic studies of human complex traits. After we discuss the current state of the field for IBD, celiac disease and AIH, we will return to the role that the HapMap Project and other genetic advances will play in the future of these diseases.
Inflammatory Bowel Diseases
Definition, Classification and Symptoms
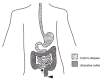
Inflammatory bowel diseases are characterized by chronic relapsing inflammation of the gastrointestinal tract. Crohn's disease (MIM 266600) and ulcerative colitis (MIM 191390) are the two main subtypes of IBD. Crohn's disease most frequently manifests itself with abdominal pain and diarrhea and is often complicated by intestinal fistulization (an abnormal passage between an injured organ and a healthy organ), intestinal obstruction or both. Tissue damage in Crohn's patients may involve any part of the gastrointestinal tract but is frequently localized to the terminal ileum and/or colon (fig.1); inflammation is transmural and discontinuous and may contain granulomas.21 The major symptoms of ulcerative colitis (UC) include diarrhea, rectal bleeding, the passage of mucus and abdominal pain. Inflammation in UC patients is continuous and limited to the rectal and colonic mucosal layers (fig.1); no fistulas and granulomas are observed.22 Approximately 5–10% of IBD cases cannot be unequivocally assigned to Crohn's disease or UC and so are diagnosed with indeterminate colitis (IC). IC is often a temporary classification until a final diagnosis can be made. In two independent prospective studies, 32–50% of IC patients were classified as having either Crohn's disease or UC at follow-up that ranged from 1 month to 26 years after the diagnosis of IC.23,24
Autoimmune Features
Whether Crohn's disease and UC are true autoimmune disorders is an open question, although production of autoantibodies is observed in IBD patients. Sources of the targeted autoantigens include the colon, DNA, thyroglobulin, tropomyosin, smooth muscle, gastric parietal cell, erythrocyte, pancreas, and neutrophil cytoplasm (i.e., anti-neutrophil cytoplasmic antibodies or ANCA).25 Production of anti-colonic, anti-tropomyosin and ANCA is observed more often in UC than in Crohn's disease; however, not all patients are seropositive. Moreover, none of these autoantibodies has been shown to directly cause gut inflammation. Therefore, whether they are central to disease pathogenesis remains to be established. An alternate hypothesis is that IBD is an immune reaction to antigens in the intestinal microflora. We will revisit this possibility when we discuss the association between Crohn's disease and the caspase recruitment domain-15 (CARD15) gene below.
Epidemiology: Inheritance and Environment
The combined prevalence of all IBD diseases is estimated to affect about 1 per 1,000 individuals. The incidence of IBD has been suggested to be highest in developed industrialized countries, but few studies have been done in developing countries. IBDs are most prevalent in Ashkenazi Jews and are rare in Asian and Hispano-American populations.1,5 Despite conflicting data on the prevalence of IBD among African-Americans, there has been a steady increase in reported cases among this ethnic group.5 Overall, the prevalence and incidence of IBD, and Crohn's disease in particular, seem to have increased significantly in the last few decades.5
Genetic determinants are important in both UC and Crohn's disease. The range for the estimated familial recurrence among patients with IBD is 5–30%, and the risk to first-degree relatives of affected individuals is estimated to be 5–32 fold.2,26 Moreover, the concordance rate in monozygotic twins compared with dizygotic twins is significantly greater for both Crohn's disease (58–63% vs 0–4%) and UC (18–19% vs 0–5%),27,28 establishing a stronger genetic influence in Crohn's than in UC.
The environmental factor most convincingly associated with IBD is smoking. Smoking increases the risk for Crohn's disease by a factor of 2–5 fold and decreases the risk for UC by a similar magnitude.29,30 Nicotine is speculated to be the principal component responsible for this effect, possibly via the dysregulation of the T-helper (Th) immune response pathways. Th1 cells activate cellular immunity to fight viruses and other intracellular pathogens, eliminate cancerous cells, and stimulate delayed-type hypersensitivity skin reactions, resulting in inflammatory response. In contrast, Th2 cells induce humoral and allergic responses, up-regulate antibody production to fight extracellular organisms, and suppress inflammation. In vivo, nicotine inhibits Th2 in nonadherent mononuclear cells of healthy nonsmokers undergoing nicotine patch treatment, perhaps explaining the increased Crohn's disease risk as Crohn's phenotypes are thought to be Th1 mediated.31 Conversely, UC protection may result from a mucin deficiency that smoking acts to reverse.32 Studies showed that nicotine significantly increased mucin synthesis in colonic cultured epithelial biopsies from both UC patients and control individuals,33 and inhibited the synthesis of the pro-inflammatory cytokines IL-1β and TNFα in the mouse colonic mucosa.34 In addition, nicotine was found to reduce circular colonic smooth muscle activity, which appears to be up-regulated in active UC.35 Finally, clinical trials have shown nicotine to be of some benefit in UC;36 however, further research is required to establish its therapeutic role and the mechanisms responsible for its action.
There is little correlation of dietary habits with IBD, in contrast with celiac disease (discussed later). High consumption of refined sugar and little dietary fiber seems to increase the risk for IBD;37,38 similarly, zinc deficiency in Crohn's disease patients is associated with an increased absorption dysfunction.39 Diet may affect disease susceptibility by modulating the intestinal permeability or the intestinal flora milieu, and this possibility is the underlying principle for prebiotics therapy. Prebiotics are nondigestible edibles that are proposed to selectively stimulate the growth and activity of beneficial gut bacteria. Prebiotics currently under study for treatment of IBD include lactosucrose, oligofructose, inulin, bran, psyllium, and germinated barley.40
Genetics
Linkage Studies
Seven genomewide scans and several targeted scans in families with Crohn's disease and/or UC have identified eight regions potentially conferring susceptibility to IBD (Table 2). The first genomewide search in Crohn's disease reported a potential locus in the pericentromeric region of chromosome 16 (LOD score = 5.79).41 This locus (IBD1) was also the first replicated through pooling genotype data from over 600 families collected by 11 centers distributed throughout North America, Europe, and Australia.42 This supports the hypothesis that common diseases can be explained by genetic risk factors that are common to many populations, but exist at different frequencies.
Table 2
Summary of IBD susceptibility loci.
Potential IBD susceptibility loci identified through genome scans are localized on chromosomes 3p, 7 and 12;43 5q35, 14q11, and 17q21;45 1, 6, 10, 22, and X;46 14q11;47 and 3p, 6p, 5q31, and 19p13.48 Some of the reported loci were replicated in follow-up studies, but no one locus was identified in every study.49-57 This apparent lack of consistency can be explained by the modest effect of most genetic loci in complex human traits and by the modest power and significant effect of sampling variance on linkage results in a single cohort. Nonetheless, a recent meta-analysis of data from nearly 500 Crohn's families and five genomewide scans identified five top-ranking Crohn's loci: IBD1 (16q12), IBD5 (5q31), IBD3 (MHC), IBD2 (12p), and IBD6 (19p13).58
While causal variation was identified under the IBD1 and IBD5 linkage peaks (discussed below), the etiological alleles underlying the other loci have yet to be identified. This is likely because many of the remaining linkage peaks have not yet experienced the same level of scrutiny as IBD1 and IBD5. Extensive analysis of the MHC locus on 6p has been performed, but no consistent association with disease has been found, in spite of the fact that the attention given to the MHC is similar to that in other diseases where clear associations have been defined (e.g., multiple sclerosis, systemic lupus, type 1 diabetes).
Association: MHC Genes
The interest in the MHC region of 6p resulted in ∼100 association studies that examined the role of specific MHC variation in UC and Crohn's disease. Yet, with the exception of studies in Japanese UC patients, no single gene or allele has emerged as consistently associated with disease (ECW, personal communication and ref. 68). All five Japanese studies found statistically significant positive associations between UC and either DRB1*1502 or the DR2 serotype that includes this variant.63-67 However, the DRB1*1502 allele is not found in high frequency in Caucasians. This observation may suggest genetic heterogeneity between Caucasian and Japanese populations. Another possibility is that DRB1 is not causal and that the association reflects a common causal variant that is in linkage disequilibrium with different alleles in these two populations.
Association: Non-MHC Genes
CARD15
Parallel studies using positional mapping59 and candidate gene analysis60 allowed the identification of the underlying genetic variation associated with Crohn's disease at the IBD1 (16q12) locus. Strong association of three variants in CARD15 (previously known as NOD2) is observed in Crohn's patients of European descent.59,60,69-72 Two of the three variants are missense mutations (Arg702Trp and Gly908Arg) and one is a frameshift mutation resulting in early truncation of the CARD15 protein (Leu1007fsinsC). The genotype relative risk (GRR) for heterozygotes of any of the three variants is ∼2–6 fold, while homozygotes and compound heterozygotes have a GRR of >20 fold.73 These mutations are found at an appreciable frequency in European-derived Crohn's cohorts where between 30–40% of all individuals have at least one copy of one of these three variants compared with 1–7% of control individuals.74 Interestingly, these variants are very rare in the Japanese, Chinese and Korean populations, possibly explaining the decreased disease prevalence in these populations.72,75-78
CARD15 belongs to a large family of genes involved in the innate immune response.79 Members of this family are also orthologues of defense genes found in a wealth of species, including plants. Specifically, CARD proteins bear sequence similarity to plant disease resistance proteins (R proteins) that detect pathogens and initiate defense mechanisms, including MAP kinase activation, oxygen radical formation, salicylate production, induced transcription of kinases and transcription factors, and rapid cell death.80 One potential function of CARD15 is as a similar interface between pathogens and the human immune system, thus raising the possibility that Crohn's is not autoimmune per se, but rather the result of an abnormal immune response triggered by gut pathogens.
In addition to its expression in peripheral blood monocytes, CARD15 mRNA is found in primary intestinal cells,81 and specifically detected in terminal ileum Paneth cells.82 Overexpression of wild-type CARD15 in intestinal epithelial cells reduces bacterial survival, possibly serving as a key component of the innate mucosal responses to luminal bacteria, while the 3020insC truncation variant fails to exhibit such antibacterial properties.81 Interestingly, both CARD15 mRNA and protein are up-regulated by TNFα in colonic epithelial cell lines.81 Further understanding of CARD15 function may help reveal an aspect of the underlying etiology of Crohn's disease and clarify whether this disease is the result of a pathogenic immune reaction to antigens derived from the intestinal microflora.
IBD5
Substantial effort was invested in the identification of causal variation at the IBD5 locus.62 This effort represents the first successful mapping of a susceptibility locus for a complex genetic disease based on haplotype analysis. Reiterative mapping with a large number of microsatellite markers allowed the definition of a 500-kb critical region. Thorough mutation screening of the genes in the region revealed no likely causal sequence variants, so a comprehensive sequence analysis of the entire critical region was performed (eight individuals sequenced for 470 kb). In this study, 301 of the 651 single nucleotide polymorphisms (SNPs) discovered were typed in Crohn's simplex families. Analysis of these data led to the discovery of a block-like haplotype structure of the genome that was reviewed in the introduction of this chapter.17,18 A single risk haplotype (transmission ratio = 2.5:1) was identified with a frequency of 37% in controls and 75% in Crohn's patients. Current simulations show that the disease locus has a 90% probability of being within a 250-kb region where the relative risk to developing Crohn's disease is ∼2.82a SNPs that are unique to this overtransmitted haplotype have been shown to be associated with disease in four independent studies.83-86 Once this finding has been confirmed extensively through replication, the challenge is to demonstrate the functionality that is relevant for IBD pathogenesis and is perturbed in individuals bearing the mutated haplotype.
Candidate Genes
Additional association studies have examined gene candidates that were chosen based on their relevant immunological function. A small number of variants in these genes have been examined in multiple studies. For example, positive association was observed for identical variants of the DNA mismatch repair (MLH1) gene by two independent groups.87,88 Conversely, seemingly significant disease associations have been challenged by subsequent studies, including those with the CD11 gene cluster,89,90 interleukin 1 receptor antagonist (IL-1RN),91-104 IL-1B,92,96,98,103 IL-4R,90,105,106IL-10,107,108 immunoglobulin (Ig) G1 heavy chain (Gm),109,110 vitamin D receptor (VDR),111,112 and intercellular adhesion molecule-1 (ICAM-1).113-116
The association with the C3435T polymorphism in the multi-drug resistance-1 (MDR1) gene identifies important caveats for the interpretation of genetic association results, therefore we discuss it in some detail. MDR1 is an interesting candidate gene since MDR1 knockout mice spontaneously develop colitis due to an intestinal epithelial barrier dysfunction117 (Table 3). The C3435T polymorphism was first associated with UC in a German cohort,118 but four independent cohorts of German, English, Greek or North American origin119-121 could not replicate the finding (the significance of the association seen in a fifth Caucasian cohort depended on the choice of control group122). C3435T is in strong linkage disequilibrium with a second polymorphism (Ala893Ser/Thr),123 which was associated with IBD in a North American cohort.120 Therefore, some of the controversy may reflect population differences in haplotype structure at the MDR1 locus. Further studies are necessary to fully delineate the MDR1 haplotype structure and whether any variation at this locus influences risk to IBD.
Table 3
Animal models for IBD.
