

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Introduction
During an action potential calcium (Ca2+) ions enter the cell through voltage-gated Ca2+ channels (Cav). Cav channels first open and subsequently close before recovering to the resting state (fig. 1A). The process of channel closure during maintained membrane depolarization is called “inactivation”. During inactivation Cav channels undergo several conformational changes: one is mediated by intracellular Ca2+ interacting with a channel-calmodulin complex (Ca2+-dependent inactivation), two other conformational changes are voltage-dependent (fast and slow voltage-dependent inactivation, fig. 1A, see refs. 1, 2 for review, and other chapters in this book).
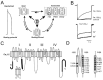
Evolution has designed high-voltage-activated (HVA) Cav channel families (also known as L-type [Cav1.1, Cav1.2, Cav1.3, Cav1.4], P/Q-type [Cav2.1], N-type [Cav2.2] and R-type [Cav2.3] channels) and low-voltage-activated (LVA) Ca2+ channel family Cav3.3 The different family members display different inactivation properties.1,2 HVA Cav channels are heterooligomeric protein complexes composed of an a1-subunit, auxiliary β-, α2-δ- and in some channels of an additional γ-subunit.4 The α1-subunits sense the membrane voltage, form the pore and define the basic pharmacological properties of the channels5-8 (fig. 1C, D). Four β-subunit genes (β1, β2, β3, β4) and various splice variants have been identified. These subunits modulate the inactivation properties of HVA Cav (fig. 1B).6 HVA and LVA Cav channels undergo fast and slow voltage-dependent inactivation.9-12 Ca2+/calmodulin-dependent inactivation is exclusively observed in HVA (Cav1.2, Cav2.1 and other HVA Cav channels).13-17
The blocking action of many Ca2+ antagonists such as the 1,4-dihydropyridines (DHPs), phenylalkylamines (PAAs), diltiazem (DIL) and mibefradil (MIB) in myocardial, smooth muscle and neuronal cells is enhanced by membrane depolarizations.18-23 This is conventionally explained by a higher affinity of these drugs for open or inactivated channels (fig. 1A). The precise mechanisms of channel block by these compounds remains to be elucidated.
Recent studies on chimeric and mutant Cav channels provided new insights into the underlying molecular events. In particular the design of channels with different inactivation properties may help us to understand the molecular role of inactivation in channel inhibition by Ca2+ antagonists.
Voltage-Dependent Inactivation and Drug Sensitivity of Chimeric Cav
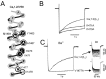
First evidence for a link between structural determinants of Cav channel inactivation and drug-sensitivity came from studies on chimeric channels. In order to localise the drug-binding determinants for Ca2+ antagonists Grabner et al24 inserted various sequence stretches of a PAA, DHP and DIL-sensitive α1 subunit of an L-type channel into the poorly sensitive α12.1 (fig. 2A). Transfer of Cav1.2 segments IIIS5, IIIS6 and IVS6 to Cav2.1 confered PAA- and DIL-sensitivity to the resulting construct AL12 (fig.2B).25

Analysis of the inactivation of some of the chimeric channels revealed a correlation between inactivation kinetics and drug sensitivity. Substitution of domains IIIS5, IIIS6 and the connecting linker (chimera AL20, fig. 2C) diminished both inactivation and block by DIL and PAA.25 This was surprising because AL20 carries more than half of the PAA- and DIL-determinants (fig. 1D). The almost complete loss of drug sensitivity raised the suspicion that the process of channel inactivation might interfere with drug action. This hypothesis was further supported by chimera AL22 lacking all PAA- and DIL-determinants in segment IIIS6 but inactivating significantly faster than AL12 and Cav2.1 (fig. 2D). This channel exhibited PAA- and DIL-sensitivity comparable to (or even higher) than chimera AL12 and wild type Cav1.2.25
A further example of a correlation between inactivation and drug-sensitivity in a chimeric channel comes from the work of Motoike et al26 who substituted a small sequence stretch outside the putative drug binding region of Cav1.2 in the intracellular loop between IVS4 and IVS5 by the homologous residues from the rat brain Na+ channel (chimera HHT-5371). As illustrated in Figure 2E, this substitution simultaneously diminished channel inactivation and DIL-sensitivity.
Amino Acid Residues Located in the Putative Drug-Binding Region Affect Drug-Sensitivity and Channel Inactivation
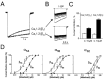
Deeper insights into the interdependence between inactivation and channel block were enabled by the design of mutant Cav channels with different inactivation properties. Hering et al25 demonstrated that transfer of only three Cav1.2-specific amino acids in segment IVS6 to Cav2.1 (Y1804, A1808 and I1811 a1A numbering, named chimera AL2525, fig. 3A) confers high sensitivity for (-)gallopamil and DIL (fig. 3B, compare top left and top right traces). The authors paid, however, attention to the fact that these mutations significantly accelerated channel inactivation (fig. 3B, top left and right traces). Furthermore, mutating either A1808 back to serine or I1811 back to methionine (fig. 3B, middle panels) reduced not only PAA- and DIL-sensitivities but, simultaneously, the rate of channel inactivation.

On the other hand, insertion of a fourth Cav1.2 residue (M1805) into the corresponding position of AL25 further enhanced inactivation and sensitivity for (-)gallopamil and DIL (fig. 3B, bottom left). These data confirmed a close relation between the rate of intrinsic inactivation and the apparent sensitivity for Ca2+ antagonists. We hypothesise that the augmentation of the use-dependent block at least for some of the mutants is mediated by enhanced inactivation rather than by transfer of specific binding determinants.
Interestingly, mutation Y1804I decreased DIL- (and PAA-) sensitivity without significantly slowing the rate of channel inactivation (fig.3B bottom right). Y1804 is, therefore, more likely to contribute drug-binding energy than the other IVS6 residues that could equally modulate drug sensitivity in an indirect manner by affecting inactivation. A possible key role of Y1804 in drug binding is also supported by data from Degtiar et al who demonstrated low PAA-sensitivity of a chimeric construct (AL30) inactivating even faster than AL25 but lacking Y1804.27
It was therefore interesting to study if the putative drug binding determinants in the drug sensitive wild type Cav1.2 would affect inactivation. A first analysis of the impact of IVS6 amino acids in equivalent positions (fig.4) revealed that substitution of I1470 (corresponding to the Cav2.1 M1811, (fig.3A) and the adjacent DHP-determinant I1471 by alanine both substantially slow channel inactivation (fig. 4A,B).
Drug-Sensitivity Is Affected by Inactivation Determinants Located Outside the Putative Drug-Binding Region
A similar correlation between channel inactivation and apparent sensitivity for Ca2+ antagonists was observed for amino acids substitutions outside the putative drug-binding region. As illustrated in Figure 4C, substitution of valine (V1478) in the lower part of segment IVS6 of Cav1.2 by alanine significantly reduced both inactivation and diltiazem sensitivity. A systematic analysis of amino acids in segment IIIS6 of a Cav1.1/Cav2.1 chimera revealed a strong correlation between the two parameters (fig. 5C).28 Substitution of two residues in segment IIIS6 localised close to the inner mouth of the channel pore by alanine (I1163A and F1164A, fig. 5) and to an even greater extent the corresponding double mutation (IF1163,1164AA) gradually reduced the rate of current inactivation and simultaneously diminished use-dependent block by (-)gallopamil (fig. 5B).

By investigating the effect of a single amino acid substitution in the I-II linker (R387E in Cav2.1) Sokolov et al29 observed a similar trend: the slower the channel inactivated the lower was the apparent sensitivity for (-)gallopamil. In addition, Jimenez et al30 reported that ultraslow inactivation generated by a single valine insertion in to the I-II linker of the Cav1.2 α1 subunit strongly reduced use-dependent channel block by MIB. Together these data illustrate that Ca2+ channel block is modulated by inactivation determinants that are located inside as well as outside of the putative drug-binding region.
β-Subunits Modulate Inactivation and Channel Inhibition
It is well established that the β-subunit composition of HVA Cav channels affects their inactivation properties.6 According to a widely accepted hypothesis many Ca2+ antagonists bind predominately (with high affinity) to the inactivated channel state.20,21 Since β-subunits either enhance or reduce Cav channel inactivation, they have emerged as important tools for the analysis of state dependent drug action. Coexpression of the α1 subunit with ‘accelerating’ β-subunits (β1- or β3-) was, therefore, expected to enhance channel block.
In line with this hypothesis Lacinova et al31 reported an almost 14-fold higher apparent sensitivity of Cav1.2 for the PAA verapamil if the Cav1.2 α1-subunit was coexpressed with a β3-subunit (Cav1.2(β3)).
(-)Gallopamil inhibited Cav1.2(β3) 3.4-fold stronger than Cav1.2 α1 subunits expressed without β-subunits.31 Sokolov et al32 studied the underlying molecular mechanism and compared PAA-sensitivity of Cav1.2 composed of either an ‘accelerating’ β3-subunits or a ‘decelerating’ β2a. These studies revealed a relation between the rate of β-mediated inactivation and PAA-sensitivity (fig.6A-C).
Jimenez et al30 demonstrated that inhibition of Cav2.1, Cav2.3 and Cav1.2 by MIB is significantly stronger if the corresponding α1-subunits are coexpressed with an ‘accelerating’ β1b rather than with a ‘decelerating’ β2a-subunit. The concentration of drug required to block Cav2.1 and Cav2.3 composed of β2a was approximately 10-fold higher (fig. 6D, left, middle). A smaller difference was observed for Cav1.2 (fig. 6D, right). Further evidence for a role of the β-subunit interaction in Ca2+ channel block comes from Zamponi et al33 who reported that coexpression of the Cav2.3 α1 subunit with β2a impaired channel block by piperidines compared to Cav2.3 coexpressed with the ‘accelerating’ β1b. In summary, these data suggest that a β-subunit-mediated increase or reduction in voltage-dependent inactivation modulates Cav channel inhibition by PAAs, MIB and piperidines.
Inactivation Determinants and DHP Sensitivity
Evidence for a key role of inactivation in channel inhibition by DHPs came from the drug-induced shift of Cav1.2 availability to hyperpolarized voltages in myocardial and smooth muscle cells. This is conventionally explained as high affinity binding of drugs such as nifedipine or isradipine to the inactivated channel conformation.20,21 Cav1.2 composed of ‘accelerating’ β1a- (Cav1.2(β1a)) or β3-subunits (Cav1.2(β3)) were, therefore, expected to enhance DHP-binding and channel block.
However, Lacinova et al31 reported almost identical sensitivities of Cav1.2(β3) and Cav1.2 channels lacking the β subunit for (+)isradipine, despite the considerable differences in inactivation.
Berjukow et al34 came to a similar conclusion by comparing the DHP sensitivities of Cav channels containing either β1a or β2a subunits (fig.7). A DHP-sensitive Cav2.1 mutant composed of the ‘accelerating’ β1a (α1A-DHP/β1a) displayed at —80 mV a 3-fold higher steady-state fraction of channels in an inactivated state compared to slowly inactivating α1A-DHP/β2a. This larger fraction of hypothetically high affinity drug receptors did, however, not promote additional channel block by (+)isradipine (fig. 7A). Analogous observations were made for Cav1.2, the prime target of DHP in vascular smooth muscle cells (fig. 7).34
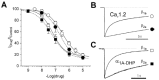
There is, nevertheless, evidence for an important role of the inactivation machinery in channel block by DHPs. The more rapidly inactivating mutant α1A-DHP displays a higher sensitivity for (+)isradipine than the slower inactivating wild-type Cav1.2, irrespective whether an ‘accelerating’ β1a or a ‘decelerating’ β2a-subunit were coexpressed (fig. 7A). It is, therefore, tempting to speculate that the higher DHP-sensitivity of α1A-DHP is related to their faster inactivation compared to Cav1.2 (fig. 7B,C).
Furthermore, Berjukow et al34 demonstrated that substitution of the putative DHP-binding determinant I1471 in segment IVS6 (fig. 1D) by alanine slowed the rate of fast voltage-dependent inactivation (fig. 8A,B) and additionally accelerated recovery of drug modified channels from inactivation. It appears that mutation I1471A destabilises the intrinsic fast inactivated state and the DHP-induced state.

A detailed study of kinetics of entry into and recovery from inactivation and DHP-inhibition of various Cav1.2 and Cav2.1 mutants led us to a hypothesis that these drugs promote fast voltage-dependent inactivation without binding selectively to this state. This idea was further supported by (+)-isradipine action on Cav1.2 mutant V1477A that is almost completely lacking fast voltage-dependent inactivation. (+)-Isradipine accelerated the current decay in a voltage-dependent manner comparable to voltage dependence of inactivation of wild type Cav1.2 (fig. 8C). The effect of (+)-isradipine on this channel can, therefore, be interpreted as a restoration of fast voltage-dependent inactivation.35
On the Role of Ca2+-Dependent Inactivation in Drug Sensitivity
Ca2+-dependent inactivation causes an additional rapid component in current decay through Cav1.2 channels (fig. 1B and 9A). Sokolov et al32 reported that accelerated inactivation with Ca2+ as charge carrier (compared to Ba2+) is accompanied by enhanced use-dependent block by (-)gallopamil (fig.9B). This effect was apparently caused by a slower recovery from channel block in the presence of Ca2+ (fig. 9C). Its molecular basis has yet to be elucidated.

Simulation of the Drug-Channel Interaction
Different versions of the “modulated receptor paradigm”36 explain the cumulative Ca2+ channel block by PAA, DIL and MIB during a train of test pulses (“use-dependent block”, fig. 10) by selective drug binding to either open or inactivated channels.20,21,37

Such a scenario would imply that during a single pulse the drug “has not enough time” to bind to all channels. Thus, due to the slow drug binding only a small fraction of channels would be blocked. Upon repolarization the channels recover to the low affinity resting state where the drug dissociates from its receptor site (fig. 10). Drug unbinding is, however, slow as well and a certain fraction of channels will remain blocked until the next pulse is applied leading finally to cumulative (use-dependent) channel inhibition.
Alternatively, drug binding could occur independently of the channel state. In such a scenario that we call “drug inactivation synergism,”38 Ca2+ antagonists accelerate channel transitions into the fast inactivated state like ‘accelerating’ β-subunits or point mutations.
Figure 10 illustrates the two competing models. Both of them satisfactory reproduce the experimental data. In the “drug inactivation synergism” model the effect of (-)gallopamil on Cav1.2(β3) and Cav1.2(β2a) is simulated by an about 7-fold increase in the on-rate of fast voltage-dependent inactivation (rate α(PAA)) and an 8-fold decrease in recovery from drug-induced inactivation (rate β(PAA)). The difference in use-dependent inhibition of the two β-subunit compositions arise, therefore, solely due to the differences in intrinsic inactivation in Cav1.2(β3) and Cav1.2(β2a), i.e., the “drug-inactivation synergism” model reproduces the differences in apparent drug sensitivity without the necessity to assume different drug affinities for Cav1.2(β3) and Cav1.2(β2a).
In the “open channel block” model we had to presume a higher affinity of (-)gallopamil for rapidly inactivating Cav1.2(β3) compared to Cav1.2(β2a) (kon[D](Cav(β3)) = 0.5 s-1 vs. kon[D](Cav(β2a)) = 0.15 s-1). This model reproduces the data only if we postulate a direct effect of the β-subunit on the drug binding process. Moreover, if we hypothesise equal affinities this model predicts enhanced inhibition of slowly inactivating (open) Cav channels and not—as observed in most experiments—stronger block of faster inactivating channels (figs. 2-7).
Summary and Outlook
Point mutations in different parts of the Cav α1-subunits that either enhance or impair channel inactivation simultaneously increase or reduce channel inhibition by PAA, DIL, MIB and to a certain extent by DHPs. Fast inactivating channels tend to be more efficiently inhibited than slow-inactivating ones. This simple correlation has been confirmed by independent groups for wild type, chimeric or mutant HVA Cav.25-33 A similar conclusion can be drawn the drug action on Cav channels containing either ‘accelerating’ (e.g., β1 or β3) or ‘decelerating’ (β2a) β-subunits.30,32 Subunit compositions promoting faster channel inactivation increase the apparent drug sensitivity.
A possible scenario of a “drug inactivation synergism” is illustrated in (fig. 10) This model does not exclude state-dependent drug access for partially charged compounds. However, once these drugs dock within their binding pockets they are more likely to promote inactivation than to plug the open channel pore (fig.10). Some amino acids (e.g., I1470, I1471 in segment IVS6 and the IFV motif close to the inner channel mouth in segment IIIS6 of Cav1.2 as well as M1464 and A1467 in Cav2.1 environment (fig. 1D)) are strong inactivation determinants.
It is currently not entirely clear which of the determinants of drug sensitivity in segments IIIS5, IIIS6 and IVS6 form part of the drug binding pocket and which of them affect the drug-channel interaction in an indirect manner via inactivation (fig. 1D). In functional studies the effects of mutations on PAA-, DIL- and MIB-sensitivity is indistinguishable from mutations localised outside the putative drug-binding region. The respective impact of IIIS5, IIIS6 and IVS6 amino acids in inactivation and drug binding has yet to be thoroughly elucidated.
Acknowledgements
We Thank Jörg Mitterdorfer for Comments on the Manuscript. Supported by FWF grant 15914.
References
- 1.
- Stotz SC, Zamponi GW. Structural determinants of fast inactivation of high voltage-activated Ca2+ channels. Trends Neurosci. 2001;24:176–181. [PubMed: 11182458]
- 2.
- Hering S, Berjukow S, Sokolov S. et al. Molecular determinants of inactivation in voltage-gated Ca2+ channels. J Physiol. 2000;528:237–249. [PMC free article: PMC2270139] [PubMed: 11034614]
- 3.
- Ertel EA, Campbell KP, Harpold MM. et al. Nomenclature of voltage-gated calcium channels. Neuron. 2000;25:533–535. [PubMed: 10774722]
- 4.
- Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–555. [PubMed: 11031246]
- 5.
- Striessnig J, Grabner M, Mitterdorfer J. et al. Structural basis of drug binding to L Ca2+ channels. Trends Pharmacol Sci. 1998;9:108–115. [PubMed: 9584627]
- 6.
- Walker D, De WaardM. Subunit interaction sites in voltage-dependent Ca2+ channels: Role in channel function. Trends Neurosci. 1998;21(4):148–154. [PubMed: 9554724]
- 7.
- Stea A, Tomlinson WJ, Soong TW. et al. Localization and functional properties of a rat brain alpha 1A calcium channel reflect similarities to neuronal Q- and P-type channels. Proc Natl Acad Sci USA. 1994;91:10576–10580. [PMC free article: PMC45064] [PubMed: 7524096]
- 8.
- Birnbaumer L, Qin N, Olcese R. et al. Structures and functions of calcium channel beta subunits. J Bioenerg Biomembr. 1998;30:357–375. [PubMed: 9758332]
- 9.
- Boyett MR, Honjo H, Harrison SM. et al. Ultra-slow voltage-dependent inactivation of the calcium current in guinea-pig and ferret ventricular myocytes. Pflugers Arch. 1994;428(1):39–50. [PubMed: 7971160]
- 10.
- Hering J, Maulet Y, Feltz A. et al. Slow inactivation of the Cav3.1 isoform of the T-type channel Biophys J 200180(1)p2621a.
- 11.
- Sokolov S, Weiss RG, Timin EN. et al. Modulation of slow inactivation in class A Ca2+ channels by beta-subunits. J Physiol. 2000;527(3):445–454. [PMC free article: PMC2270100] [PubMed: 10990532]
- 12.
- Degtiar VE, Scheller RH, Tsien RW. Syntaxin modulation of slow inactivation of N-type calcium channels. J Neurosci. 2000;20(12):4355–4367. [PMC free article: PMC6772443] [PubMed: 10844004]
- 13.
- Qin N, Olcese R, Bransby M. et al. Ca2+-induced inhibition of the cardiac Ca2+ channel depends on calmodulin. Proc Natl Acad Sci USA. 1999;96:2435–2438. [PMC free article: PMC26802] [PubMed: 10051660]
- 14.
- Lee A, Wong ST, Gallagher D. et al. Ca2+/Calmodulin binds to and modulates P/Q-type calcium channels. Nature. 1999;399:155–159. [PubMed: 10335845]
- 15.
- Peterson BZ, DeMaria CD, Yue DT. Calmodulin is the Ca2+ sensor for Ca2+-dependet inactivation of L-type calcium channels. Neuron. 1999;22:549–558. [PubMed: 10197534]
- 16.
- Zuhlke RD, Pitt GS, Deisseroth K. et al. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 1999;399:159–162. [PubMed: 10335846]
- 17.
- Erickson MG, Alseikhan BA, Peterson BZ. et al. Preassociation of calmodulin with voltage-gated Ca2+ channels revealed by FRET in single living cells. Neuron. 2001;31(6):973–985. [PubMed: 11580897]
- 18.
- Lee KS, Tsien RW. Mechanism of calcium channel blockade by verapamil, D600, diltiazem and nitrendipine in single dialysed heart cells. Nature. 1983;302(5911):790–794. [PubMed: 6302512]
- 19.
- McDonald TF, Pelzer D, Trautwein W. Cat ventricular muscle treated with D600: Characteristics of calcium channel block and unblock. J Physiol. 1984;352:217–241. [PMC free article: PMC1193208] [PubMed: 6086908]
- 20.
- Bean BP. Nitrendipine block of cardiac calcium channels: High-affinity binding to the inactivated state. Proc Natl Acad Sci USA. 1984;81:6388–6392. [PMC free article: PMC391929] [PubMed: 6093100]
- 21.
- Sanguinetti MC, Kass RS. Voltage-dependent block of calcium channel current in the calf cardiac purkinje fibre by dihydropyridine calcium channel antagonists. Circ Res. 1984;55:336–348. [PubMed: 6088117]
- 22.
- Bezprozvanny I, Tsien RW. Voltage-dependent blockade of diverse types of voltage-gated Ca2+ channels expressed in Xenopus oocytes by the Ca2+ channel antagonist mibefradil (Ro 40-5967). Mol Pharmacol. 1995;48(3):540–549. [PubMed: 7565636]
- 23.
- Aczel S, Kurka B, Hering S. Mechanism of voltage- and use-dependent block of class A Ca2+ channels by mibefradil. Br J Pharmacol. 1998;125(3):447–454. [PMC free article: PMC1565645] [PubMed: 9806326]
- 24.
- Grabner M, Wang Z, Hering S. et al. Transfer of 1,4-dihydropyridine sensitivity from L-type to class A (BI) calcium channels. Neuron. 1996;16(1):207–218. [PubMed: 8562085]
- 25.
- Hering S, Aczel S, Grabner M. et al. Transfer of high sensitivity for benzothiazepines from L-type to class A (BI) calcium channels. J Biol Chem. 1996;271(40):24471–24475. [PubMed: 8798706]
- 26.
- Motoike HK, Bodi I, Nakayama H. et al. A region in IVS5 of the human cardiac L-type calcium channel is required for the use-dependent block by phenylalkylamines and benzothiazepines. J Biol Chem. 1999;274(14):9409–9420. [PubMed: 10092621]
- 27.
- Degtiar VE, Aczel S, Doring F. et al. Calcium channel block by (-)devapamil is affected by the sequence environment and composition of the phenylalkylamine receptor site. Biophys J. 1997;73(1):157–167. [PMC free article: PMC1180917] [PubMed: 9199780]
- 28.
- Hering S, Aczel S, Kraus RL. et al. Molecular mechanism of use-dependent calcium channel block by phenylalkylamines: Role of inactivation. Proc Natl Acad Sci USA. 1997;94(24):13323–13328. [PMC free article: PMC24307] [PubMed: 9371844]
- 29.
- Sokolov S, Weiss RG, Kurka B. et al. Inactivation determinant in the I-II loop of the Ca2+ channel α1-subunit and beta-subunit interaction affect sensitivity for the phenylalkylamine (-)gallopamil. J Physiol. 1999;519(2):315–322. [PMC free article: PMC2269510] [PubMed: 10457051]
- 30.
- Jimenéz C, Bourinet E, Leuranguer V. et al. Determinants of voltage-dependent inactivation affect mibefradil block of calcium channels. Neuropharmacology. 2000;39:1–10. [PubMed: 10665814]
- 31.
- Lacinova L, Ludwig A, Bosse E. et al. The block of the expressed L-type calcium channel is modulated by the beta 3 subunit. FEBS Lett. 1995;373:103–107. [PubMed: 7589444]
- 32.
- Sokolov S, Timin E, Hering S. On the role of Ca2+- and voltage-dependent inactivation in Cav1.2 sensitivity for the phenylalkylamine (-)gallopamil. Circ Res. 2001;89:700–708. [PubMed: 11597993]
- 33.
- Zamponi GW, Soong TW, Bourinet E. et al. Beta subunit coexpression and the alpha1 subunit domain I-II linker affect piperidine block of neuronal calcium channels. J Neurosci. 1996;6:2430–2443. [PMC free article: PMC6578750] [PubMed: 8786420]
- 34.
- Berjukow S, Marksteiner R, Gapp F. et al. Molecular mechanism of calcium channel block by isradipine. Role of a drug-induced inactivated channel conformation. J Biol Chem. 2000;275:22114–22120. [PubMed: 10766758]
- 35.
- Berjukow S, Hering S. Voltage-dependent acceleration of Cav1.2 channel current decay by (+)- and (-)-isradipine. Br J Pharmacol. 2001;133(7):959–966. [PMC free article: PMC1572885] [PubMed: 11487504]
- 36.
- Hille B. Local anaesthetics: Hydrophilic and hydrophobic pathways for drug-receptor action. J Gen Physiol. 1977;69:497–515. [PMC free article: PMC2215053] [PubMed: 300786]
- 37.
- Timin EN, Hering S. A method for estimation of drug affinity constants to the open conformational state of calcium channels. Biophys J. 1992;63(3):808–814. [PMC free article: PMC1262213] [PubMed: 1330037]
- 38.
- Hering S. β-Subunits: Fine tuning of calcium channel block. Trends Pharmacol Sci. 2002 in press [PubMed: 12413805]
- 39.
- Hering S, Berjukow S, Aczel S. et al. Ca2+ channel block and inactivation: Common molecular determinants. Trends Pharmacol Sci. 1998;19:439–443. [PubMed: 9850606]
- 40.
- Hockerman GH, Dilmac N, Scheuer T. et al. Molecular determinants of diltiazem block in domains IIIS6 and IVS6 of L-type Ca2+ channels. Mol Pharmacol. 2000;58(6):1264–1270. [PubMed: 11093762]
- 41.
- Kraus RL, Hering S, Grabner M. et al. Molecular mechanism of diltiazem interaction with L-type Ca2+ channels. J Biol Chem. 1998;273(42):27205–27212. [PubMed: 9765241]
- 42.
- Berjukow S, Gapp F, Aczel S. et al. Sequence differences between alpha1C and alpha1S Ca2+ channel subunits reveal structural determinants of a guarded and modulated benzothiazepine receptor. J Biol Chem. 1999;274(10):6154–6160. [PubMed: 10037699]
- 43.
- Shi C, Soldatov NM. Molecular determinants of voltage-dependent slow inactivation of the Ca2+ channel. J Biol Chem. 2002;277(9):6813–6821. [PubMed: 11751866]
- 44.
- Berjukow S, Marksteiner R, