

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Dermatomyositis (DM), polymyositis (PM) and inclusion body myositis (IBM) belong to the heterogeneous group of the inflammatory myopathies and are characterized by muscle cell infiltrations and specific alterations of the muscle fibers. In DM it is evident a perifascicular atrophy of muscle tissue due to the activation and deposition of complement on capillaries; in PM and IBM there is a prominent endomysial infiltration of clonally expanded CD8+ T lymphocytes that surround and eventually invade single nonnecrotic muscle fibers, positive for MHC class I molecules. Muscle fibers in PM/IBM die for the action of cytotoxic enzymes (perforin and granzymes) released by the invading CD8+ T lymphocytes. In IBM, beside the autoimmune attack, there is an abnormal accumulation of proteins in vacuoles within muscle fibers. Triggering factors of myositis as well as the processes by which the immunological attack induces muscle weakness are still unknown. Upregulation of adhesion molecules, cytokines, chemokines contribute to recruit cells of the immune system and to maintain a chronic inflamed area. In vivo and in vitro studies on muscle cells have assessed their functions as target cells or antigen presenting cells. Combined studies on gene profiles and cellular immunology of disease-associated muscle biopsies will be of great help in clarifying the pathogenetic mechanisms underlying these inflammatory myopathies.
Introduction
The idiopathic inflammatory myopathies (IIM) are a heterogeneous group of diseases characterized by muscle inflammation.1,2 The principal clinical variants of IIM are: dermatomyositis (DM), polymyositis (PM), and inclusion body myositis (IBM).1,2 The latter is divided into: sporadic-IBM (s-IBM), the most common muscle disease that starts after age 50 years and leads to severe disability, and hereditary inclusion body myopathies (h-IBM), characterized by pathologic features that strikingly resemble those of s-IBM except for lack of lymphocyte inflammation (hence the term “myopathy” instead of “myositis”).3 Inflammatory myopathies are included in the clinicopathological interest of different medical specialties (e.g., neurology, rheumatology, dermatology, etc.) resulting in different diagnostic evaluation and treatment work-up. A recent meeting, under the auspices of the ENMC (European Neuromuscular Centre) in which European and American neurologists and rheumatologists convened, put a tremendous effort in establishing common diagnostic criteria and measuring outcomes in the perspective of international randomized clinical trials.4
DM is a humorally mediated microangiopathy, while PM is a T-cell mediated disorder in which a cytotoxic attack against single nonnecrotic muscle fibers occurs. The pathogenesis of IBM is unknown.3 DM and PM are considered to be responsive to immunosuppressive and immunomodulating therapies, in contrast to IBM, which is refractory to all treatment.4 The triggering factors of IIM are still unknown; a growing body of evidence suggests that genetically susceptible individuals probably develop an idiopathic inflammatory myopathy in response to particular environment stimuli.
Clinical Aspects
Dermatomyositis
DM is a rare multisystemic autoimmune disease that affects children and adults of both genders and all ethnic groups (Table 1). It primarily involves skin and skeletal muscle. Cutaneous manifestations may precede the onset of myositis by several months or up to 2 years and more; Gottron's papules, heliotrope rash, and macular erythemas are the most typical manifestations.5 Skin lesions can be worsened by UVA and UVB light; this increased photosensitivity may be due to a polymorphism in tumor necrosis factor-α (TNF-α)-308A allele, detected with high frequency in adult and juvenile DM Caucasian patients (reviewed in ref. 5). Muscle weakness can vary from mild to severe (quadriparesis). Clinical manifestations other than those involving muscle tissue can occur: subcutaneous calcifications, joint contractures, dysphagia, fever, malaise, weight loss, arthralgia, Raynaud's phenomenon, tumor.1,2 DM diagnosis is confirmed by muscle biopsy (see paragraph regarding histopathology).
Table 1
Idiopathic inflammatory myopathies: clinical features.
Polymyositis
PM, as a difference with DM, has less distinguished clinical features (Table 1).1,2 However, PM can be suspected in all cases presenting as a subacute proximal myopathy without evidence of inherited transmission. Incidence and prevalence are reported to be similar to those of DM, but PM is extremely rare in infancy. Female to male ratio is 3:1. The clinical course of PM is usually subacute. In the typical affected adult patient anamnesis is negative for: cutaneous symptoms, involvement of ocular and facial muscles, presence of hereditary muscular diseases and exposure to myotoxic drugs or toxins. Onset of the disease can be difficult to ascertain because a subclinical disease may persist over months before the patients refer to the physician. Apart from cutaneous alterations, the degree of severity and distribution of muscle weakness and wasting are similar to those described for DM, except for myalgia and muscle tenderness, which are less frequent than in DM.
Inclusion Body Myositis
IBM has clinical-pathological features well differentiated from PM or DM (Table 1).1,3 IBM is tipically a chronic evolutive muscle disorders whose onset is usually after the age of 50. Because onset is extremely insidious and disease course so slow, the time of beginning and the incidence of the disease is very difficult to establish. IBM is more frequent in males (male to female ratio 3:1) and in whites than in blacks. Muscle weakness and atrophy affect more frequently distal muscles: deficit of the foot extensors might be evident in more than 50% of the cases and represent the clue of early diagnosis. Selective involvement of triceps, biceps, ileopsoas and quadriceps is frequently evident and responsible for sudden falls of these patients. A noticeable evidence of asymmetric involvement of muscles is a typical feature of IBM. Tendon reflexes are usually lost and because of distal atrophy and weakness a neurogenic disease can be misdiagnosed. Though IBM is considered an acquired IM, familial cases have been described, some associated with leukoencephalopathy. An empyrical criterion to suspect IBM is the unresponsiveness to immunosuppressive therapy of suspected PM patients.
Histopathology
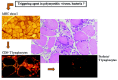
PM and s-IBM are characterized by an endomysial mononuclear cell infiltrate, mainly composed of cytotoxic CD8+ T lymphocytes and macrophages, which surrounds and eventually invades single nonnecrotic muscle fibers. CD8+ T cells are activated (HLA-DR,+ LFA-1+), have a memory phenotype (CD45RO+) and released perforin when in close contact with muscle fiber.1,2 Besides inflammation, in s-IBM muscle fibers abnormally accumulated proteins are observed (fig.1, fig.2).6 In DM the most prominent cells are CD4+ T lymphocytes localized in the perivascular site which might provide help to B cells to produce antibodies that, fixing complement, induce a vascular damage. The deposition of the lytic membrane attack complex (MAC) on capillaries induces perivascular inflammation, capillary depletion, muscle fiber necrosis and perifascicular atrophy, diagnostic for DM even in the absence of inflammation (fig.3).1,2


Immunopathogenesis
We will focus on the genetic characteristics of inflammatory myopathy patients and the phenotypes of effector and target cells involved in the immune response. All the information are summarized in Table 2, Table 3, and Table 4.
Table 2
Association between human leukocyte antigen genes and idiopathic inflammatory myopathies.
Table 3
Phenotypes of effector cells in inflammatory myopathies (updated 9-1-2004).
Table 4
Immunobiological features of muscle cells in inflammatory myopathies (updated 9-1-2004).
Major Histocompatibility Complex (MHC)
As for other autoimmune diseases, a strong association between human leukocyte antigen (HLA) genes and all clinical forms of IIM has been found (Table 2). At first the genetic marker associated with IIM was HLA B8 (studied in patients with juvenile DM), then the studies were extended including a large number of patients and the major genetic risk factors for the development of myositis were identified in HLA DRB1*0301 and DQA1*0501 in whites.7 DRB1*0301 is a common genetic risk factor for familial and sporadic IIM, but contributes in a lesser extent in the familial IIM; while, the unique genetic risk factor to familial IIM is homozygosity at the HLA-DQA1 locus.8 Hausmanowa-Petrusewicz et al9 reported that the HLA-DRB1*0301; DQA1*0501 haplotype was found to be significantly increased in Polish IIM population as a whole and in those IIM patients positive for anti-synthetase, anti-PM-Scl, and anti-Ku autoantibodies. Other groups observed that HLA-DRB1*0301 (DR3), DQA1*0501, and DQB1*0201 (DQ2) alleles were each increased in white patients with myositis, especially those with PM, and most strikingly in those with myositis-specific autoantibodies. In other ethnic groups, except the Japanese, only frequencies of HLA-DQA1*0501 and the structurally similar DQA1*0401 alleles were significantly increased and most significantly associated with anti-Jo-1, anti-PL-12, and other autoantibodies, compared with IIM patients without autoantibodies. HLA-DQA1*0102 and *0103 alleles predominated in those IIM patients, including Japanese, positive for myositis-specific antibodies but negative for HLA-DQA1*0501 and *0401. A negative association of the HLA-DR2 alleles (DRB1*1501 and *1503) with PM but not with DM was found.10
Recently, HLA I and II haplotypes have been analyzed in a cohort of s-IBM patients:11 the previously mentioned association with B8 and DR37,8,10 was detected and a new HLA association, A*03, DQ5/DQB1*05, was observed.12 Three hypothesis about the cause of HLA association with s-IBM were put forward by the authors: (1) s-IBM is caused by a viral infection (even if in s-IBM it appears unlikely); (2) s-IBM is an autoimmune disease (putative antigens can be auto and viral antigens); (3) s-IBM is due to genes, so far unidentified, in linkage disequilibrium with HLA alleles (for example, in MHC locus are located genes for TNF-α and β, the complement factors 2 and 4, heat shock protein 70).11
The importance of HLA molecules in the pathogenesis of IIM is strongly supported by the observation that, while normal muscle fibers do not express MHC class I molecules on their surface, IIM muscle fibers are strongly positive for MHC class I and class II expression, even in cells apparently distant from cell infiltrates.1,12,13 It remains to be elucidated whether MHC molecule expression is induced by infectious agents or by proinflammatory cytokines14,15 or by a nonspecific response to tissue injury and regeneration. Lundberg et al16 observed that in chronic PM and DM clinical symptoms persist even in the absence of inflammatory infiltrates together with an increased expression of IL-1α in the capillaries and MHC class I on muscle fibers, mainly confined to type II muscle fibers.17 The authors hypothesized that infiltrating cells might not be the primary factors of muscle damage. As observed in an animal model,18 overexpression of IL-1α and MHC class I might be sufficient to induce clinical myositis, muscle damage and eventually muscle inflammation.
In vitro myoblasts and myotubes constitutively express low levels of MHC class I and adhesion molecules such as LFA-3. After myoblast stimulation with IFN-γ or IFN-γ and TNF-α increased expression of MHC class I and de novo synthesis of MHC class II and ICAM-1 has been observed.19-25 When myoblasts are allowed to fuse into myotubes and these cells are innervated MHC class II molecules disappear on cell surface even after IFN-γ stimulation, suggesting that MHC class II synthesis is developmentally regulated during myogenesis and that overexpression of this molecule on pathological muscle fibers might be independent by proinflammatory cytokine production.22
In the last years, it is emerging the role of non classic MHC class I molecules, in particular of HLA-G, in IIM pathogenesis. This molecule is similar to the MHC class I (β2-microglobulin association, CD8 binding, presentation of a restricted peptide repertoire) but with peculiar characteristics: less polymorphic, highly restricted tissue distribution, seven different isoforms (membrane-associated, HLA-G1, -G2, -G3, -G4, and soluble, HLA-G5, -G6, -G7).26 HLA-G is a key molecule in fetal-maternal tolerance and in the adult life protects target cells from cytotoxic T and natural killer cell attack.27 Normally muscle fibers do not express HLA-G, while a highly positive signal has been observed on IIM muscle fibers, also positive for MHC class I molecules, and on many inflammatory cells.28 Moreover, the authors demonstrated that in vitro IFN-γ was able to up regulate mRNA transcripts corresponding to different isoforms of HLA-G and their surface expression in cultured myoblasts isolated from control subjects and patients.28 Transfection of myoblasts or muscle cell line (TE671) with HLA-G molecules (HLA-G1 and -G5) rendered these cells resistant to alloreactive lysis, reduced alloproliferation, interfered with priming of antigen-specific cytotoxic T cells or inhibited antigen-specific effector lysis.29 In inflammatory myopathies and in other conditions of inflamed muscles (e.g., myoblast transplantation, vaccination) HLA-G might be a muscle cell effort to protect themselves from immune cell-mediated attack.30
Besides MHC class I and II molecules, costimulatory molecules are necessary to stimulate T lymphocytes. Three different costimulatory pathways have been discovered: the B7-1/B7-2 (CD80/CD86) and their receptors CD28/CTLA-4, the best characterized; the inducible costimulatory ligand (ICOSL) and its receptor ICOS (a T cell specific costimulatory molecule homologous to CD28/CTLA4); the receptor PD-1 (programmed death gene 1), which interacts with two novel B7 family members, PD-L1 (B7-H1) and PD-L2 (B7-DC). All these coreceptors can enhance or attenuate T cell activation.31