Preliminary associations to IBD, for which replication has not yet been reported, include NRAMP-1,124 IL-4,106 IL-11,125 IL-16,126 Factor V (Leiden mutation),127 microsomal epoxide hydrolase,128 kinin receptor β1,129 manose-binding lectin (MBL),130 mucin-3,131 epidermal growth factor receptor (EGFR),112 and NFκB.132 Preliminary studies for other genes show no association with IBD risk, including Ig superfamily 6,133 prothrombin G20210A,134 IL-12β,135 IL-25,136 interferon-γ,137 chemokine receptor 5,103,138 NRAMP-2,96 β7integrin,139 CTLA-4,140 CARD4/NOD1,141 and STAT6.90 However, only after the existing variation has been thoroughly sampled should a gene be confidently excluded as a susceptibility candidate.
Genotype–Phenotype and Genotype–Genotype Interactions
The identification of causal variation is by no means the end of the genetic investigation. Subsequent studies are necessary to determine whether specific variants preferentially influence discrete disease subphenotypes. In the case of IBD, CARD15 variants are associated with ileal disease localization,42,69,71,73,74,142-145 fibrostenosis,146,147 and fistulization.147 In addition, CARD15 variation may explain the opposite effects of smoking—which promotes Crohn's disease but prevents UC29,30 —since the risk for ileal disease was found to be increased in Crohn's disease patients with a smoking history.144
Moreover, as complex genetic diseases are thought to be the synthesis of positively and negatively acting variation, one must determine whether a causal variant influences disease independently or synergistically. For example, once identified, IBD5 and CARD15 variation could be assessed for interaction. In multiple studies, these variants seem to independently influence risk for Crohn's disease.83-86 Linkage analyses stratified on genotype have provided additional insight into genotype–genotype interactions. CARD15-stratified genomewide scans identified suggestive linkage at 6p and 10p,56 implicating specific interaction between these loci. Similarly, stratification by CARD15 and IBD5 variation together demonstrated linkage to chromosomes 3 and X.57 However, much more analysis is needed to fully understand the relationship between these two variants and disease.
Animal Models
Numerous animal models of colitis have been examined, however none precisely recapitulates the chronic and relapsing expression of IBD. These models can be classified by five categories: spontaneously occurring, induced by microbial infection, cell transfer, chemically induced, and genetically engineered models (Table 3). Each of these models gives special insight into the specific pathways that may play roles in human disease. On one hand, evidence from cell transfer models suggests that the observed inflammatory response is actively inhibited by CD4+ regulatory T-cells and immunosuppressive cytokines such as IL-10 and TGFβ1.148 Chemically induced models, on the other hand, have identified cytokines that may lessen disease symptoms. Specifically, DNBS-induced colitis can be prevented by IL-10 gene transfer149 and TNBS-induced colitis can be ameliorated by IL-4 150 or anti-IL-12 antibodies.151 Lastly, genetically engineered models have demonstrated that while disruption of both the Th1 and Th2 pathways induces colitis, there are differences in the inflammatory response that mimic the differences observed between Crohn's and UC. By example, TCRα knockout mice exhibit colitis that shares many features with UC, including dominant Th2 response in the colonic inflammation.152 Intriguingly, in many of these genetic models, inflammation did not develop if the mice were maintained in germ-free conditions, suggesting that the disease symptoms are an abnormal inflammatory response to components of the intestinal flora.
It is worth mentioning that, despite the association of CARD15 variants and human disease, mice bearing a targeted deletion of the CARD domains of this gene showed no signs of intestinal pathology.153 One possible explanation for this lack of phenotype is functional overlap with another murine CARD domain protein (NOD1) also involved in bacterial recognition. 154 , 155 Regardless, the lack of intestinal phenotype in the CARD15-deficient mice illustrates that IBD is a complex disease resulting from a combinatorial effect of multiple genetic variants and environmental factors.
Celiac Disease
Definition, Classification and Symptoms
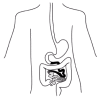
Celiac disease (also known as celiac sprue or gluten-sensitive enteropathy; MIM 212750) is a chronic gastrointestinal disease in which exposure to proteins from wheat, rye, barley and possibly oats leads to villous atrophy in the small intestine and consequent nutrient malabsorption. In wheat, such proteins are collectively known as gliadins and constitute the toxic component of gluten. Symptoms include diarrhea, general weakness, anemia and weight loss. The disease affects the mucosa of the proximal small intestine with damage gradually decreasing in severity distally (fig.2). However, in severe cases, the lesions extend to the ileum. Diagnosis of the disease is ultimately confirmed by small intestinal biopsy showing a flat mucosa that is reversed on a gluten-free diet.195
Autoimmune Features
In the past 6 years, valuable discoveries were made with respect to celiac disease mechanism; however, many questions remain. Deamidation of the gliadin component of gluten196 and its resultant aggregation in the gut is thought to be an important disease trigger.197 , 198 Deamidation is required for HLA-DQ2 and HLA-DQ8 presentation.199 - 201 Recognition of the gliadin/HLA complex by T-cells leads to, among other consequences, the production of anti-gliadin antibodies. These anti-gliadin antibodies are indicators of the disease; however, they are not detected in all celiac cases.198 Rather, the presence of autoantibodies targeting various submucosal connective tissue (endomysium) antigens is the most accurate serological marker for celiac disease.198 Recently, antibodies to tissue transglutaminase (tTG) were identified as a major component of these anti-endomysial antibodies.202 Presence of these anti-tTG specific antibodies is also an accurate diagnostic measure of disease (95–100% sensitivity; 94–97% specificity).203 - 205
Normally an intracellular enzyme, it appears tTG is released by cells upon wounding. Intriguingly, such extracellular calcium-dependent tTG was shown to be sufficient to catalyze gluten deamidation.200 Moreover, it was shown through immunoprecipitation that tTG is more abundant in gliadin complexes in the duodenal mucosa of celiac patients compared with controls.198 While unlikely to be coincidental given the serological characteristics of celiac disease, a direct connection between these observations has not yet been defined. It remains to be determined whether anti-tTG antibodies are actually causal in the flattening of the intestinal mucosa (i.e., whether celiac disease is truly autoimmune). Future studies should aim to dissect the mechanism by which gluten, tTG, and the immune system conspire to cause celiac disease.
Epidemiology: Inheritance and Environment
Using data from post-biopsy confirmed celiac cases, the estimated prevalence for celiac disease ranges from 147–3,000 per 100,000 individuals, including reports in North and South American, European, Indian, Arab, and South Asian populations (Table 1). There is a slight predominance of celiac disease in females. The risk for first-degree relatives to manifest the disease ranges from 5–20%.3 The concordance rate for HLA-identical siblings is 30%206 while that of monozygous twins is 70–86%,207 suggesting that the contribution of nonHLA risk factors in the etiology of this disease is substantial. As mentioned above, the main environmental etiological factor for celiac disease is wheat gluten.
Genetics
Linkage Studies
Genomewide searches for genetic risk factors have identified numerous putative loci (Table 4). Confirmed linkage of the MHC region (CELIAC 1) in celiac disease exists).10,208-214 In addition, significant linkage was shown for four non-MHC regions: 5q31-33 (CELIAC 2),213 2q23-33 (CELIAC 3),214 19p13 (CELIAC 4),10 and 15q12.215 These findings need to be confirmed by replication in independent data sets. Unlike for IBD, no genes have been identified for linkage studies for celiac disease.
Table 4
Summary of celiac disease genetic linkage studies.
Association: MHC Genes
Consistent with their ability to present epitopes from deamidated gluten molecules, susceptibility to celiac disease is associated primarily with HLA-DQ2 and HLA-DQ8.228 Association has also been reported with various DR serotypes, including DR3, DR5, and DR7,229-233 as well as variation in tumor necrosis factor-α (TNFα),234-238 heat-shock protein 70 (HSP70-1 and HSP70-2),239,240 inhibitor of kB-like (IKBL),241 and the MHC class I chain-related (MICA) genes.242,243 However, recent studies suggest that these variants are simply in linkage disequilibrium with the causal variation.244
Association: Non-MHC Genes
The power of a study to definitively exclude a locus of a particular strength of effect depends on two things: sample size and marker coverage. Thus, negative association studies must be interpreted cautiously since it is difficult to exclude a locus absolutely. For celiac disease, a number of genes have been reported as unassociated at various levels of statistical significance: T-cell receptors genes TCRα, TCR β, TCRγ,TCRδ,245 nitride oxide synthase (NOS),246 matrix metalloproteinase genes MMP-1 and MMP-3,247 IL-12B,248,249 interferon regulatory factor 1 (IRF1),249 insulin (INS),250 and tissue transglutaminase (TGM2).251,252 However, positive evidence for association has been observed for genes encoding Ig Gm allotypes,253 cytotoxic T-lymphocyte associated antigen 4 (CTLA-4; D2S2216,220 D2S2214,226 CT-60,254 and +49*A/G see below), MBL2,257 inducible costimulator (ICOS),255 and for microsatellite markers at locus 19p13.10 Only the +49*A/G dimorphism of CTLA-4 has been examined in numerous studies. Therefore we will restrict our discussion to this variant.
The evidence for association with +49*A is not consistent across studies,220-222,226,227,256- 260 but a meta-analysis shows modest association for CTLA-4+49*A in celiac disease.214 Importantly, variation contained in the CTLA-4 gene has been reported to confer susceptibility to many autoimmune genetic diseases, including insulin-dependent diabetes mellitus (IDDM), Grave's disease, and Hashimoto's hypothyroidism.261 However, a recent positional mapping association study of 109 polymorphisms in the 330-kb region surrounding the CTLA-4 gene strongly suggests that a yet unidentified common variant in the 6.1-kb region 3' of CTLA-4 is responsible for the association with IDDM, Grave's and autoimmune hypothyrodism.262 Moreover, these data firmly rejected +49A/G as IDDM's causal SNP,262 a result which raises the possibility that +49*A is simply linked to the causal variant in celiac disease as well.
Animal Models
Presently, there are no adequate animal models for the systemic complications of celiac disease. A model of gluten-sensitive enteropathy occurs spontaneously in a strain of Irish setter dogs.263 Few studies have used this model system to address the etiopathology of celiac disease in the past. One possible reason for this is that no linkage was seen between the enteropathy of these dogs and the canine MHC.264 Moreover, there is limited interest in developing animal models for celiac disease, which may be in part due to the fact that biological samples derived from celiac patients, such as blood and small intestine T-cells, constitute an advantageous experimental system where the major environmental component (i.e., gluten and related proteins) can be easily controlled through dieting. Nonetheless, it is most likely that gene knock-out models will be engineered as disease susceptibility-conferring gene variants are revealed, allowing for the exploration of in vivo factors that modulate intestinal permeability, mechanisms for extraintestinal alterations, interactions between gluten and other metabolic, nutritional and environmental factors involved in the disease, as well as genetically-based (i.e., pharmacogenomic) therapies.
Autoimmune Hepatitis
Definition, Classification and Symptoms
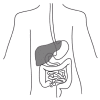
Autoimmune hepatitis (AIH) is a chronic inflammation of the liver (fig.3) for which early symptoms are fatigue, jaundice and anorexia.265 AIH accounts for 10–20% of chronic hepatitis cases in North America, but less than 4% of patients in India.266,267 AIH is diagnosed based on criteria defined by the International Autoimmune Hepatitis Group.268 A scoring system for these criteria allows the classification of cases as definite AIH or probable AIH. These criteria include the absence of infection with hepatitis viruses (i.e., exclusion of viral nucleic acids, antigens and antibodies), the presence of circulating autoantibodies (see below), hypergammaglobulinemia, and being of the female sex.
Autoimmune Features
The loss of tolerance to autologous liver tissue is the likely cause of inflammation in AIH, but the autoantibodies present in AIH patients have yet to be functionally implicated in the pathogenesis of AIH.269 In the absence of this functional knowledge, two distinct forms of AIH have been identified based on the patient's particular autoantibody set: AIH type 1 (AIH-1) and AIH type 2 (AIH-2).
AIH-1 is characterized by anti-nuclear (ANA) and anti-smooth muscle (SMA) antibodies. These patients account for 70–80% of AIH patients. Although the frequency of AIH-2 is lower (3–4%), autoimmune characteristics are better characterized for this subtype. For instance, the target of anti-liver/kidney microsome type 1 (LKM1) antibodies, which are the hallmark of AIH-2, is cytochrome P450-2D6.270 AIH-2 patients also experience an earlier onset and more aggressive course of disease, a higher prevalence of autoimmunity directed against other organs, and progress to cirrhosis more frequently.271 In addition, the serum of about 10% of these patients contains autoantibodies that detect specific UDP-glucuronosyltransferases (UGTs). A third form, AIH-3, which is clinically indistinguishable from AIH-1, was proposed based on the presence of antibodies against cytosolic liver or liver-pancreas antigens.272
Epidemiology: Inheritance and Environment
There are few epidemiological studies for AIH. Prevalence of the disease is estimated to be 4, 16.9 and 42.9 per 100,000 individuals in populations from Singapore, Norway and Alaska, respectively.11-13 The AIH-2 subtype is much less common than AIH-1 and is more frequent in southern Europe than in northern Europe, the United States or Japan.14
Various drugs and viral infections are environmental factors associated with the onset of hepatitis with autoimmune involvement (see, for example, refs. 273, 274). However, no infectious agent, metabolic defect or toxin has been determined to be a risk factor for AIH.
Genetics
Linkage Studies
Given the limited number of families with multiple members affected with AIH, no whole-genome linkage scans have been performed to date, and all genetic studies for AIH are based on case–control association analysis of candidate genes with known immunoregulatory functions.
Association: MHC Locus
Larger cohorts and more complete analysis of the variation at the MHC locus will be required to precisely identify the genetic variation that influences risk for AIH. However, some studies provide preliminary insight into the search for susceptibility loci.
MHC variants that have been associated with risk to AIH-1 include HLA-DR3, HLA-DR4, and DRB1*1301.275,276 Interestingly, the particular DR4 suballeles associated with AIH-1 appear to differ in different populations, suggesting that risk is associated with the larger DR4 superclass and not a particular allele. Genetic studies in AIH-2 are limited by its rarity and regional occurrence. However, the DRB1*07, DRB1*15, DQB1*06, and DRB1*03 alleles have been shown to be associated with risk for disease.275
Association: Non-MHC Loci
Among the potential non-MHC susceptibility factors (Table 5) are the CTLA-4 +49G allele,277,278 the VDR Fok polymorphism,279 and the CD45 tyrosine phosphatase +77C/G mutation.280 In addition, genetic variants for the heavy chain constant regions of both TCRβ281 and IgG1282 were reportedly associated with AIH. Interestingly, the association with TCRβ was strongest in patients without HLA-DR3 and DR4, and is significantly decreased in early onset cases.281 These associations remain to be confirmed in larger samples. Other studies of candidate genes, such as IL-1B, IL-1RN, and IL-10,283,284 and the autoimmune regulator AIRE,285 failed to identify an association with disease susceptibility. Caveats for the apparent lack of association in negative studies might be the limited number of samples available for study and the heterogeneity of the sample population (e.g., in the AIRE study of 85 AIH cases, 14% of individuals were seropositive for AIH-2, while the remaining 86% were diagnosed as AIH-1).
Table 5
AIH non-MHC association studies.
Animal Models
Most experimental models for AIH result from the immunization of rodents or rabbits with liver antigens in complete Freund's adjuvant (CFA), and have been recently reviewed.288-290 No model recapitulates all the features of the disease, and hepatic lesions are also observed in control animals injected with CFA. However, adoptive transfer of the disease into syngeneic recipients by splenocytes and lymph node cells from immunized animals (see, for example, ref. 291) support the autoimmune causal nature of the disease in these models. There are no published reports of experimental induction of AIH with purified cytochrome P450 IID6 or any other AIH-related autoantigen. Transgenic models allow examination of liver-specific immune responses, with the disadvantage that most develop tolerance for the transgene (reviewed in ref. 288). Proposed knockout models include TGFβ- and IL-2-deficient mice; however, although these mice develop spontaneous hepatitis with autoimmune features, various additional complications are not specific to AIH, and this phenotype is absolutely dependent on the genetic background of the BALB/c murine strain.292,293
Conclusions
Association Studies
Association studies are a powerful approach for identifying common loci of modest effect. However, positive association studies are often not replicated in subsequent data sets. This is seen for some of the studies presented in this chapter, for example, the UC association to IL-1RN for which an initial positive result failed to replicate. While it is possible that this reflects a lack of association, it is also possible that the replication studies were not appropriately powered to detect an association. Careful attention to original and replication study designs can help increase the reproducibility of results. Specific steps to improve the reliability of data include fully assessing variation at a locus, obtaining appropriate sample size given the estimated frequency and effect of the target variant, and evaluating cohort stratification, for example, by comparing allele frequencies of randomly chosen markers in suspected subpopulations. These steps will provide higher confidence in positively associated variation as well as allow unassociated loci to be more definitively excluded.
An additional challenge to association studies is extended linkage disequilibrium. This is acutely illustrated in the case of MHC studies.294-296 To surmount this obstacle it is particularly important to fully assess all variation for association with disease. Historically, the MHC has been studied by typing a handful of genes (usually the classical HLA loci, TNFα and C4). However, as seen for IBD, celiac disease and AIH, association studies using these methods generally implicate more than one allele as influencing disease susceptibility. While this may indicate a multi-gene or multi-allelic disease etiology, it may simply reflect an inability to discriminate between causal alleles and variation that is merely in linkage disequilibrium. For instance, preliminary data suggest that celiac disease associations to DR types are in fact secondary due to linkage disequilibrium with DQ.244
Recently, a preliminary integrated map of the SNP, HLA, and microsatellite variation in the MHC was reported.296 Analysis of these data showed that the haplotype structure of the MHC is no different than that of the genome as a whole, and, also, that a higher density of markers would provide a powerful resource for disease studies. In combination with larger cohort sizes, this map may help narrow associated regions through mapping ancestral recombination events. Such efforts may permit the definitive identification of causal variation in many diseases, including IBD, AIH, and celiac disease.
Functional Studies
Once a susceptibility-conferring haplotype is identified, the specific variation responsible for the association needs to be determined. As mentioned, a major obstacle in this regard is linkage disequilibrium, which makes it difficult to isolate the effect of causal variation from that of one which is simply linked. However, determination of causality is the end goal of any human complex genetic disease study. Therefore, researchers turn to functional studies to provide definitive proof.
Candidate genes should be expressed in cells that may play roles in disease etiology. For instance, IBD researchers hope to see expression in immune tissues or the gut—or in both, as is the case for CARD15. Depending on the location of the hypothesized causal variation (promoter, intronic, coding), distinct approaches are taken. If the variant is located in the gene's promoter or in a splice junction, expression levels or tissue localization patterns of specific isoforms may differ in individuals bearing the putative causal variant. If the variation is in the coding sequence, one might turn to in vitro biochemistry to determine whether the associated protein variant had different properties.
These experiments only determine that the variant of interest causes changes in gene expression or protein function; they do not elucidate the mechanism by which disease results. Animal models and in vitro disease models can be useful to bridge the gap between function and disease mechanism. For instance, if researchers can replicate disease phenotypes by “knocking-in” the human variant into a mouse model, they can be fairly certain that the variation plays a role in disease mechanism. However, an inability to show involvement in a model system may simply reflect the limitations of the model. Such a result does not rule out that the variant contributes to human disease. This is perfectly illustrated in the case of CARD15.153 The targeted disruption in the mouse homologue of the CARD15 gene shows no intestinal pathology, however the human genetic evidence (three independent mutations with compound heterozygotes conferring similar risk to homozygotes73) is conclusive. While the challenges of identifying disease-causing variation are great, determining function of those variants and establishing definitive roles in disease will likely prove even more difficult.
Future Directions
As detailed in this chapter, significant progress has been made in recent years toward understanding the etiology of IBD, celiac disease and AIH. Yet, the genetic variation that influences each disease is not fully understood. Recent accomplishments have provided the community a greater understanding of the genetics of complex disease; however, well-powered, well-phenotyped cohorts are required to further improve our knowledge of disease mechanism. These studies will likely require multi-center collaborative efforts such as those that have already begun to benefit our insight into IBD and celiac disease.
Acknowledgments
We thank Cisca Wijmenga, Leslie Gaffney, Philip De Jager, Lisa Farwell and Tracey Petryshen for critical reading of this manuscript. ECW is supported by a Cancer Research Institute Fellowship. This work was supported by NIH-R01#DK64869 (JDR).
References
- 1.
- Yang SK, Loftus JrEV, Sandborn WJ. Epidemiology of inflammatory bowel disease in Asia. Inflamm Bowel Dis. 2001;7(3):260–270. [PubMed: 11515854]
- 2.
- Yang H, Taylor KD, Rotter JI. Inflammatory bowel disease. I. Genetic epidemiology. Mol Genet Metab. 2001;74(1-2):1–21. [PubMed: 11592800]
- 3.
- Book L, Zone JJ, Neuhausen SL. Prevalence of celiac disease among relatives of sib pairs with celiac disease in U.S. families. Am J Gastroenterol. 2003;98(2):377–381. [PubMed: 12591058]
- 4.
- Beck S, Trowsdale J. The human major histocompatability complex: Lessons from the DNA sequence. Annu Rev Genomics Hum Genet. 2000;1:117–137. [PubMed: 11701627]
- 5.
- Farrokhyar F, Swarbrick ET, Irvine EJ. A critical review of epidemiological studies in inflammatory bowel disease. Scand J Gastroenterol. 2001;36(1):2–15. [PubMed: 11218235]
- 6.
- Vyse TJ, Todd JA. Genetic analysis of autoimmune disease. Cell. 1996;85(3):311–318. [PubMed: 8616887]
- 7.
- AGA technical review on celiac sprue. Gastroenterology. 2001;120(6):1526–1540. [PubMed: 11313324]
- 8.
- Fasano A, Catassi C. Current approaches to diagnosis and treatment of celiac disease: An evolving spectrum. Gastroenterology. 2001;120(3):636–651. [PubMed: 11179241]
- 9.
- Bevan S, Popat S, Braegger CP. et al. Contribution of the MHC region to the familial risk of coeliac disease. J Med Genet. 1999;36(9):687–690. [PMC free article: PMC1734425] [PubMed: 10507725]
- 10.
- Van BelzenMJ, Meijer JW, Sandkuijl LA. et al. A major nonHLA locus in celiac disease maps to chromosome 19. Gastroenterology. 2003;125(4):1032–1041. [PubMed: 14517787]
- 11.
- Boberg KM. Prevalence and epidemiology of autoimmune hepatitis. Clin Liver Dis. 2002;6(3):347–359. [PubMed: 12362572]
- 12.
- Hurlburt KJ, McMahon BJ, Deubner H. et al. Prevalence of autoimmune liver disease in Alaska natives. Am J Gastroenterol. 2002;97(9):2402–2407. [PubMed: 12358264]
- 13.
- Lee YM, Teo EK, Ng TM. et al. Autoimmune hepatitis in Singapore: A rare syndrome affecting middle-aged women. J Gastroenterol Hepatol. 2001;16(12):1384–1389. [PubMed: 11851837]
- 14.
- Feld JJ, Heathcote EJ. Epidemiology of autoimmune liver disease. J Gastroenterol Hepatol. 2003;18(10):1118–1128. [PubMed: 12974897]
- 15.
- Risch N. Assessing the role of HLA-linked and unlinked determinants of disease. Am J Hum Genet. 1987;40(1):1–14. [PMC free article: PMC1684002] [PubMed: 3468804]
- 16.
- Lander E, Kruglyak L. Genetic dissection of complex traits: Guidelines for interpreting and reporting linkage results. Nat Genet. 1995;11(3):241–247. [PubMed: 7581446]
- 17.
- Daly MJ, Rioux JD, Schaffner SF. et al. High-resolution haplotype structure in the human genome. Nat Genet. 2001;29(2):229–232. [PubMed: 11586305]
- 18.
- Gabriel SB, Schaffner SF, Nguyen H. et al. The structure of haplotype blocks in the human genome. Science. 2002;296(5576):2225–2229. [PubMed: 12029063]
- 19.
- Jeffreys AJ, Kauppi L, Neumann R. Intensely punctate meiotic recombination in the class II region of the major histocompatibility complex. Nat Genet. 2001;29(2):217–222. [PubMed: 11586303]
- 20.
- Gibbs RA, Belmont JW, Hardenbol P. et al. The international HapMap project. Nature. 2003;426(6968):789–796. [PubMed: 14685227]
- 21.
- Kornbluth A, Sachar DB, Salomon P. Crohn's disease In: Feldman M, Friedman LS, Sleisenger MH, eds.Sleisenger and Fordtran's Gastrointestinal and Liver Disease 6th ed. Philadelphia: WB Saunders,199821708–1734.
- 22.
- Jewell DP. Ulcerative colitis In: Feldman M, Friedman LS, Sleisenger MH, eds.Sleisenger and Fordtran's Gastrointestinal and Liver Disease 6th ed. Philadelphia: WB Saunders,199821735–1761.
- 23.
- Joossens S, Reinisch W, Vermeire S. et al. The value of serologic markers in indeterminate colitis: A prospective follow-up study. Gastroenterology. 2002;122(5):1242–1247. [PubMed: 11984510]
- 24.
- Moum B, Ekbom A, Vatn MH. et al. Inflammatory bowel disease: Reevaluation of the diagnosis in a prospective population based study in south eastern Norway. Gut. 1997;40(3):328–332. [PMC free article: PMC1027081] [PubMed: 9135520]
- 25.
- Reumaux D, Sendid B, Poulain D. et al. Serological markers in inflammatory bowel diseases. Best Pract Res Clin Gastroenterol. 2003;17(1):19–35. [PubMed: 12617880]
- 26.
- Binder V. Genetic epidemiology in inflammatory bowel disease. Dig Dis. 1998;16(6):351–355. [PubMed: 10207221]
- 27.
- Orholm M, Binder V, Sorensen TI. et al. Concordance of inflammatory bowel disease among Danish twins. Results of a nationwide study. Scand J Gastroenterol. 2000;35(10):1075–1081. [PubMed: 11099061]
- 28.
- Halfvarson J, Bodin L, Tysk C. et al. Inflammatory bowel disease in a Swedish twin cohort: A long-term follow-up of concordance and clinical characteristics. Gastroenterology. 2003;124(7):1767–1773. [PubMed: 12806610]
- 29.
- Cottone M, Rosselli M, Orlando A. et al. Smoking habits and recurrence in Crohn's disease. Gastroenterology. 1994;106(3):643–648. [PubMed: 8119535]
- 30.
- Reif S, Lavy A, Keter D. et al. Lack of association between smoking and Crohn's disease but the usual association with ulcerative colitis in Jewish patients in Israel: A multicenter study. Am J Gastroenterol. 2000;95(2):474–478. [PubMed: 10685753]
- 31.
- Madretsma S, Wolters LM, van Dijk JP. et al. In-vivo effect of nicotine on cytokine production by human nonadherent mononuclear cells. Eur J Gastroenterol Hepatol. 1996;8(10):1017–1020. [PubMed: 8930570]
- 32.
- Pullan RD. Colonic mucus, smoking and ulcerative colitis. Ann R Coll Surg Engl. 1996;78(2):85–91. [PMC free article: PMC2502541] [PubMed: 8678464]
- 33.
- Finnie IA, Campbell BJ, Taylor BA. et al. Stimulation of colonic mucin synthesis by corticosteroids and nicotine. Clin Sci (Lond). 1996;91(3):359–364. [PubMed: 8869420]
- 34.
- Van DijkJP, Madretsma GS, Keuskamp ZJ. et al. Nicotine inhibits cytokine synthesis by mouse colonic mucosa. Eur J Pharmacol. 1995;278(1):R11–12. [PubMed: 7664807]
- 35.
- Green JT, Richardson C, Marshall RW. et al. Nitric oxide mediates a therapeutic effect of nicotine in ulcerative colitis. Aliment Pharmacol Ther. 2000;14(11):1429–1434. [PubMed: 11069313]
- 36.
- Wolf JM, Lashner BA. Inflammatory bowel disease: Sorting out the treatment options. Cleve Clin J Med. 2002;69(8):621–626 629-631. [PubMed: 12184470]
- 37.
- Katschinski B, Logan RF, Edmond M. et al. Smoking and sugar intake are separate but interactive risk factors in Crohn's disease. Gut. 1988;29(9):1202–1206. [PMC free article: PMC1434365] [PubMed: 3197994]
- 38.
- Persson PG, Ahlbom A, Hellers G. Diet and inflammatory bowel disease: A case-control study. Epidemiology. 1992;3(1):47–52. [PubMed: 1313310]
- 39.
- Ainley C, Cason J, Slavin BM. et al. The influence of zinc status and malnutrition on immunological function in Crohn's disease. Gastroenterology. 1991;100(6):1616–1625. [PubMed: 1902189]
- 40.
- Kanauchi O, Mitsuyama K, Araki Y. et al. Modification of intestinal flora in the treatment of inflammatory bowel disease. Curr Pharm Des. 2003;9(4):333–346. [PubMed: 12570821]
- 41.
- Hugot JP, Laurent-Puig P, Gower-Rousseau C. et al. Mapping of a susceptibility locus for Crohn's disease on chromosome 16. Nature. 1996;379(6568):821–823. [PubMed: 8587604]
- 42.
- Cavanaugh J. International collaboration provides convincing linkage replication in complex disease through analysis of a large pooled data set: Crohn disease and chromosome 16. Am J Hum Genet. 2001;68(5):1165–1171. [PMC free article: PMC1226097] [PubMed: 11309682]
- 43.
- Satsangi J, Parkes M, Louis E. et al. Two stage genome-wide search in inflammatory bowel disease provides evidence for susceptibility loci on chromosomes 3, 7 and 12. Nat Genet. 1996;14(2):199–202. [PubMed: 8841195]
- 44.
- Cho JH, Nicolae DL, Gold LH. et al. Identification of novel susceptibility loci for inflammatory bowel disease on chromosomes 1p, 3q, and 4q: Evidence for epistasis between 1p and IBD1. Proc Natl Acad Sci USA. 1998;95(13):7502–7507. [PMC free article: PMC22666] [PubMed: 9636179]
- 45.
- Ma Y, Ohmen JD, Li Z. et al. A genome-wide search identifies potential new susceptibility loci for Crohn's disease. Inflamm Bowel Dis. 1999;5(4):271–278. [PubMed: 10579120]
- 46.
- Hampe J, Schreiber S, Shaw SH. et al. A genomewide analysis provides evidence for novel linkages in inflammatory bowel disease in a large European cohort. Am J Hum Genet. 1999;64(3):808–816. [PMC free article: PMC1377799] [PubMed: 10053016]
- 47.
- Duerr RH, Barmada MM, Zhang L. et al. Linkage and association between inflammatory bowel disease and a locus on chromosome 12. Am J Hum Genet. 1998;63(1):95–100. [PMC free article: PMC1377250] [PubMed: 9634527]
- 48.
- Rioux JD, Silverberg MS, Daly MJ. et al. Genomewide search in Canadian families with inflammatory bowel disease reveals two novel susceptibility loci. Am J Hum Genet. 2000;66:1863–1870. [PMC free article: PMC1378042] [PubMed: 10777714]
- 49.
- Cavanaugh JA, Callen DF, Wilson SR. et al. Analysis of Australian Crohn's disease pedigrees refines the localization for susceptibility to inflammatory bowel disease on chromosome 16. Ann Hum Genet. 1998;62(Pt 4):291–298. [PubMed: 9924607]
- 50.
- Brant SR, Fu Y, Fields CT. et al. American families with Crohn's disease have strong evidence for linkage to chromosome 16 but not chromosome 12. Gastroenterology. 1998;115(5):1056–1061. [PubMed: 9797357]
- 51.
- Rioux JD, Daly MJ, Green T. et al. Absence of linkage between inflammatory bowel disease and selected loci on chromosomes 3, 7, 12, and 16. Gastroenterology. 1998;115(5):1062–1065. [PubMed: 9797358]
- 52.
- Annese V, Latiano A, Bovio P. et al. Genetic analysis in Italian families with inflammatory bowel disease supports linkage to the IBD1 locus-a GISC study. Eur J Hum Genet. 1999;7(5):567–573. [PubMed: 10439963]
- 53.
- Vermeire S, Peeters M, Vlietinck R. et al. Exclusion of linkage of Crohn's disease to previously reported regions on chromosomes 12, 7, and 3 in the Belgian population indicates genetic heterogeneity. Inflamm Bowel Dis. 2000;6(3):165–170. [PubMed: 10961588]
- 54.
- Hampe J, Lynch NJ, Daniels S. et al. Fine mapping of the chromosome 3p susceptibility locus in inflammatory bowel disease. Gut. 2001;48(2):191–197. [PMC free article: PMC1728207] [PubMed: 11156639]
- 55.
- Paavola P, Helio T, Kiuru M. et al. Genetic analysis in Finnish families with inflammatory bowel disease supports linkage to chromosome 3p21. Eur J Hum Genet. 2001;9(5):328–334. [PubMed: 11378820]
- 56.
- Shaw SH, Hampe J, White R. et al. Stratification by CARD15 variant genotype in a genome-wide search for inflammatory bowel disease susceptibility loci. Hum Genet. 2003;113(6):514–521. [PubMed: 13680363]
- 57.
- Van HeelDA, Dechairo BM, Dawson G. et al. The IBD6 Crohn's disease locus demonstrates complex interactions with CARD15 and IBD5 disease-associated variants. Hum Mol Genet. 2003;12(20):2569–2575. [PubMed: 12928481]
- 58.
- Williams CN, Kocher K, Lander ES. et al. Using a genome-wide scan and meta-analysis to identify a novel IBD locus and confirm previously identified IBD loci. Inflamm Bowel Dis. 2002;8(6):375–381. [PubMed: 12454612]
- 59.
- Hugot JP, Chamaillard M, Zouali H. et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. 2001;411(6837):599–603. [PubMed: 11385576]
- 60.
- Ogura Y, Bonen DK, Inohara N. et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 2001;411(6837):603–606. [PubMed: 11385577]
- 61.
- Duerr RH, Barmada MM, Zhang L. et al. High-density genome scan in Crohn disease shows confirmed linkage to chromosome 14q11-12. Am J Hum Genet. 2000;66(6):1857–1862. [PMC free article: PMC1378032] [PubMed: 10747815]
- 62.
- Rioux JD, Daly MJ, Silverberg MS. et al. Genetic variation in the 5q31 cytokine gene cluster confers susceptibility to Crohn disease. Nat Genet. 2001;29(2):223–228. [PubMed: 11586304]
- 63.
- Kobayashi K, Atoh M, Konoeda Y. et al. HLA-DR, DQ and T cell antigen receptor constant beta genes in Japanese patients with ulcerative colitis. Clin Exp Immunol. 1990;80(3):400–403. [PMC free article: PMC1535193] [PubMed: 1973647]
- 64.
- Asakura H, Tsuchiya M, Aiso S. et al. Association of the human lymphocyte-DR2 antigen with Japanese ulcerative colitis. Gastroenterology. 1982;82(3):413–418. [PubMed: 6947923]
- 65.
- Sugimura K, Asakura H, Mizuki N. et al. Analysis of genes within the HLA region affecting susceptibility to ulcerative colitis. Hum Immunol. 1993;36(2):112–118. [PubMed: 8096500]
- 66.
- Futami S, Aoyama N, Honsako Y. et al. HLA-DRB1*1502 allele, subtype of DR15, is associated with susceptibility to ulcerative colitis and its progression. Dig Dis Sci. 1995;40(4):814–818. [PubMed: 7720475]
- 67.
- Yoshitake S, Kimura A, Okada M. et al. HLA class II alleles in Japanese patients with inflammatory bowel disease. Tissue Antigens. 1999;53(4 Pt 1):350–358. [PubMed: 10323339]
- 68.
- Stokkers PC, Reitsma PH, Tytgat GN. et al. HLA-DR and -DQ phenotypes in inflammatory bowel disease: A meta-analysis. Gut. 1999;45(3):395–401. [PMC free article: PMC1727649] [PubMed: 10446108]
- 69.
- Hampe J, Cuthbert A, Croucher PJ. et al. Association between insertion mutation in NOD2 gene and Crohn's disease in German and British populations. Lancet. 2001;357(9272):1925–1928. [PubMed: 11425413]
- 70.
- Lesage S, Zouali H, Cezard JP. et al. CARD15/NOD2 mutational analysis and genotype-phenotype correlation in 612 patients with inflammatory bowel disease. Am J Hum Genet. 2002;70(4):845–857. [PMC free article: PMC379113] [PubMed: 11875755]
- 71.
- Vermeire S, Wild G, Kocher K. et al. CARD15 genetic variation in a Quebec population: Prevalence, genotype-phenotype relationship, and haplotype structure. Am J Hum Genet. 2002;71(1):74–83. [PMC free article: PMC384994] [PubMed: 12019468]
- 72.
- Croucher PJ, Mascheretti S, Hampe J. et al. Haplotype structure and association to Crohn's disease of CARD15 mutations in two ethnically divergent populations. Eur J Hum Genet. 2003;11(1):6–16. [PubMed: 12529700]
- 73.
- Cuthbert AP, Fisher SA, Mirza MM. et al. The contribution of NOD2 gene mutations to the risk and site of disease in inflammatory bowel disease. Gastroenterology. 2002;122(4):867–874. [PubMed: 11910337]
- 74.
- Helio T, Halme L, Lappalainen M. et al. CARD15/NOD2 gene variants are associated with familially occurring and complicated forms of Crohn's disease. Gut. 2003;52(4):558–562. [PMC free article: PMC1773614] [PubMed: 12631669]
- 75.
- Inoue N, Tamura K, Kinouchi Y. et al. Lack of common NOD2 variants in Japanese patients with Crohn's disease. Gastroenterology. 2002;123(1):86–91. [PubMed: 12105836]
- 76.
- Yamazaki K, Takazoe M, Tanaka T. et al. Absence of mutation in the NOD2/CARD15 gene among 483 Japanese patients with Crohn's disease. J Hum Genet. 2002;47(9):469–472. [PubMed: 12202985]
- 77.
- Marsh S, McLeod HL. Crohn's disease: Ethnic variation in CARD15 genotypes. Gut. 2003;52(5):770. [PMC free article: PMC1773649] [PubMed: 12692072]
- 78.
- Leong RW, Armuzzi A, Ahmad T. et al. NOD2/CARD15 gene polymorphisms and Crohn's disease in the Chinese population. Aliment Pharmacol Ther. 2003;17(12):1465–1470. [PubMed: 12823148]
- 79.
- Inohara N, Nunez G. NODs: Intracellular proteins involved in inflammation and apoptosis. Nat Rev Immunol. 2003;3(5):371–382. [PubMed: 12766759]
- 80.
- Dangl JL, Jones JD. Plant pathogens and integrated defence responses to infection. Nature. 2001;411(6839):826–833. [PubMed: 11459065]
- 81.
- Hisamatsu T, Suzuki M, Reinecker HC. et al. CARD15/NOD2 functions as an antibacterial factor in human intestinal epithelial cells. Gastroenterology. 2003;124(4):993–1000. [PubMed: 12671896]
- 82.
- Ogura Y, Lala S, Xin W. et al. Expression of NOD2 in Paneth cells: A possible link to Crohn's ileitis. Gut. 2003;52(11):1591–1597. [PMC free article: PMC1773866] [PubMed: 14570728]
- 82a.
- Daly MJ, Rioux JD. New approaches to gene hunting in IBD. Inflamm Bowel Dis. 2004;10(3):312–317. [PubMed: 15290928]
- 83.
- Giallourakis C, Stoll M, Miller K. et al. IBD5 is a general risk factor for inflammatory bowel disease: Replication of association with Crohn disease and identification of a novel association with ulcerative colitis. Am J Hum Genet. 2003;73(1):205–211. [PMC free article: PMC1180582] [PubMed: 12776251]
- 84.
- Negoro K, McGovern DP, Kinouchi Y. et al. Analysis of the IBD5 locus and potential gene-gene interactions in Crohn's disease. Gut. 2003;52(4):541–546. [PMC free article: PMC1773608] [PubMed: 12631666]
- 85.
- Mirza MM, Fisher SA, King K. et al. Genetic evidence for interaction of the 5q31 cytokine locus and the CARD15 gene in Crohn disease. Am J Hum Genet. 2003;72(4):1018–1022. [PMC free article: PMC1180331] [PubMed: 12618963]
- 86.
- Armuzzi A, Ahmad T, Ling KL. et al. Genotype-phenotype analysis of the Crohn's disease susceptibility haplotype on chromosome 5q31. Gut. 2003;52(8):1133–1139. [PMC free article: PMC1773736] [PubMed: 12865271]
- 87.
- Pokorny RM, Hofmeister A, Galandiuk S. et al. Crohn's disease and ulcerative colitis are associated with the DNA repair gene MLH1. Ann Surg. 1997;225(6):718–723 discussion 723-715. [PMC free article: PMC1190876] [PubMed: 9230812]
- 88.
- Annese V, Piepoli A, Andriulli A. et al. Association of Crohn's disease and ulcerative colitis with haplotypes of the MLH1 gene in Italian inflammatory bowel disease patients. J Med Genet. 2002;39(5):332–334. [PMC free article: PMC1735109] [PubMed: 12011151]
- 89.
- Frenzel H, Hampe J, Huse K. et al. Mutation detection and physical mapping of the CD11 gene cluster in association with inflammatory bowel disease. Immunogenetics. 2002;53(10-11):835–842. [PubMed: 11862384]
- 90.
- de JongDJ, Franke B, Naber AH. et al. No evidence for involvement of IL-4R and CD11B from the IBD1 region and STAT6 in the IBD2 region in Crohn's disease. Eur J Hum Genet. 2003;11(11):884–887. [PubMed: 14571275]
- 91.
- Mansfield JC, Holden H, Tarlow JK. et al. Novel genetic association between ulcerative colitis and the anti-inflammatory cytokine interleukin-1 receptor antagonist. Gastroenterology. 1994;106(3):637–642. [PubMed: 8119534]
- 92.
- Bioque G, Crusius JB, Koutroubakis I. et al. Allelic polymorphism in IL-1 beta and IL-1 receptor antagonist (IL-1Ra) genes in inflammatory bowel disease. Clin Exp Immunol. 1995;102(2):379–383. [PMC free article: PMC1553429] [PubMed: 7586694]
- 93.
- Roussomoustakaki M, Satsangi J, Welsh K. et al. Genetic markers may predict disease behavior in patients with ulcerative colitis. Gastroenterology. 1997;112(6):1845–1853. [PubMed: 9178675]
- 94.
- Heresbach D, Alizadeh M, Dabadie A. et al. Significance of interleukin-1beta and interleukin-1 receptor antagonist genetic polymorphism in inflammatory bowel diseases. Am J Gastroenterol. 1997;92(7):1164–1169. [PubMed: 9219791]
- 95.
- Hacker UT, Bidlingmaier C, Gomolka M. et al. Inflammatory bowel disease: No association between allele combinations of the interleukin (IL) I beta and IL-I receptor antagonist gene polymorphisms. Eur J Clin Invest. 1998;28(3):214–219. [PubMed: 9568467]
- 96.
- Stokkers PC, van AkenBE, Basoski N. et al. Five genetic markers in the interleukin 1 family in relation to inflammatory bowel disease. Gut. 1998;43(1):33–39. [PMC free article: PMC1727194] [PubMed: 9771403]
- 97.
- Gonzalez SarmientoR, Araoz P, Rodriguez R. et al. Polymorphism of the IL1RN gene in Spanish patients with ulcerative colitis. Med Clin (Barc). 1999;112(20):778–779. [PubMed: 10422059]
- 98.
- Nemetz A, Kope A, Molnar T. et al. Significant differences in the interleukin-1beta and interleukin-1 receptor antagonist gene polymorphisms in a Hungarian population with inflammatory bowel disease. Scand J Gastroenterol. 1999;34(2):175–179. [PubMed: 10192196]
- 99.
- Bouma G, Crusius JB, Garcia-Gonzalez MA. et al. Genetic markers in clinically well defined patients with ulcerative colitis (UC). Clin Exp Immunol. 1999;115(2):294–300. [PMC free article: PMC1905167] [PubMed: 9933456]
- 100.
- Tountas NA, Casini-Raggi V, Yang H. et al. Functional and ethnic association of allele 2 of the interleukin-1 receptor antagonist gene in ulcerative colitis. Gastroenterology. 1999;117(4):806–813. [PubMed: 10500062]
- 101.
- Papo M, Quer JC, Gutierrez C. et al. Genetic heterogeneity within ulcerative colitis determined by an interleukin-1 receptor antagonist gene polymorphism and antineutrophil cytoplasmic antibodies. Eur J Gastroenterol Hepatol. 1999;11(4):413–420. [PubMed: 10321759]
- 102.
- Carter MJ, di GiovineFS, Jones S. et al. Association of the interleukin 1 receptor antagonist gene with ulcerative colitis in Northern European Caucasians. Gut. 2001;48(4):461–467. [PMC free article: PMC1728235] [PubMed: 11247888]
- 103.
- Craggs A, Welfare M, Donaldson PT. et al. The CC chemokine receptor 5 delta32 mutation is not associated with inflammatory bowel disease (IBD) in NE England. Genes Immun. 2001;2(2):114–116. [PubMed: 11393656]
- 104.
- Nohara H, Saito Y, Higaki S. et al. Polymorphisms of the IL-1beta and IL-1beta-inducible genes in ulcerative colitis. J Gastroenterol. 2002;37(Suppl 14):107–110. [PubMed: 12572877]
- 105.
- Olavesen MG, Hampe J, Mirza MM. et al. Analysis of single-nucleotide polymorphisms in the interleukin-4 receptor gene for association with inflammatory bowel disease. Immunogenetics. 2000;51(1):1–7. [PubMed: 10663555]
- 106.
- Aithal GP, Day CP, Leathart J. et al. Association of single nucleotide polymorphisms in the interleukin-4 gene and interleukin-4 receptor gene with Crohn's disease in a British population. Genes Immun. 2001;2(1):44–47. [PubMed: 11294568]
- 107.
- Tagore A, Gonsalkorale WM, Pravica V. et al. Interleukin-10 (IL-10) genotypes in inflammatory bowel disease. Tissue Antigens. 1999;54(4):386–390. [PubMed: 10551422]
- 108.
- Mitchell SA, Grove J, Spurkland A. et al. Association of the tumour necrosis factor alpha -308 but not the interleukin 10 -627 promoter polymorphism with genetic susceptibility to primary sclerosing cholangitis. Gut. 2001;49(2):288–294. [PMC free article: PMC1728404] [PubMed: 11454808]
- 109.
- Kagnoff MF, Brown RJ, Schanfield MS. Association between Crohn's disease and immunoglobulin heavy chain (Gm) allotypes. Gastroenterology. 1983;85(5):1044–1047. [PubMed: 6578156]
- 110.
- Gudjonsson H, Schanfield MS, Albertini RJ. et al. Association and linkage studies of immunoglobulin heavy chain allotypes in inflammatory bowel disease. Tissue Antigens. 1988;31(5):243–249. [PubMed: 3135639]
- 111.
- Simmons JD, Mullighan C, Welsh KI. et al. Vitamin D receptor gene polymorphism: Association with Crohn's disease susceptibility. Gut. 2000;47(2):211–214. [PMC free article: PMC1728007] [PubMed: 10896912]
- 112.
- Martin K, Radlmayr M, Borchers R. et al. Candidate genes colocalized to linkage regions in inflammatory bowel disease. Digestion. 2002;66(2):121–126. [PubMed: 12428072]
- 113.
- Yang H, Vora DK, Targan SR. et al. Intercellular adhesion molecule 1 gene associations with immunologic subsets of inflammatory bowel disease. Gastroenterology. 1995;109(2):440–448. [PubMed: 7615193]
- 114.
- Braun C, Zahn R, Martin K. et al. Polymorphisms of the ICAM-1 gene are associated with inflammatory bowel disease, regardless of the p-ANCA status. Clin Immunol. 2001;101(3):357–360. [PubMed: 11726228]
- 115.
- Matsuzawa J, Sugimura K, Matsuda Y. et al. Association between K469E allele of intercellular adhesion molecule 1 gene and inflammatory bowel disease in a Japanese population. Gut. 2003;52(1):75–78. [PMC free article: PMC1773528] [PubMed: 12477764]
- 116.
- Papa A, Danese S, Armuzzi A. et al. Association between K469E allele of intercellular adhesion molecule 1 gene and inflammatory bowel disease in different populations. Gut. 2003;52(8):1227– 1228 author reply 1228. [PMC free article: PMC1773734] [PubMed: 12865290]
- 117.
- Panwala CM, Jones JC, Viney JL. A novel model of inflammatory bowel disease: Mice deficient for the multiple drug resistance gene, mdr1a, spontaneously develop colitis. J Immunol. 1998;161(10):5733–5744. [PubMed: 9820555]
- 118.
- Schwab M, Schaeffeler E, Marx C. et al. Association between the C3435T MDR1 gene polymorphism and susceptibility for ulcerative colitis. Gastroenterology. 2003;124(1):26–33. [PubMed: 12512026]
- 119.
- Croucher PJ, Mascheretti S, Foelsch UR. et al. Lack of association between the C3435T MDR1 gene polymorphism and inflammatory bowel disease in two independent Northern European populations. Gastroenterology. 2003;125(6):1919–1920 author reply 1920-1921. [PubMed: 14727637]
- 120.
- Brant SR, Panhuysen CI, Nicolae D. et al. MDR1 Ala893 polymorphism is associated with inflammatory bowel disease. Am J Hum Genet. 2003;73(6):1282–1292. [PMC free article: PMC1180394] [PubMed: 14610718]
- 121.
- Gazouli M, Zacharatos P, Gorgoulis P. The C3435T MDR1gene polymorphism is not associated with susceptibility for ulcerative colitis in a Greek population. Gastroenterology. 2004;126(1):367–369. [PubMed: 14755848]
- 122.
- Glas J, Torok HP, Schiemann U. et al. MDR1 gene polymorphism in ulcerative colitis. Gastroenterology. 2004;126(1):367. [PubMed: 14724799]
- 123.
- Kim RB, Leake BF, Choo EF. et al. Identification of functionally variant MDR1 alleles among European Americans and African Americans. Clin Pharmacol Ther. 2001;70(2):189–199. [PubMed: 11503014]
- 124.
- Hofmeister A, Neibergs HL, Pokorny RM. et al. The natural resistance-associated macrophage protein gene is associated with Crohn's disease. Surgery. 1997;122(2):173–178 discussion 178-179. [PubMed: 9288120]
- 125.
- Klein W, Tromm A, Griga T. et al. A polymorphism in the IL11 gene is associated with ulcerative colitis. Genes Immun. 2002;3(8):494–496. [PubMed: 12486609]
- 126.
- Glas J, Torok HP, Unterhuber H. et al. The -295T-to-C promoter polymorphism of the IL-16 gene is associated with Crohn's disease. Clin Immunol. 2003;106(3):197–200. [PubMed: 12706406]
- 127.
- Haslam N, Standen GR, Probert CS. An investigation of the association of the factor V Leiden mutation and inflammatory bowel disease. Eur J Gastroenterol Hepatol. 1999;11(11):1289–1291. [PubMed: 10563542]
- 128.
- de Jong DJ, van der Logt EM, van Schaik A. et al. Genetic polymorphisms in biotransformation enzymes in Crohn's disease: Association with microsomal epoxide hydrolase. Gut. 2003;52(4):547–551. [PMC free article: PMC1773587] [PubMed: 12631667]
- 129.
- Bachvarov DR, Landry M, Houle S. et al. Altered frequency of a promoter polymorphic allele of the kinin B1 receptor gene in inflammatory bowel disease. Gastroenterology. 1998;115(5):1045–1048. [PubMed: 9797354]
- 130.
- Rector A, Lemey P, Laffut W. et al. Mannan-binding lectin (MBL) gene polymorphisms in ulcerative colitis and Crohn's disease. Genes Immun. 2001;2(6):323–328. [PubMed: 11607788]
- 131.
- Kyo K, Muto T, Nagawa H. et al. Associations of distinct variants of the intestinal mucin gene MUC3A with ulcerative colitis and Crohn's disease. J Hum Genet. 2001;46(1):5–20. [PubMed: 11289722]
- 132.
- Karban AS, Okazaki T, Panhuysen CI. et al. Functional annotation of a novel NFKB1 promoter polymorphism that increases risk for ulcerative colitis. Hum Mol Genet. 2004;13(1):35–45. [PubMed: 14613970]
- 133.
- King K, Moody A, Fisher SA. et al. Genetic variation in the IGSF6 gene and lack of association with inflammatory bowel disease. Eur J Immunogenet. 2003;30(3):187–190. [PubMed: 12786995]
- 134.
- Haslam N, Standen GR, Probert CS. An investigation of the association of the prothrombin G20210A gene mutation and inflammatory bowel disease: Factor II and IBD. Inflamm Bowel Dis. 2001;7(2):133–135. [PubMed: 11383586]
- 135.
- Seegers D, Zwiers A, Strober W. et al. A TaqI polymorphism in the 3'UTR of the s gene correlates with increased IL-12 secretion. Genes Immun. 2002;3(7):419–423. [PubMed: 12424624]
- 136.
- Buning C, Genschel J, Weltrich R. et al. The interleukin-25 gene located in the inflammatory bowel disease (IBD) 4 region: No association with inflammatory bowel disease. Eur J Immunogenet. 2003;30(5):329–333. [PubMed: 14641539]
- 137.
- Hampe J, Hermann B, Bridger S. et al. The interferon-gamma gene as a positional and functional candidate gene for inflammatory bowel disease. Int J Colorectal Dis. 1998;13(5-6):260–263. [PubMed: 9870173]
- 138.
- Rector A, Vermeire S, Thoelen I. et al. Analysis of the CC chemokine receptor 5 (CCR5) delta-32 polymorphism in inflammatory bowel disease. Hum Genet. 2001;108(3):190–193. [PubMed: 11354628]
- 139.
- Van HeelDA, Carey AH, Jewell DP. Identification of novel polymorphisms in the beta7 integrin gene: Family-based association studies in inflammatory bowel disease. Genes Immun. 2001;2(8):455–460. [PubMed: 11781713]
- 140.
- Xia B, Crusius JB, Wu J. et al. CTLA4 gene polymorphisms in Dutch and Chinese patients with inflammatory bowel disease. Scand J Gastroenterol. 2002;37(11):1296–1300. [PubMed: 12465728]
- 141.
- Zouali H, Lesage S, Merlin F. et al. CARD4/NOD1 is not involved in inflammatory bowel disease. Gut. 2003;52(1):71–74. [PMC free article: PMC1773516] [PubMed: 12477763]
- 142.
- Murillo L, Crusius JB, van Bodegraven AA. et al. CARD15 gene and the classification of Crohn's disease. Immunogenetics. 2002;54(1):59–61. [PubMed: 11976792]
- 143.
- Bairead E, Harmon DL, Curtis AM. et al. Association of NOD2 with Crohn's disease in a homogenous Irish population. Eur J Hum Genet. 2003;11(3):237–244. [PubMed: 12673278]
- 144.
- Brant SR, Picco MF, Achkar JP. et al. Defining complex contributions of NOD2/CARD15 gene mutations, age at onset, and tobacco use on Crohn's disease phenotypes. Inflamm Bowel Dis. 2003;9(5):281–289. [PubMed: 14555911]
- 145.
- Tomer G, Ceballos C, Concepcion E. et al. NOD2/CARD15 variants are associated with lower weight at diagnosis in children with Crohn's disease. Am J Gastroenterol. 2003;98(11):2479–2484. [PubMed: 14638352]
- 146.
- Abreu MT, Taylor KD, Lin YC. et al. Mutations in NOD2 are associated with fibrostenosing disease in patients with Crohn's disease. Gastroenterology. 2002;123(3):679–688. [PubMed: 12198692]
- 147.
- Radlmayr M, Torok HP, Martin K. et al. The c-insertion mutation of the NOD2 gene is associated with fistulizing and fibrostenotic phenotypes in Crohn's disease. Gastroenterology. 2002;122(7):2091–2092. [PubMed: 12055616]
- 148.
- Strober W, Fuss IJ, Blumberg RS. The immunology of mucosal models of inflammation. Annu Rev Immunol. 2002;20:495–549. [PubMed: 11861611]
- 149.
- Barbara G, Xing Z, Hogaboam CM. et al. Interleukin 10 gene transfer prevents experimental colitis in rats. Gut. 2000;46(3):344–349. [PMC free article: PMC1727851] [PubMed: 10673295]
- 150.
- Hogaboam CM, Vallance BA, Kumar A. et al. Therapeutic effects of interleukin-4 gene transfer in experimental inflammatory bowel disease. J Clin Invest. 1997;100(11):2766–2776. [PMC free article: PMC508481] [PubMed: 9389741]
- 151.
- Neurath MF, Fuss I, Kelsall BL. et al. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J Exp Med. 1995;182(5):1281–1290. [PMC free article: PMC2192205] [PubMed: 7595199]
- 152.
- Mombaerts P, Mizoguchi E, Grusby MJ. et al. Spontaneous development of inflammatory bowel disease in T cell receptor mutant mice. Cell. 1993;75(2):274–282. [PubMed: 8104709]
- 153.
- Pauleau AL, Murray PJ. Role of nod2 in the response of macrophages to toll-like receptor agonists. Mol Cell Biol. 2003;23(21):7531–7539. [PMC free article: PMC207570] [PubMed: 14560001]
- 154.
- Chamaillard M, Hashimoto M, Horie Y. et al. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat Immunol. 2003;4(7):702–707. [PubMed: 12796777]
- 155.
- Girardin SE, Boneca IG, Carneiro LA. et al. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science. 2003;300(5625):1584–1587. [PubMed: 12791997]
- 156.
- Madara JL, Podolsky DK, King NW. et al. Characterization of spontaneous colitis in cotton-top tamarins (Saguinus oedipus) and its response to sulfasalazine. Gastroenterology. 1985;88(1 Pt 1):13–19. [PubMed: 2856876]
- 157.
- Sundberg JP, Elson CO, Bedigian H. et al. Spontaneous, heritable colitis in a new substrain of C3H/HeJ mice. Gastroenterology. 1994;107(6):1726–1735. [PubMed: 7958684]
- 158.
- Matsumoto S, Okabe Y, Setoyama H. et al. Inflammatory bowel disease-like enteritis and caecitis in a senescence accelerated mouse P1/Yit strain. Gut. 1998;43(1):71–78. [PMC free article: PMC1727165] [PubMed: 9771408]
- 159.
- Kosiewicz MM, Nast CC, Krishnan A. et al. Th1-type responses mediate spontaneous ileitis in a novel murine model of Crohn's disease. J Clin Invest. 2001;107(6):695–702. [PMC free article: PMC208944] [PubMed: 11254669]
- 160.
- Morris GP, Beck PL, Herridge MS. et al. Hapten-induced model of chronic inflammation and ulceration in the rat colon. Gastroenterology. 1989;96(3):795–803. [PubMed: 2914642]
- 161.
- Boirivant M, Fuss IJ, Chu A. et al. Oxazolone colitis: A murine model of T helper cell type 2 colitis treatable with antibodies to interleukin 4. J Exp Med. 1998;188(10):1929–1939. [PMC free article: PMC2212414] [PubMed: 9815270]
- 162.
- MacPherson BR, Pfeiffer CJ. Experimental production of diffuse colitis in rats. Digestion. 1978;17(2):135–150. [PubMed: 627326]
- 163.
- Yamada T, Sartor RB, Marshall S. et al. Mucosal injury and inflammation in a model of chronic granulomatous colitis in rats. Gastroenterology. 1993;104(3):759–771. [PubMed: 7680014]
- 164.
- Hodgson HJ, Potter BJ, Skinner J. et al. Immune-complex mediated colitis in rabbits. An experimental model. Gut. 1978;19(3):225–232. [PMC free article: PMC1411904] [PubMed: 564806]
- 165.
- Watt J, Marcus R. Ulcerative colitis in the guinea-pig caused by seaweed extract. J Pharm Pharmacol. 1969;21(Suppl):187S+. [PubMed: 4391155]
- 166.
- Okayasu I, Hatakeyama S, Yamada M. et al. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. 1990;98(3):694–702. [PubMed: 1688816]
- 167.
- Stewart TH, Hetenyi C, Rowsell H. et al. Ulcerative enterocolitis in dogs induced by drugs. J Pathol. 1980;131(4):363–378. [PubMed: 6107336]
- 168.
- Bucy RP, Xu XY, Li J. et al. Cyclosporin A-induced autoimmune disease in mice. J Immunol. 1993;151(2):1039–1050. [PubMed: 8335890]
- 169.
- Rachmilewitz D, Okon E, Karmeli F. Sulphydryl blocker induced small intestinal inflammation in rats: A new model mimicking Crohn's disease. Gut. 1997;41(3):358–365. [PMC free article: PMC1891507] [PubMed: 9378392]
- 170.
- Quinn TC, Taylor HR, Schachter J. Experimental proctitis due to rectal infection with Chlamydia trachomatis in nonhuman primates. J Infect Dis. 1986;154(5):833–841. [PubMed: 3534107]
- 171.
- Shomer NH, Dangler CA, Schrenzel MD. et al. Helicobacter bilis-induced inflammatory bowel disease in scid mice with defined flora. Infect Immun. 1997;65(11):4858–4864. [PMC free article: PMC175697] [PubMed: 9353076]
- 172.
- Cahill RJ, Foltz CJ, Fox JG. et al. Inflammatory bowel disease: An immunity-mediated condition triggered by bacterial infection with Helicobacter hepaticus. Infect Immun. 1997;65(8):3126–3131. [PMC free article: PMC175441] [PubMed: 9234764]
- 173.
- Hammer RE, Maika SD, Richardson JA. et al. Spontaneous inflammatory disease in transgenic rats expressing HLA-B27 and human beta 2m: An animal model of HLA-B27-associated human disorders. Cell. 1990;63(5):1099–1112. [PubMed: 2257626]
- 174.
- Watanabe M, Ueno Y, Yajima T. et al. Interleukin 7 transgenic mice develop chronic colitis with decreased interleukin 7 protein accumulation in the colonic mucosa. J Exp Med. 1998;187(3):389–402. [PMC free article: PMC2212121] [PubMed: 9449719]
- 175.
- Hermiston ML, Gordon JI. Inflammatory bowel disease and adenomas in mice expressing a dominant negative N-cadherin. Science. 1995;270(5239):1203–1207. [PubMed: 7502046]
- 176.
- Clegg CH, Rulffes JT, Haugen HS. et al. Thymus dysfunction and chronic inflammatory disease in gp39 transgenic mice. Int Immunol. 1997;9(8):1111–1122. [PubMed: 9263008]
- 177.
- Bush TG, Savidge TC, Freeman TC. et al. Fulminant jejuno-ileitis following ablation of enteric glia in adult transgenic mice. Cell. 1998;93(2):189–201. [PubMed: 9568712]
- 178.
- Hahm KB, Im YH, Parks TW. et al. Loss of transforming growth factor beta signalling in the intestine contributes to tissue injury in inflammatory bowel disease. Gut. 2001;49(2):190–198. [PMC free article: PMC1728415] [PubMed: 11454793]
- 179.
- Shull MM, Ormsby I, Kier AB. et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359(6397):693–699. [PMC free article: PMC3889166] [PubMed: 1436033]
- 180.
- Takeda K, Clausen BE, Kaisho T. et al. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999;10(1):39–49. [PubMed: 10023769]
- 181.
- Wirtz S, Finotto S, Kanzler S. et al. Cutting edge: Chronic intestinal inflammation in STAT-4 transgenic mice: Characterization of disease and adoptive transfer by TNF- plus IFN-gamma-producing CD4+ T cells that respond to bacterial antigens. J Immunol. 1999;162(4):1884–1888. [PubMed: 9973454]
- 182.
- Mashimo H, Wu DC, Podolsky DK. et al. Impaired defense of intestinal mucosa in mice lacking intestinal trefoil factor. Science. 1996;274(5285):262–265. [PubMed: 8824194]
- 183.
- Sadlack B, Merz H, Schorle H. et al. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell. 1993;75(2):253–261. [PubMed: 8402910]
- 184.
- Willerford DM, Chen J, Ferry JA. et al. Interleukin-2 receptor alpha chain regulates the size and content of the peripheral lymphoid compartment. Immunity. 1995;3(4):521–530. [PubMed: 7584142]
- 185.
- Kuhn R, Lohler J, Rennick D. et al. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75(2):263–274. [PubMed: 8402911]
- 186.
- Kontoyiannis D, Pasparakis M, Pizarro TT. et al. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: Implications for joint and gut-associated immunopathologies. Immunity. 1999;10(3):387–398. [PubMed: 10204494]
- 187.
- Fox JG, Rogers AB, Whary MT. et al. Gastroenteritis in NF-kappaB-deficient mice is produced with wild-type Camplyobacter jejuni but not with C. jejuni lacking cytolethal distending toxin despite persistent colonization with both strains. Infect Immun. 2004;72(2):1116–1125. [PMC free article: PMC321575] [PubMed: 14742559]
- 188.
- Rudolph U, Finegold MJ, Rich SS. et al. Gi2 alpha protein deficiency: A model of inflammatory bowel disease. J Clin Immunol. 1995;15(6 Suppl):101S–105S. [PubMed: 8613481]
- 189.
- Baribault H, Penner J, Iozzo RV. et al. Colorectal hyperplasia and inflammation in keratin 8-deficient FVB/ N mice. Genes Dev. 1994;8(24):2964–2973. [PubMed: 7528156]
- 190.
- Spencer SD, Di MarcoF, Hooley J. et al. The orphan receptor CRF2-4 is an essential subunit of the interleukin 10 receptor. J Exp Med. 1998;187(4):571–578. [PMC free article: PMC2212143] [PubMed: 9463407]
- 191.
- Snapper SB, Rosen FS, Mizoguchi E. et al. Wiskott-Aldrich syndrome protein-deficient mice reveal a role for WASP in T but not B cell activation. Immunity. 1998;9(1):81–91. [PubMed: 9697838]
- 192.
- Morrissey PJ, Charrier K, Braddy S. et al. CD4+ T cells that express high levels of CD45RB induce wasting disease when transferred into congenic severe combined immunodeficient mice. Disease development is prevented by cotransfer of purified CD4+ T cells. J Exp Med. 1993;178(1):237–244. [PMC free article: PMC2191069] [PubMed: 8100269]
- 193.
- Hollander GA, Simpson SJ, Mizoguchi E. et al. Severe colitis in mice with aberrant thymic selection. Immunity. 1995;3(1):27–38. [PubMed: 7621076]
- 194.
- Steinhoff U, Brinkmann V, Klemm U. et al. Autoimmune intestinal pathology induced by hsp60-specific CD8 T cells. Immunity. 1999;11(3):349–358. [PubMed: 10514013]
- 195.
- AGA technical review on celiac sprue. Gastroenterology. 2001;120(6):1526–1540. [PubMed: 11313324]
- 196.
- Shan L, Molberg O, Parrot I. et al. Structural basis for gluten intolerance in celiac sprue. Science. 2002;297(5590):2275–2279. [PubMed: 12351792]
- 197.
- Teshigawara K, Kannagi R, Noro N. et al. Possible involvement of transglutaminase in endocytosis and antigen presentation. Microbiol Immunol. 1985;29(8):737–750. [PubMed: 2866438]
- 198.
- Ciccocioppo R, Di SabatinoA, Ara C. et al. Gliadin and tissue transglutaminase complexes in normal and coeliac duodenal mucosa. Clin Exp Immunol. 2003;134(3):516–524. [PMC free article: PMC1808891] [PubMed: 14632760]
- 199.
- Lundin KE, Scott H, Hansen T. et al. Gliadin-specific, HLA-DQ(alpha 1*0501,beta 1*0201) restricted T cells isolated from the small intestinal mucosa of celiac disease patients. J Exp Med. 1993;178(1):187–196. [PMC free article: PMC2191064] [PubMed: 8315377]
- 200.
- Molberg O, McAdam SN, Korner R. et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat Med. 1998;4(6):713–717. [PubMed: 9623982]
- 201.
- Molberg O, McAdam S, Lundin KE. et al. T cells from celiac disease lesions recognize gliadin epitopes deamidated in situ by endogenous tissue transglutaminase. Eur J Immunol. 2001;31(5):1317–1323. [PubMed: 11465088]
- 202.
- Dieterich W, Ehnis T, Bauer M. et al. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat Med. 1997;3(7):797–801. [PubMed: 9212111]
- 203.
- Sulkanen S, Halttunen T, Laurila K. et al. Tissue transglutaminase autoantibody enzyme-linked immunosorbent assay in detecting celiac disease. Gastroenterology. 1998;115(6):1322–1328. [PubMed: 9834257]
- 204.
- Dieterich W, Laag E, Schopper H. et al. Autoantibodies to tissue transglutaminase as predictors of celiac disease. Gastroenterology. 1998;115(6):1317–1321. [PubMed: 9834256]
- 205.
- Llorente MJ, Sebastian M, Fernandez-Acenero MJ. et al. IgA antibodies against tissue transglutaminase in the diagnosis of celiac disease: Concordance with intestinal biopsy in children and adults. Clin Chem. 2004;50(2):451–453. [PubMed: 14752021]
- 206.
- Sollid LM, Thorsby E. HLA susceptibility genes in celiac disease: Genetic mapping and role in pathogenesis. Gastroenterology. 1993;105(3):910–922. [PubMed: 8359659]
- 207.
- Greco L, Romino R, Coto I. et al. The first large population based twin study of coeliac disease. Gut. 2002;50(5):624–628. [PMC free article: PMC1773191] [PubMed: 11950806]
- 208.
- Zhong F, McCombs CC, Olson JM. et al. An autosomal screen for genes that predispose to celiac disease in the western counties of Ireland. Nat Genet. 1996;14(3):329–333. [PubMed: 8896565]
- 209.
- Greco L, Corazza G, Babron MC. et al. Genome search in celiac disease. Am J Hum Genet. 1998;62(3):669–675. [PMC free article: PMC1376948] [PubMed: 9497251]
- 210.
- Naluai AT, Nilsson S, Gudjonsdottir AH. et al. Genome-wide linkage analysis of Scandinavian affected sib-pairs supports presence of susceptibility loci for celiac disease on chromosomes 5 and 11. Eur J Hum Genet. 2001;9(12):938–944. [PubMed: 11840196]
- 211.
- Neuhausen SL, Feolo M, Farnham J. et al. Linkage analysis of HLA and candidate genes for celiac disease in a North American family-based study. BMC Med Genet. 2001;2(1):12. [PMC free article: PMC60993] [PubMed: 11737870]
- 212.
- Liu J, Juo SH, Holopainen P. et al. Genomewide linkage analysis of celiac disease in Finnish families. Am J Hum Genet. 2002;70(1):51–59. [PMC free article: PMC384903] [PubMed: 11715113]
- 213.
- Babron MC, Nilsson S, Adamovic S. et al. Meta and pooled analysis of European coeliac disease data. Eur J Hum Genet. 2003;11(11):828–834. [PubMed: 14571266]
- 214.
- Rioux JD, Karinen H, Kocher K. et al. Genomewide search and association studies in a Finnish celiac disease population: Identification of a novel locus and replication of the HLA and CTLA4 loci Am J Med Genet 2004. (epub ahead of print Sep 22) [PubMed: 15386476]
- 215.
- Woolley N, Holopainen P, Ollikainen V. et al. A new locus for coeliac disease mapped to chromosome 15 in a population isolate. Hum Genet. 2002;111(1):40–45. [PubMed: 12136234]
- 216.
- King AL, Yiannakou JY, Brett PM. et al. A genome-wide family-based linkage study of coeliac disease. Ann Hum Genet. 2000;64(Pt 6):479–490. [PubMed: 11281212]
- 217.
- Popat S, Bevan S, Braegger CP. et al. Genome screening of coeliac disease. J Med Genet. 2002;39(5):328–331. [PMC free article: PMC1735127] [PubMed: 12011149]
- 218.
- Neuhausen SL, Feolo M, Camp NJ. et al. Genome-wide linkage analysis for celiac disease in North American families. Am J Med Genet. 2002;111(1):1–9. [PubMed: 12124726]
- 219.
- Houlston RS, Tomlinson IP, Ford D. et al. Linkage analysis of candidate regions for coeliac disease genes. Hum Mol Genet. 1997;6(8):1335–1339. [PubMed: 9259281]
- 220.
- Holopainen P, Arvas M, Sistonen P. et al. CD28/CTLA4 gene region on chromosome 2q33 confers genetic susceptibility to celiac disease. A linkage and family-based association study. Tissue Antigens. 1999;53(5):470–475. [PubMed: 10372542]
- 221.
- Clot F, Fulchignoni-Lataud MC, Renoux C. et al. Linkage and association study of the CTLA-4 region in coeliac disease for Italian and Tunisian populations. Tissue Antigens. 1999;54(5):527–530. [PubMed: 10599894]
- 222.
- Naluai AT, Nilsson S, Samuelsson L. et al. The CTLA4/CD28 gene region on chromosome 2q33 confers susceptibility to celiac disease in a way possibly distinct from that of type 1 diabetes and other chronic inflammatory disorders. Tissue Antigens. 2000;56(4):350–355. [PubMed: 11098935]
- 223.
- Holopainen P, Mustalahti K, Uimari P. et al. Candidate gene regions and genetic heterogeneity in gluten sensitivity. Gut. 2001;48(5):696–701. [PMC free article: PMC1728294] [PubMed: 11302971]
- 224.
- Greco L, Babron MC, Corazza GR. et al. Existence of a genetic risk factor on chromosome 5q in Italian coeliac disease families. Ann Hum Genet. 2001;65(Pt 1):35–41. [PubMed: 11415521]
- 225.
- King AL, Fraser JS, Moodie SJ. et al. Coeliac disease: Follow-up linkage study provides further support for existence of a susceptibility locus on chromosome 11p11. Ann Hum Genet. 2001;65(Pt 4):377–386. [PubMed: 11592927]
- 226.
- King AL, Moodie SJ, Fraser JS. et al. CTLA-4/CD28 gene region is associated with genetic susceptibility to coeliac disease in UK families. J Med Genet. 2002;39(1):51–54. [PMC free article: PMC1734962] [PubMed: 11826026]
- 227.
- Popat S, Hearle N, Hogberg L. et al. Variation in the CTLA4/CD28 gene region confers an increased risk of coeliac disease. Ann Hum Genet. 2002;66(Pt 2):125–137. [PubMed: 12174216]
- 228.
- Louka AS, Sollid LM. HLA in coeliac disease: Unravelling the complex genetics of a complex disorder. Tissue Antigens. 2003;61(2):105–117. [PubMed: 12694579]
- 229.
- Keuning JJ, Pena AS, van Leeuwen A. et al. HLA-DW3 associated with coeliac disease. Lancet. 1976;1(7958):506–508. [PubMed: 55781]
- 230.
- Ek J, Albrechtsen D, Solheim BG. et al. Strong association between the HLA-Dw3-related B cell alloantigen -DRw3 and coeliac disease. Scand J Gastroenterol. 1978;13(2):229–233. [PubMed: 76332]
- 231.
- Mearin ML, Biemond I, Pena AS. et al. HLA-DR phenotypes in Spanish coeliac children: Their contribution to the understanding of the genetics of the disease. Gut. 1983;24(6):532–537. [PMC free article: PMC1420005] [PubMed: 6602084]
- 232.
- Tosi R, Vismara D, Tanigaki N. et al. Evidence that celiac disease is primarily associated with a DC locus allelic specificity. Clin Immunol Immunopathol. 1983;28(3):395–404. [PubMed: 6192959]
- 233.
- Trabace S, Giunta A, Rosso M. et al. HLA-ABC and DR antigens in celiac disease. A study in a pediatric Italian population. Vox Sang. 1984;46(2):102–106. [PubMed: 6422636]
- 234.
- McManus R, Wilson AG, Mansfield J. et al. TNF2, a polymorphism of the tumour necrosis-alpha gene promoter, is a component of the celiac disease major histocompatibility complex haplotype. Eur J Immunol. 1996;26(9):2113–2118. [PubMed: 8814255]
- 235.
- Polvi A, Maki M, Collin P. et al. TNF microsatellite alleles a2 and b3 are not primarily associated with celiac disease in the Finnish population. Tissue Antigens. 1998;51(5):553–555. [PubMed: 9672155]
- 236.
- de la Concha EG, Fernandez-Arquero M, Vigil P. et al. Celiac disease and TNF promoter polymorphisms. Hum Immunol. 2000;61(5):513–517. [PubMed: 10773355]
- 237.
- Hahn-Zoric M, Hytonen AM, Hanson LA. et al. Association of -1087 IL10 and -308 TNFA gene polymorphisms with serological markers of coeliac disease. J Clin Immunol. 2003;23(4):291–296. [PubMed: 12959221]
- 238.
- Louka AS, Lie BA, Talseth B. et al. Coeliac disease patients carry conserved HLA-DR3-DQ2 haplotypes revealed by association of TNF alleles. Immunogenetics. 2003;55(5):339–343. [PubMed: 12845502]
- 239.
- Partanen J, Milner C, Campbell RD. et al. HLA-linked heat-shock protein 70 (HSP70-2) gene polymorphism and celiac disease. Tissue Antigens. 1993;41(1):15–19. [PubMed: 8096093]
- 240.
- Ramos-Arroyo MA, Feijoo E, Sanchez-Valverde F. et al. Heat-shock protein 70-1 and HLA class II gene polymorphisms associated with celiac disease susceptibility in Navarra (Spain). Hum Immunol. 2001;62(8):821–825. [PubMed: 11476906]
- 241.
- de la Concha EG, Fernandez-Arquero M, Lopez-Nava G. et al. Susceptibility to severe ulcerative colitis is associated with polymorphism in the central MHC gene IKBL. Gastroenterology. 2000;119(6):1491–1495. [PubMed: 11113070]
- 242.
- Fernandez L, Fernandez-Arquero M, Gual L. et al. Triplet repeat polymorphism in the transmembrane region of the MICA gene in celiac disease. Tissue Antigens. 2002;59(3):219–222. [PubMed: 12074713]
- 243.
- Rueda B, Pascual M, Lopez-Nevot MA. et al. Association of MICA-A5.1 allele with susceptibility to celiac disease in a family study. Am J Gastroenterol. 2003;98(2):359–362. [PubMed: 12591055]
- 244.
- Van BelzenMJ, Koeleman BP, Crusius JB. et al. Defining the contribution of the HLA region to DQ2-positive coeliac disease patients. Genes Immun. 2004;5(3):215–220. [PubMed: 15014431]
- 245.
- Roschmann E, Wienker TF, Volk BA. Role of T cell receptor delta gene in susceptibility to celiac disease. J Mol Med. 1996;74(2):93–98. [PubMed: 8820404]
- 246.
- Rueda B, Lopez-Nevot MA, Pascual M. et al. Polymorphism of the inducible nitric oxide synthase gene in celiac disease. Hum Immunol. 2002;63(11):1062–1065. [PubMed: 12392860]
- 247.
- Louka AS, Stensby EK, Ek J. et al. Coeliac disease candidate genes: No association with functional polymorphisms in matrix metalloproteinase 1 and 3 gene promoters. Scand J Gastroenterol. 2002;37(8):931–935. [PubMed: 12229968]
- 248.
- Louka AS, Torinsson NaluaiA, D'Alfonso S. et al. The IL12B gene does not confer susceptibility to coeliac disease. Tissue Antigens. 2002;59(1):70–72. [PubMed: 11972887]
- 249.
- Seegers D, Borm ME, van Belzen MJ. et al. IL12B and IRF1 gene polymorphisms and susceptibility to celiac disease. Eur J Immunogenet. 2003;30(6):421–425. [PubMed: 14675396]
- 250.
- Perez De Nanclares G, Bilbao JR, Calvo B. et al. 5'-Insulin gene VNTR polymorphism is specific for type 1 diabetes: No association with celiac or Addison's disease. Ann NY Acad Sci. 2003;1005:319–323. [PubMed: 14679083]
- 251.
- Aldersley MA, Hamlin PJ, Jones PF. et al. No polymorphism in the tissue transglutaminase gene detected in coeliac disease patients. Scand J Gastroenterol. 2000;35(1):61–63. [PubMed: 10672836]
- 252.
- Van BelzenMJ, Mulder CJ, Pearson PL. et al. The tissue transglutaminase gene is not a primary factor predisposing to celiac disease. Am J Gastroenterol. 2001;96(12):3337–3340. [PubMed: 11774946]
- 253.
- Bouguerra F, Dugoujon JM, Babron MC. et al. Susceptibility to coeliac disease in Tunisian children and GM immunoglobulin allotypes. Eur J Immunogenet. 1999;26(4):293–297. [PubMed: 10457894]
- 254.
- Van BelzenMJ, Mulder CJJ, Zhernakova A. et al. CTLA4 +49A/G and CT60 polymorphisms in Dutch coeliac disease patients. Eur J Hum Genet. 2004;12(9):782–785. [PubMed: 15199380]
- 255.
- Boniotto M, Braida L, Spano A. et al. Variant mannose-binding lectin alleles are associated with celiac disease. Immunogenetics. 2002;54(8):596–598. [PubMed: 12439623]
- 256.
- Haimila K, Smedberg T, Mustalahti K. et al. Genetic association of coeliac disease susceptibility to polymorphisms in the ICOS gene on chromosome 2q33. Genes Immun. 2004;5(2):85–92. [PubMed: 14712308]
- 257.
- Djilali-Saiah I, Schmitz J, Harfouch-Hammoud E. et al. CTLA-4 gene polymorphism is associated with predisposition to coeliac disease. Gut. 1998;43(2):187–189. [PMC free article: PMC1727221] [PubMed: 10189842]
- 258.
- Popat S, Hearle N, Wixey J. et al. Analysis of the CTLA4 gene in Swedish coeliac disease patients. Scand J Gastroenterol. 2002;37(1):28–31. [PubMed: 11843030]
- 259.
- Mora B, Bonamico M, Indovina P. et al. CTLA-4 +49 A/G dimorphism in Italian patients with celiac disease. Hum Immunol. 2003;64(2):297–301. [PubMed: 12559633]
- 260.
- King AL, Moodie SJ, Fraser JS. et al. Coeliac disease: Investigation of proposed causal variants in the CTLA4 gene region. Eur J Immunogenet. 2003;30(6):427–432. [PubMed: 14675397]
- 261.
- Kristiansen OP, Larsen ZM, Pociot F. CTLA-4 in autoimmune diseases-a general susceptibility gene to autoimmunity? Genes Immun. 2000;1(3):170–184. [PubMed: 11196709]
- 262.
- Ueda H, Howson JM, Esposito L. et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature. 2003;423(6939):506–511. [PubMed: 12724780]
- 263.
- Batt RM, McLean L, Carter MW. Sequential morphologic and biochemical studies of naturally occurring wheat-sensitive enteropathy in Irish setter dogs. Dig Dis Sci. 1987;32(2):184–194. [PubMed: 3026759]
- 264.
- Polvi A, Garden OA, Houlston RS. et al. Genetic susceptibility to gluten sensitive enteropathy in Irish setter dogs is not linked to the major histocompatibility complex. Tissue Antigens. 1998;52(6):543–549. [PubMed: 9894853]
- 265.
- Manns MP, Luttig B, Obermayer-Straub P. Autoimmune diseases: The liver In: Rose NR, McKay IR, eds.The Autoimmune Diseases 3rd ed. New York: Academic Press,1998511–544.
- 266.
- Gupta R, Agarwal SR, Jain M. et al. Autoimmune hepatitis in the Indian subcontinent: 7 years experience. J Gastroenterol Hepatol. 2001;16(10):1144–1148. [PubMed: 11686842]
- 267.
- Yachha SK, Srivastava A, Chetri K. et al. Autoimmune liver disease in children. J Gastroenterol Hepatol. 2001;16(6):674–677. [PubMed: 11422621]
- 268.
- Johnson PJ, McFarlane IG. Meeting report: International Autoimmune Hepatitis Group. Hepatology. 1993;18(4):998–1005. [PubMed: 8406375]
- 269.
- Manns MP, Obermayer-Straub P. Cytochromes P450 and uridine triphosphate-glucuronosyltransferases: Model autoantigens to study drug-induced, virus-induced, and autoimmune liver disease. Hepatology. 1997;26(4):1054–1066. [PubMed: 9328334]
- 270.
- Zanger UM, Hauri HP, Loeper J. et al. Antibodies against human cytochrome P-450db1 in autoimmune hepatitis type II. Proc Natl Acad Sci USA. 1988;85(21):8256–8260. [PMC free article: PMC282408] [PubMed: 3186722]
- 271.
- Czaja AJ, Manns MP. The validity and importance of subtypes in autoimmune hepatitis: A point of view. Am J Gastroenterol. 1995;90(8):1206–1211. [PubMed: 7639216]
- 272.
- Manns M, Gerken G, Kyriatsoulis A. et al. Characterisation of a new subgroup of autoimmune chronic active hepatitis by autoantibodies against a soluble liver antigen. Lancet. 1987;1(8528):292–294. [PubMed: 2880112]
- 273.
- Strassburg CP, Obermayer-Straub P, Manns MP. Autoimmunity in hepatitis C and D virus infection. J Viral Hepat. 1996;3(2):49–59. [PubMed: 8811638]
- 274.
- Goldstein NS, Bayati N, Silverman AL. et al. Minocycline as a cause of drug-induced autoimmune hepatitis. Report of four cases and comparison with autoimmune hepatitis. Am J Clin Pathol. 2000;114(4):591–598. [PubMed: 11026106]
- 275.
- Czaja AJ, Donaldson PT. Genetic susceptibilities for immune expression and liver cell injury in autoimmune hepatitis. Immunol Rev. 2000;174:250–259. [PubMed: 10807521]
- 276.
- Czaja AJ, Souto EO, Bittencourt PL. et al. Clinical distinctions and pathogenic implications of type 1 autoimmune hepatitis in Brazil and the United States. J Hepatol. 2002;37(3):302–308. [PubMed: 12175624]
- 277.
- Agarwal K, Jones DE, Daly AK. et al. CTLA-4 gene polymorphism confers susceptibility to primary biliary cirrhosis. J Hepatol. 2000;32(4):538–541. [PubMed: 10782900]
- 278.
- Djilali-Saiah I, Ouellette P, Caillat-Zucman S. et al. CTLA-4/CD 28 region polymorphisms in children from families with autoimmune hepatitis. Hum Immunol. 2001;62(12):1356–1362. [PubMed: 11756004]
- 279.
- Vogel A, Strassburg CP, Manns MP. Genetic association of vitamin D receptor polymorphisms with primary biliary cirrhosis and autoimmune hepatitis. Hepatology. 2002;35(1):126–131. [PubMed: 11786968]
- 280.
- Vogel A, Strassburg CP, Manns MP. 77 C/G mutation in the tyrosine phosphatase CD45 gene and autoimmune hepatitis: Evidence for a genetic link. Genes Immun. 2003;4(1):79–81. [PubMed: 12595907]
- 281.
- Manabe K, Hibberd ML, Donaldson PT. et al. T-cell receptor constant beta germline gene polymorphisms and susceptibility to autoimmune hepatitis. Gastroenterology. 1994;106(5):1321–1325. [PubMed: 7909781]
- 282.
- Whittingham S, Mathews JD, Schanfield MS. et al. Interaction of HLA and Gm in autoimmune chronic active hepatitis. Clin Exp Immunol. 1981;43(1):80–86. [PMC free article: PMC1537131] [PubMed: 7249397]
- 283.
- Cookson S, Constantini PK, Clare M. et al. Frequency and nature of cytokine gene polymorphisms in type 1 autoimmune hepatitis. Hepatology. 1999;30(4):851–856. [PubMed: 10498633]
- 284.
- Czaja AJ, Cookson S, Constantini PK. et al. Cytokine polymorphisms associated with clinical features and treatment outcome in type 1 autoimmune hepatitis. Gastroenterology. 1999;117(3):645–652. [PubMed: 10464141]
- 285.
- Vogel A, Liermann H, Harms A. et al. Autoimmune regulator AIRE: Evidence for genetic differences between autoimmune hepatitis and hepatitis as part of the autoimmune polyglandular syndrome type 1. Hepatology. 2001;33(5):1047–1052. [PubMed: 11343230]
- 286.
- Agarwal K, Czaja AJ, Jones DE. et al. Cytotoxic T lymphocyte antigen-4 (CTLA-4) gene polymorphisms and susceptibility to type 1 autoimmune hepatitis. Hepatology. 2000;31(1):49–53. [PubMed: 10613727]
- 287.
- Bittencourt PL, Palacios SA, Cancado EL. et al. Cytotoxic T lymphocyte antigen-4 gene polymorphisms do not confer susceptibility to autoimmune hepatitis types 1 and 2 in Brazil. Am J Gastroenterol. 2003;98(7):1616–1620. [PubMed: 12873588]
- 288.
- Jaeckel E. Animal models of autoimmune hepatitis. Semin Liver Dis. 2002;22(4):325–338. [PubMed: 12447705]
- 289.
- Peters MG. Animal models of autoimmune liver disease. Immunol Cell Biol. 2002;80(1):113–116. [PubMed: 11869369]
- 290.
- Howell CD. Animal models of autoimmunity. Clin Liver Dis. 2002;6(3):487–495. [PubMed: 12362580]
- 291.
- Lohse AW, Brunner S, Kyriatsoulis A. et al. Autoantibodies in experimental autoimmune hepatitis. J Hepatol. 1992;14(1):48–53. [PubMed: 1737915]
- 292.
- Sadlack B, Lohler J, Schorle H. et al. Generalized autoimmune disease in interleukin-2-deficient mice is triggered by an uncontrolled activation and proliferation of CD4+ T cells. Eur J Immunol. 1995;25(11):3053–3059. [PubMed: 7489743]
- 293.
- Gorham JD, Lin JT, Sung JL. et al. Genetic regulation of autoimmune disease: BALB/c background TGF-beta 1-deficient mice develop necroinflammatory IFN-gamma-dependent hepatitis. J Immunol. 2001;166(10):6413–6422. [PubMed: 11342667]
- 294.
- Alper CA, Awdeh Z, Yunis EJ. Conserved, extended MHC haplotypes. Exp Clin Immunogenet. 1992;9(2):58–71. [PubMed: 1489551]
- 295.
- Cullen M, Perfetto SP, Klitz W. et al. High-resolution patterns of meiotic recombination across the human major histocompatibility complex. Am J Hum Genet. 2002;71(4):759–776. [PMC free article: PMC378534] [PubMed: 12297984]
- 296.
- Walsh EC, Mather KA, Schaffner SF. et al. An integrated haplotype map of the human major histocompatibility complex. Am J Hum Genet. 2003;73(3):580–590. [PMC free article: PMC1180682] [PubMed: 12920676]
- Gastroenterologic and Hepatic Diseases - Madame Curie Bioscience DatabaseGastroenterologic and Hepatic Diseases - Madame Curie Bioscience Database
- Human, Mouse and Yeast Telomerase - Madame Curie Bioscience DatabaseHuman, Mouse and Yeast Telomerase - Madame Curie Bioscience Database
- In Situ Activation of Caspases Revealed by Affinity Labeling Their Enzymatic Sit...In Situ Activation of Caspases Revealed by Affinity Labeling Their Enzymatic Sites - Madame Curie Bioscience Database
- The Origins of Species - Madame Curie Bioscience DatabaseThe Origins of Species - Madame Curie Bioscience Database
- Telomerase Activity as a Marker of Tumor Cell Survival to Evaluate Sensitivity o...Telomerase Activity as a Marker of Tumor Cell Survival to Evaluate Sensitivity of Neoplastic Cells to Cancer Treatment - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...