Muscle fibers do not express constitutively or under pro-inflammatory stimuli, detectable levels of CD80/CD86 molecules both in vivo and in vitro.32-35 Nevertheless, they are able to activate antigen-specific T cell response.36 It is not yet clear whether they are able to prime naïve T cells. Other molecules have been postulated to be expressed on muscle fibers such as a yet unidentified B7-related protein (BB-1) that interacts with CD28/CTLA4 and stimulates T lymphocytes.34,35 Recently, important advances in the field of muscle capacity to stimulate an immune response have been obtained by analyzing the costimulatory pathways alternative to CD80/CD86-CD28/CTLA4 pathway. ICOSL was expressed at low levels on muscle fibers and to be up regulated in IIM muscle tissue, in particular in PM, on the muscle fibers surrounded and invaded by T lymphocytes ICOS+37 (a marker of T cell activation).38 In DM, a strong positivity for ICOSL was observed on endothelial cells of blood vessels.37 Furthermore, ICOSL was observed on cultured myoblasts in basal condition and enhanced after TNF-α stimulation. Cocultures of MHC class II+ myoblasts with CD4+ T cells together with superantigen demonstrated that ICOSL is active since it modulates Th1 and Th2 cytokine synthesis by activated T cells.37 These observations paralleled those of B7-H1 molecule. This protein was expressed in IIM muscle biopsies and not in normal or nonmyopathic muscle tissues; it was localized to areas of strong inflammation either on muscle and mononuclear cells.39 In vitro myoblasts became positive for B7-H1 only after IFN-γ stimulation. Anti-B7-H1 monoclonal antibody strongly augmented the Th1 and Th2 cytokines in cocultures of IFN-γ stimulated myoblasts, CD4+ or CD8+ T cells and superantigens.39 The authors speculate that B7-H1 could interfere with the activity of cytotoxic T cells and, hence, this expression is another effort of muscle fiber to protect itself from the autoimmune attack.39
T Cell Receptor (TCR)
T lymphocytes recognize the antigen, presented by the MHC class I or II, via T cell receptor (TCR), a heterodimer composed by two chains, α/β or γ/δ (less frequent), encoded by different gene families combined to form the variable (V), diversity (D), joining (J) and constant (C) regions.40,41 The contact point between TCR and the antigen-MHC complex lies in the complementarity-determining region 3 (CDR3), composed by the V-(D)-J combination. If TCR recognizes an antigen the amino acid sequence of the CDR3 region should be conserved in the recruited T cells.40,41 In PM and s-IBM patients, but not in DM, T lymphocytes with a restricted TCR repertoire are recruited from the blood stream to the muscle tissue. Sequence analysis of the TCR families revealed a restricted use of Jβ genes and a CDR3 consensus motif. These data are suggestive of the presence of a conventional antigen on muscle fibers, which attracts specifically CD8+ T cells.42-50 In selected s-IBM patients the TCR repertoire has been analyzed in sequential muscle biopsies during a period of 19-22 months.51 A persistent clonal expansion of CD8+ T cells with the same TCRBV families and a persistent CDR3 amino acid sequence were observed, supporting the hypothesis that endomysial T cells are recruited by a continuous presentation by muscle fibers of the same antigen(s), even in the late stages of the disease.51 Analysis of peripheral T cells from IIM patients and age-matched controls showed in the patients a more frequent CD8+ T cell clonal expansion than CD4+ T cells. The expanded T cells persisted as large populations over time and some of the expanded clones were found in the affected muscles from the same patients.52 These results provide the evidence that a local autoimmune reaction can directly influence the periphery. Moreover, to have the possibility to isolate pathological T cell clones from the periphery will be of great help in understanding the evolution of the disease and the efficacy of specific therapeutic treatments. Benveniste et al53 demonstrated that TCR repertoire was perturbated in the peripheral blood of PM patients but not of DM patients. Analysis of TCR repertoire in the periphery might be useful in differential diagnosis between PM and DM. However, a study like this does not allow proving that clonally expanded T cells are those that invade the single nonnecrotic muscle fibers. The use of a laser microdissector has overcome this problem. Hofbauer et al54 combined CDR3 spectratyping analysis with single cell PCR performed on cells localized in direct contact with the muscle fiber and isolated by laser microdissector. With this approach they were able to identify and track autoaggressive T lymphocytes. It is accepted that in PM/IBM specific antigens, presented by muscle fibers, recruit T cells, what is the antigen remains a mistery.
Myositis-Specific Autoantibodies
Most of IIM patients' sera, approximately 50%, are positive for myositis-specific autoantibodies (MSAs). The targeted antigens are not specific for muscle tissue, the majority of them are aminoacyl-tRNA synthetases, components of the signal recognition particle, translation factors, components of a nucleosome remodelling complex (for a comprehensive review see ref. 55). MSAs are associated with specific clinical characteristics, for example anti-Jo-1 (anti-histidyl tRNA synthetase, HisRS) antibodies and the antisynthetase syndrome (DM or PM, idiopathic interstitial lung disease, arthritis and Raynaud phenomenon).55 The exact role of MSAs in IIM immunopathogenesis is still unknown. Nagaraju et al18 demonstrated that conditional overexpression of MHC class I molecules in the skeletal muscles of young mice was able to induce an inflammatory disease, limited to skeletal muscles, self-sustaining, more severe in females, and often accompanied by autoantibodies, including, in some mice, anti-Jo-1 autoantibodies (the most frequent antibodies in IIM patients, 15-25% of cases). The authors suggested that an apparently non specific event, such as the up-regulation of MHC class I in a tissue, might generate a highly specific autoimmune disease and that specific autoantibodies derive not from the specificity of the stimulus, but from the context, location, and probably the duration of the stimulus.
The majority of autoantigens, including the aminoacyl-tRNA synthetases, targets of the immune attack in different systemic autoimmune diseases, have in their sequence the cleavage site recognized by granzyme B, a highly specific protease released by activated immune cells that cuts target molecule after aspartate residues.56-58 Nonautoantigens are refractory to granzyme B cleavage.57 The HisRS is cleaved by granzyme B in the N-terminal domain and the presence of anti-Jo-1 antibodies inhibited the granzyme B cleavage, suggesting that the immunodominant epitope and the cleavage site are very close.58 HisRS and asparaginyl-tRNA synthetase (AsnRS) have chemoattractant properties versus CCR3- and CCR5-expressing cells, and can recruit immature dendritic cells.59 Moreover, T cells isolated from peripheral blood of PM patients, positive for anti-Jo-1 antibodies, and from control subjects proliferated in response to Jo-1 full-length, or peptides, in the presence of dendritic cells with a predominant response versus the N-terminal domain (the dominant B cell epitope). This response was MHC class II dependent.60 Altogether these results suggest an active role of aminoacyl-tRNA synthetase in initiating and perpetuating the immune response within IIM muscle tissue. A still unknown event (e.g., viral infection) in the appropriate host might damage muscle tissue, aminoacyl-tRNA synthetases might undergo conformational changes becoming susceptible to granzyme B cleavage and be released in the microenvironment. The fragmented aminoacyl-tRNA synthetase might recruit mononuclear cells initiating a cascade of immune events such as antigen presentation to T lymphocytes, production of B cell stimulating cytokines that results in autoantibody synthesis, further muscle damage via release of cytotoxic enzymes.58,59
Cytokines and Chemokines
Cytokines play a crucial role in inflammatory reaction. These molecules are soluble, short-lived proteins produced, constitutively or under proper stimulation, by several cell types.61 In muscle biopsies of patients with IIM several cytokines can be amplified or immunolocalized: interleukin (IL)-1α and 1β, IL-2, IL-6, IL-10, TNF-α, IFN-γ, TGF-β e GM-CSF.62-67 Some of them might play an important role in the pathogenesis of IIM, in particular IL-1α, TNF-α and TGF-β. IL-1α was mainly expressed in endomysial capillaries, in perifascicular arterioles and venules, even in the absence of inflammation,16 and in in vitro experiments it influenced MHC expression on cultured human myoblasts and myotubes, suggesting that an altered muscle metabolism can cause an eventually immune response.68 TNF-α, an important mediator of inflammation and cellular immune responses, was occasionally expressed in mononuclear cells and on muscle fibre membranes.66,67,69 A proportion of TNF-α positive fibers were also positive for the developmental form of myosin heavy chain, indicating that TNF-α might implicated also in the regenerative process and that muscle fibers can be the target of infiltrating cells, but also an active player in the immune response.69 In muscle fibers of juvenile DM (JDM) patients TNF-α was higher expressed in those patients positive for TNF-α-308A allele than in JDM patients negative for the allele. It has been hypothesized that TNF-α-308A allele influencing the overproduction of the cytokine in response to the stimulus, contributes to the chronicity of the disease and, if not treated, to the formation of calcifications (for a comprehensive review, see ref. 70). TGF-β1 was immunolocalized in extracellular matrix of IIM muscle biopsies and never in correspondence of mononuclear cell infiltrates.71 TGF-β1, linked to the extracellular matrix, might contribute to the recruitment of mononuclear cells within the muscle, since it increases the adhesiveness of endothelial cells for leukocytes, inhibits E-selectin expression in endothelial cells72 and induces the chemokine monocyte chemoattractant protein (MCP)-1/CCL2 synthesis.73
Chemokines are chemotactic cytokines that regulate leukocyte migration into inflamed area, as well as homeostatic trafficking of lymphocytes and dendritic cells. Their primary structure is characterized by the presence of four conserved cysteine residues. The largest and best-characterized families are the α-chemokines (CXCL) and the β-chemokines (CCL).74 Several chemokines (CCL2, CCL3, CCL5, CCL9, CXCL8, CXCL9, CXCL10) have been detected in IIM muscle tissue localized in correspondence of infiltrating inflammatory cells and in the extracellular matrix with a pattern of distribution related to the different pathogenetic processes underlying the three IIM forms.75-77 Chemokine synthesis and storage in the extracellular matrix can act as a microenviromental factor amplifying lymphocyte activation and migration, thereby maintaining the autoimmune attack and tissue degeneration.
Muscle cells might be actively involved in chemokine synthesis and release in the inflamed area. It has been demonstrated that CXCL8, CCL5 were constitutively expressed by cultured myoblasts and enhanced after pro-inflammatory stimulus;78 CCL2, CXCL9, CXCL10 were induced by IFN-γ or TNF-α stimulus.77,78 These data further support the hypothesis that muscle cells are not only the target of the immune-mediated attack but that they may directly release cytokines/chemokines necessary to initiate and perpetuate immunocompetent cell recruitment.
Mechanisms of Muscle Cell Damage
Degeneration and necrosis of muscle fibers by CD8+ T lymphocytes in PM and s-IBM is predominantly mediated by release of cytotoxic enzymes: perforin and granzymes.
When cytotoxic T cells recognize the antigen via TCR, the lytic granules polarise towards the interface with the target cell (the immunological synapse), fuse with the target cell plasma membrane, and focally release soluble lytic proteins (including perforin and granzymes) to induce target cell death.79 In DM and PM perforin and granzyme transcripts were expressed at similar levels and either CD3+ CD4+ and CD3+ CD8+ T cells were perforin positive.62,80 By confocal microscopy, in DM perforin was distributed randomly in the cytoplasm of the inflammatory T cells, while in PM the cytotoxic T cells that contacted a muscle fiber showed perforin located vectorially towards the target muscle cell. This suggests that in DM perforin distribution reflects a nonspecific T cell activation, while in PM the oriented perforin distribution reflects a specific T cell activation by an antigen present on muscle fibers.80
The Fas-FasL process does not seem to be involved in IIM muscle degeneration. IIM muscle fibers and T lymphocytes, but not the control muscles, are Fas positive and FasL has been observed on some degenerating/regenerating fibers and on most of infiltrating CD8+ T cells, however, apoptotic signs are absent.81-86 The resistance to Fas-mediated cell death seems to be due to the expression of anti-apoptotic molecules heterogeneously expressed in muscle fibers: Bcl-2, Fas-associated death domain-like IL-1-converting enzyme inhibitory protein (FLIP), which inhibits Fas-mediated death signaling, and human inhibitor of apoptosis (IAP)-like protein (hILP), which inhibits the activity of caspases, all proteins that play an important role in initiating and maintaining the apoptotic process.81,83,87
Gene Expression Profiles
As yet a limited experience on gene expression profile of IIM muscle biopsies by microarray technology has been reported. The study from Greenberg et al88 showed that muscle tissues from IIM patients expressed genes different from the normal muscles and that these gene profiles were diverse among the different IIM forms. With this approach the authors had the possibility to make diagnosis in two patients for whom the muscle biopsy did not show the classical pathological alterations. Several genes (MHC class I and II, cytokines, chemokines, granzyme proteases, adhesion molecules, matrix metalloproteinases) are overexpressed confirming previous data obtained with other approaches (PCR and/or immunohistochemistry). On the other hand, the molecular approach revealed some unexpected results: for example, keeping in mind the histopathology of the single IIM forms, a number of immunoglobulin genes were more abundantly expressed in PM/IBM than in DM, while IFN-inducible genes were more expressed in DM than PM/IBM, the latter result resembles that observed in JDM, where a viral antigen as triggering factor has been hypothesized;89 genes reported as relevant for IBM pathogenesis are also significantly overexpressed in PM and DM, suggesting that in IBM the abnormal accumulation of different proteins might be due to post-transcriptional defects.
IBM-Specific Genetics
A characteristic feature of IBM muscle is the abnormal accumulation, aggregation, and misfolding of several proteins, a scenario similar to that observed in Alzheimer disease brain.3,6 The major accumulated proteins include: amyloid-β precursor protein (AβPP) and amyloid-β; phosphorylated tau in the form of paired helical filaments (PHFs); presenilin-1. Since in s-IBM the disease onset is usually after age 50 years, it has been hypothesized that the abnormal protein accumulation might be due to a defective processing related to muscle fiber aging. The abnormally processed proteins might then make the muscle fiber a “foreign” to be attacked by the immune system. For h-IBM, in which no signs of inflammation are observed, responsible for protein accumulations might be a genetic defect. Candidate genes are: UDP-N-acetylglucosamine-2 epimerase/N-acetylmannosamine kinase (GNE) gene, myosin heavy chain IIa gene, transthyterin (for a comprehensive review see refs. 3,6). GNE is a bifunctional enzyme catalyzing the first two steps in the synthesis of N-acetylneuraminic (sialic) acid. Any dysregulation of sialic acid biosynthesis and distribution could lead to severe abnormalities of glycoconjugate biosynthesis. Missense mutations were identified in h-IBM Iranian Jews, Japanese and few other ethnic groups. The mutations might induce an uncorrect sialation/glycation of one or several muscle proteins, causing their misfolding and eventually abnormal processing. A missense mutation in the myosin heavy chain IIa gene has been reported in Swedish h-IBM; the observation of an overexpression of myosin heavy chain IIa protein in the IBM vacuoles suggests that mutations in myosin heavy chain IIa gene might influence the formation of vacuoles. Transthyterin binds β-amyloid preventing its fibrillar amyloidogenesis; a transthyterin mutation (Val122Ile), found in a patient with h-IBM and cardiac amyloidosis, might cause the abnormal β-amyloid deposits and amyloidogenesis.6
Acknowledgements
The authors wish to thank their research and clinical colleagues, and particularly Dr. Paolo Confalonieri, for their participation in various aspects of these studies and in the preparation of the present manuscript.
References
- 1.
- Mantegazza R, Bernasconi P, Confalonieri P. et al. Inflammatory myopathies and systemic disorder: A review of immunopathogenetic mechanisms and clinical features. J Neurol. 1997;244:277–287. [PubMed: 9178151]
- 2.
- Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet. 2003;362:971–982. [PubMed: 14511932]
- 3.
- Askanas V, Engel WK. Inclusion-body myositis and myopathies: Different etiologies, possibly similar pathogenic mechanisms. Curr Opin Neurol. 2002;15:525–531. [PubMed: 12351995]
- 4.
- Hoogendijk JE, Amato AA, Lecky BR. et al. 119th ENMC International Workshop: Trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis. Neuromuscul Disord. 2004;14:337–345. [PubMed: 15099594]
- 5.
- Santmyire-Rosenberger B, Dugan EM. Skin involvement in dermatomyositis. Curr Opin Rheumatol. 2003;15:714–722. [PubMed: 14569200]
- 6.
- Askanas V, Engel WK. Proposed pathogenetic cascade of inclusion-body myositis: Importance of amyloid-β, misfolded proteins, predisposing genes, and aging. Curr Opin Rheumatol. 2003;15:737–744. [PubMed: 14569203]
- 7.
- Shamin EA, Rider LG, Miller FW. Update on the genetics of the idiopathic inflammatory myopathies. Curr Opin Rheumatol. 2000;12:482–491. [PubMed: 11092196]
- 8.
- Rider LG, Gurley RC, Pandey JP. et al. Clinical, serologic, and immunogenetic features of familial idiopathic inflammatory myopathy. Arthritis Rheum. 1998;41:710–719. [PubMed: 9550481]
- 9.
- Hausmanowa-Petrusewicz I, Kowalska-Oledzka E, Miller FW. et al. Clinical, serologic, and immunogenetic features in polish patients with idiopathic inflammatory myopathies. Arthritis Rheum. 1997;40:1257–1266. [PubMed: 9214426]
- 10.
- Arnett FC, Targoff IN, Mimori T. et al. Interrelationship of major histocompatibility complex class II alleles and autoantibodies in four ethnic groups with various forms of myositis. Arthritis Rheum. 1996;39:1507–1518. [PubMed: 8814062]
- 11.
- Lampe JB, Gossrau G, Kempe A. et al. Analysis of HLA class I and II alleles in sporadic inclusion-body myositis. J Neurol. 2003;250:1313–1317. [PubMed: 14648147]
- 12.
- Karpati G, Pouiliot Y, Carpenter S. Expression of immunoreactive major histocompatibility complex products in human skeletal muscles. Ann Neurol. 1988;23:64–72. [PubMed: 3278673]
- 13.
- Englund P, Lindroos E, Nennesmo I. et al. Skeletal muscle fibers express major histocompatibility complex class II antigens independently of inflammatory infiltrates in inflammatory myopathies. Am J Pathol. 2001;159:1263–1273. [PMC free article: PMC1850491] [PubMed: 11583954]
- 14.
- Andreetta F, Bernasconi P, Torchiana E. et al. T-cell infiltration in polymyositis is characterized by coexpression of cytotoxic and T-cell-activating cytokine transcripts. Ann NY Acad Sci. 1995;756:418–420. [PubMed: 7645862]
- 15.
- Lundberg I, Ulfgren AK, Nyberg P. et al. Cytokine production in muscle tissue of patients with idiopathic inflammatory myopathies. Arthritis Rheum. 1997;40:865–874. [PubMed: 9153548]
- 16.
- Nyberg P, Wikman A-L, Nennesmo I. et al. Increased expression of interleukin-1 alpha and MHC class I in muscle tissue of patients with chronic, inactive polymyositis and dermatomyositis. J Rheumatol. 2000;27:940–948. [PubMed: 10782820]
- 17.
- Englund P, Nennesmo I, Klareskog L. et al. Interleukin-1alpha expression in capillaries and major histocompatibility complex class I expression in type II muscle fibers from polymyositis and dermatomyositis patients: Important pathogenic features independent of inflammatory cell clusters in muscle tissue. Arthritis Rheum. 2002;46:1044–1055. [PubMed: 11953983]
- 18.
- Nagaraju K, Raben N, Loeffler L. et al. Conditional up-regulation of MHC class I in skeletal muscle leads to self-sustaining autoimmune myositis and myositis-specific autoantibodies. Proc Natl Acad Sci USA. 2000;97:9209–9214. [PMC free article: PMC16847] [PubMed: 10922072]
- 19.
- Mantegazza R, Hughes SM, Mitchell D. et al. Modulation of MHC class II antigen expression in human myoblasts after treatment with IFN-γ Neurology. 1991;41:1128–1132. [PubMed: 1906147]
- 20.
- Holhfeld R, Engel AG. Induction of HLA-DR expression on human myoblasts with interferon-gamma. Am J Pathol. 1990;136:503–508. [PMC free article: PMC1877498] [PubMed: 2107747]
- 21.
- Goebels N, Michaelis D, Wekerle H. et al. Human myoblasts as antigen-presenting cells. J Immunol. 1992;149:661–667. [PubMed: 1352532]
- 22.
- Mantegazza R, Gebbia M, Mora M. et al. Major histocompatibility complex class II molecule expression on muscle cells is regulated by differentiation: Implications for the immunopathogenesis of muscle autoimmune diseases. J Neuroimmunol. 1996;68:53–60. [PubMed: 8784260]
- 23.
- Beauchamp JR, Abraham DJ, Bou-Gharios G. et al. Expression and function of heterotypic adhesion molecules during differentiation of human skeletal muscle in culture. Am J Pathol. 1992;140:387–401. [PMC free article: PMC1886444] [PubMed: 1739132]
- 24.
- Michaelis D, Goebels N, Hohlfeld R. et al. Costitutive and cytokine-induced expression of human leukocyte antigens and cell adhesion molecules by human myotubes. Am J Pathol. 1993;143:1142–1149. [PMC free article: PMC1887076] [PubMed: 8214008]
- 25.
- Hardiman O, Faustman D, Li X. et al. Expression of major histocompatibility complex antigens in cultures of clonally derived human myoblasts. Neurology. 1993;43:604–608. [PubMed: 8451007]
- 26.
- Carosella ED, Paul P, Moreau P. et al. HLA-G and HLA-E: Fundamental and pathophysiological aspects. Immunol Today. 2000;21:532–534. [PubMed: 11221681]
- 27.
- Carosella ED, Moreau P, Aractingi S. et al. HLA-G: A shield against inflammatory aggression. Trends Immunol. 2001;22:553–555. [PubMed: 11574278]
- 28.
- Wiendl H, Behrens L, Maier S. et al. Muscle fibers in inflammatory myopathies and cultured myoblasts express the nonclassical major histocompatibility antigen HLA-G. Ann Neurol. 2000;48:679–684. [PubMed: 11026456]
- 29.
- Wiendl H, Mitsdoerffer M, Hofmeister V. et al. The nonclassical MHC molecule HLA-G protects human muscle cells from immune-mediated lysis: Implications for myoblast transplantation and gene therapy. Brain. 2003;126:176–185. [PubMed: 12477705]
- 30.
- Wiendl H, Mitsdoerffer M, Weller M. Express and protect yourself: The potential role of HLA-G on muscle cells and in inflammatory myopathies. Hum Immunol. 2003;64:1050–1056. [PubMed: 14602235]
- 31.
- Carreno BM, Collins M. The B7 family of ligands and its receptors: New pathways for costimulation and inhibition of immune responses. Annu Rev Immunol. 2002;20:29–53. [PubMed: 11861596]
- 32.
- Nagaraju K, Raben N, Villalba ML. et al. Costimulatory markers in muscle of patients with idiopathic inflammatory myopathies and in cultured muscle cells. Clin Immunol. 1999;92:161–169. [PubMed: 10444360]
- 33.
- Bernasconi P, Confalonieri P, Andreetta F. et al. The expression of costimulatory and accessory molecules on cultured human muscle cells is not dependent on stimulus by pro-inflammatory cytokines: Relevance for the pathogenesis of inflammatory myopathy. J Neuroimmunol. 1998;85:52–58. [PubMed: 9626997]
- 34.
- Behrens L, Kerschensteiner M, Misgeld T. et al. Human muscle cells express a functional costimulatory molecule distinct from B7.1 (CD80) and B7.2 (CD86) in vitro and in inflammatory lesions. J Immunol. 1998;161:5943–5951. [PubMed: 9834075]
- 35.
- Murata K, Dalakas MC. Expression of the costimulatory molecule BB-1, the ligands CTLA-4 and CD28, and their mRNA in inflammatory myopathies. Am J Pathol. 1999;155:453–460. [PMC free article: PMC1866856] [PubMed: 10433938]
- 36.
- Curnow SJ, Willcox N, Vincent A. Induction of primary immune responses by allogeneic human myoblasts: Dissection of the cell types required for proliferation, IFNgamma secretion and cytotoxicity. J Neuroimmunol. 1998;86:53–62. [PubMed: 9655472]
- 37.
- Wiendl H, Mitsdoerffer M, Schneider D. et al. Muscle fibres and cultured muscle cells express the B7.1/2-related inducible costimulatory molecule, ICOSL: Implications for the pathogenesis of inflammatory myopathies. Brain. 2003;126:1026–1035. [PubMed: 12690043]
- 38.
- Beier KC, Hutloff A, Dittrich AM. et al. Induction, binding specificity and function of human ICOS. Eur J Immunol. 2000;30:3707–3717. [PubMed: 11169414]
- 39.
- Wiendl H, Mitsdoerffer M, Schneider D. et al. Human muscle cells express a B7-related molecule, B7-H1, with strong negative immune regulatory potential: A novel mechanism of counterbalancing the immune attack in idiopathic inflammatory myopathies. FASEB J. 2003;17:1892–1894. [PubMed: 12923066]
- 40.
- Garcia KC, Teyton L, Wilson IA. Structural basis of T cell recognition. Annu Rev Immunol. 1999;17:369–397. [PubMed: 10358763]
- 41.
- Davis MM, Boniface JJ, Reich Z. et al. Ligand recognition by αβ T cell receptors. Annu Rev Immunol. 1998;16:523–544. [PubMed: 9597140]
- 42.
- Mantegazza R, Andreetta F, Bernasconi P. et al. Analysis of T cell receptor repertoire of muscle-infiltrating T lymphocytes in polymyositis. J Clin Invest. 1993;91:2880–2886. [PMC free article: PMC443358] [PubMed: 8514895]
- 43.
- Bender A, Ernst N, Iglesias A. et al. T cell receptor repertoire in polymyositis: Clonal expansion of autoaggressive CD8+ T cells. J Exp Med. 1995;181:1863–1868. [PMC free article: PMC2192015] [PubMed: 7722460]
- 44.
- O'Hanlon TP, Dalakas MC, Plotz PH. et al. Predominant TCR-αβ variable and joining gene expression by muscle-infiltrating lymphocytes in the idiopathic inflammatory myopathies. J Immunol. 1994;152:2569–2576. [PubMed: 8133064]
- 45.
- Mantegazza R, Bernasconi P, Torchiana E. et al. Molecular analysis of T cell receptor repertoire of T cell infiltrates in sporadic and familial inclusion body myositis. Muscle Nerve. 1994;17(Suppl 1):117.
- 46.
- Lindberg C, Oldfors A, Tarkowski A. Restricted use of T cell receptor V genes in endomysial infiltrates of patients with inflammatory myopathies. Eur J Immunol. 1994;24:2659–2663. [PubMed: 7957558]
- 47.
- O'Hanlon TP, Dalakas MC, Plotz PH. et al. The αβ T-cell receptor repertoire in inclusion body myositis: Diverse patterns of gene expression by muscle-infiltrating lymphocytes. J Autoimmun. 1994;7:321–333. [PubMed: 7916906]
- 48.
- Fyhr IM, Moslemi AR, Mosavi AA. et al. Oligoclonal expansion of muscle infiltrating T cells in inclusion body myositis. J Neuroimmunol. 1997;79:185–189. [PubMed: 9394791]
- 49.
- Fyhr IM, Moslemi AR, Lindberg C. et al. T cell receptor β-chain repertoire in inclusion body myositis. J Neuroimmunol. 1998;91:129–134. [PubMed: 9846829]
- 50.
- Bender A, Behrens L, Engel AG. et al. T-cell heterogeneity in muscle lesions of inclusion body myositis. J Neuroimmunol. 1998;84:86–91. [PubMed: 9600712]
- 51.
- Amemiya K, Granger RP, Dalakas MC. Clonal restriction of T-cell receptor expression by infiltrating lymphocytes in inclusion body myositis persists over time: Studies in repeated muscle biopsies. Brain. 2000;123:2030–2039. [PubMed: 11004120]
- 52.
- Nishio J, Suzuki M, Miyasaka N. et al. Clonal biases of peripheral CD8 T cell repertoire directly reflect local inflammation in polymyositis. J Immunol. 2001;167:4051–4058. [PubMed: 11564826]
- 53.
- Benveniste O, Chérin P, Maisonobe T. et al. Severe perturbations of the blood T cell repertoire in polymyositis, but not in dermatomyositis patients. J Immunol. 2001;167:3521–3529. [PubMed: 11544346]
- 54.
- Hofbauer M, Wiesener S, Babbe H. et al. Clonal tracking of autoaggressive T cells in polymyositis by combining laser microdissection, single-cell PCR, and CDR3-spectratype analysis. Proc Natl Acad Sci USA. 2003;100:4090–4095. [PMC free article: PMC153053] [PubMed: 12651958]
- 55.
- Hengstman GJD, van EngelenBGM, Vree Egberts WTM. et al. Myositis-specific autoantibodies: Overview and recent developments. Curr Opin Rheumatol. 2001;13:476–482. [PubMed: 11698723]
- 56.
- Lieberman J. The ABCs of granule-mediated cytotoxicity: New weapons in the arsenal. Nat Rev Immunol. 2003;3:361–370. [PubMed: 12766758]
- 57.
- Casciola-Rosen L, Andrade F, Ulanet D. et al. Cleavage by granzyme B is strongly predictive of autoantigen status: Implications for initiation of autoimmunity. J Exp Med. 1999;190:815–825. [PMC free article: PMC2195625] [PubMed: 10499920]
- 58.
- Levine SM, Rosen A, Casciola-Rosen LA. Anti-aminoacyl tRNA synthetase immune responses: Insights into the pathogenesis of the idiopathic inflammatory myopathies. Curr Opin Rheumatol. 2003;15:708–713. [PubMed: 14569199]
- 59.
- Howard OMZ, Dong HF, Yang D. et al. Histidyl-tRNA synthetase and asparaginyl-tRNA synthetase, autoantigens in myositis, activate chemokine receptors on T lymphocytes and immature dendritic cells. J Exp Med. 2002;196:781–791. [PMC free article: PMC2194054] [PubMed: 12235211]
- 60.
- Ascherman DP, Oriss TB, Oddis CV. et al. Critical requirement for professional APCs in eliciting T cell responses to novel fragments of histidyl-tRNA synthetase (Jo-1) in Jo-1 antibody-positive polymyositis. J Immunol. 2002;169:7127–7134. [PubMed: 12471150]
- 61.
- Janeway CA, Bottomly K. Signals and signs for lymphocyte responses. Cell. 1994;76:275–285. [PubMed: 7904901]
- 62.
- Andreetta F, Bernasconi P, Torchiana E. et al. T-cell infiltration in polymyositis is characterized by coexpression of cytotoxic and T-cell-activating cytokine transcripts. Ann NY Acad Sci. 1995;756:418–420. [PubMed: 7645862]
- 63.
- Lundberg I, Brengman JM, Engel AG. Analysis of cytokine expression in muscle in inflammatory myopathies, Duchenne dystrophy, and nonweak controls. J Neuroimmunol. 1995;63:9–16. [PubMed: 8557829]
- 64.
- Lundberg I, Ulfgren AK, Nyberg P. et al. Cytokine production in muscle tissue of patients with idiopathic inflammatory myopathies. Arthritis Rheum. 1997;40:865–874. [PubMed: 9153548]
- 65.
- Tews DS, Goebel HH. Cytokine expression profile in idiopathic inflammatory myopathies. J Neuropathol Exp Neurol. 1996;55:342–347. [PubMed: 8786392]
- 66.
- De BleeckerJL, Meire VI, Declercq W. et al. Immunolocalization of tumor necrosis factor-alpha and its receptors in inflammatory myopathies. Neuromusc Disord. 1999;9:239–246. [PubMed: 10399751]
- 67.
- Tateyama M, Nagano I, Yoshioka M. et al. Expression of tumor necrosis factor-alpha in muscles in polymyositis. J Neurol Sci. 1997;146:45–51. [PubMed: 9077495]
- 68.
- Nagaraju K, Raben N, Merritt G. et al. A variety of cytokines and immunologically relevant surface molecules are expressed by normal human skeletal muscle cells under proinflammatory stimuli. Clin Exp Immunol. 1998;113:407–414. [PMC free article: PMC1905062] [PubMed: 9737670]
- 69.
- Kuru S, Inukai A, Kato T. et al. Expression of tumor necrosis factor-α in regenerating muscle fibers in inflammatory and noninflammatory myopathies. Acta Neuropathol. 2003;105:217–224. [PubMed: 12557007]
- 70.
- Uzel G, Pachman LM. Cytokines in juvenile dermatomyositis pathophysiology: Potential and challenge. Curr Opin Rheumatol. 2003;15:691–697. [PubMed: 14569197]
- 71.
- Confalonieri P, Bernasconi P, Cornelio F. et al. Transforming growth factor-beta1 in polymyositis and dermatomyositis correlates with fibrosis but not with mononuclear cell infiltrate. J Neuropathol Exp Neurol. 1997;56:479–484. [PubMed: 9143260]
- 72.
- Gamble JR, Khew-Goodall Y, Vadas MA. Transforming growth factor-beta inhibits E-selectin expression on human endothelial cells. J Immunol. 1993;150:4494–4503. [PubMed: 7683321]
- 73.
- Hurwitz AA, Lyman WD, Berman JW. Tumor necrosis factor α and transforming growth factor β up-regulate astrocyte expression of monocyte chemoattractant protein-1. J Neuroimmunol. 1995;57:193–198. [PubMed: 7706436]
- 74.
- Zlotnik A, Yoshie O. Chemokines: A new classification system and their role in immunity. Immunity. 2000;12:121–127. [PubMed: 10714678]
- 75.
- Confalonieri P, Bernasconi P, Megna P. et al. Increased expression of β-chemokines in muscle of patients with inflammatory myopathies. J Neuropathol Exp Neurol. 2000;59:164–169. [PubMed: 10749105]
- 76.
- De BleeckerJL, De PaepeB, Vanwalleghem IE. et al. Differential expression of chemokines in inflammatory myopathies. Neurology. 2002;58:1779–1785. [PubMed: 12084877]
- 77.
- Raju R, Vasconcelos O, Granger R. et al. Expression of IFN-gamma-inducible chemokines in inclusion body myositis. J Neuroimmunol. 2003;141:125–131. [PubMed: 12965263]
- 78.
- De RossiM, Bernasconi P, Baggi F. et al. Cytokines and chemokines are both expressed by human myoblasts: Possible relevance for the immune pathogenesis of muscle inflammation. Int Immunol. 2000;12:1329–1335. [PubMed: 10967028]
- 79.
- Lieberman J. Mechanisms of granule-mediated cytotoxicity. Curr Opin Immunol. 2003;15:513–515.
- 80.
- Goebels N, Michaelis D, Engelhardt M. et al. Differential expression of perforin in muscle-infiltrating T cells in polymyositis and dermatomyositis. J Clin Invest. 1996;97:2905–2910. [PMC free article: PMC507387] [PubMed: 8675705]
- 81.
- Nagaraju K, Casciola-Rosen L, Rosen A. et al. The inhibition of apoptosis in myositis and in normal muscle cells. J Immunol. 2000;164:5459–5465. [PubMed: 10799913]
- 82.
- Inukai A, Kobayashi Y, Ito K. et al. Expression of Fas antigen is not associated with apoptosis in human myopathies. Muscle Nerve. 1997;20:702–709. [PubMed: 9149077]
- 83.
- Behrens L, Bender A, Johnson MA. et al. Cytotoxic mechanisms in inflammatory myopathies. Coexpression of Fas and protective Bcl-2 in muscle fibres and inflammatory cells. Brain. 1997;120:929–938. [PubMed: 9217678]
- 84.
- Olive M, Martinez-Matos JA, Montero J. et al. Apoptosis is not the mechanism of cell death of muscle fibers in human muscular dystrophies and inflammatory myopathies. Muscle Nerve. 1997;20:1328–1330. [PubMed: 9324094]
- 85.
- Schneider C, Gold R, Dalakas MC. et al. MHC class I-mediated cytotoxicity does not induce apoptosis in muscle fibers nor in inflammatory T cells: Studies in patients with polymyositis, dermatomyositis, and inclusion body myositis. J Neuropathol Exp Neurol. 1996;55:1205–1209. [PubMed: 8957443]
- 86.
- Tews DS, Goebel HH. Cell death and oxidative damage in inflammatory myopathies. Clin Immunol Immunopathol. 1998;87:240–247. [PubMed: 9646833]
- 87.
- Li M, Dalakas MC. Expression of human IAP-like protein in skeletal muscle: A possible explanation for the rare incidence of muscle fiber apoptosis in T-cell mediated inflammatory myopathies. J Neuroimmunol. 2000;106:1–5. [PubMed: 10814776]
- 88.
- Greenberg SA, Sanoudou D, Haslett JN. et al. Molecular profiles of inflammatory myopathies. Neurology. 2002;59:1170–1182. [PubMed: 12391344]
- 89.
- Tezak Z, Hoffman EP, Lutz JL. et al. Gene expression profiling in DQA1*0501+ children with untreated dermatomyositis: A novel model of pathogenesis. J Immunol. 2002;168:4154–4163. [PubMed: 11937576]
- Inflammatory Myopathies: Dermatomyositis, Polymyositis and Inclusion Body Myosit...Inflammatory Myopathies: Dermatomyositis, Polymyositis and Inclusion Body Myositis - Madame Curie Bioscience Database
- Caspases as Targets for Drug Development - Madame Curie Bioscience DatabaseCaspases as Targets for Drug Development - Madame Curie Bioscience Database
- Insights into the Modulation of Ceramide Metabolism by Naturally Occurring and S...Insights into the Modulation of Ceramide Metabolism by Naturally Occurring and Synthetic Sphingolipid Analogs as Monitored by Electrospray Tandem Mass Spectrometry - Madame Curie Bioscience Database
- Modeling Structure-Activity Relationships - Madame Curie Bioscience DatabaseModeling Structure-Activity Relationships - Madame Curie Bioscience Database
- Regional Differences and Variability in Left Ventricular Wall Motion - Madame Cu...Regional Differences and Variability in Left Ventricular Wall Motion - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...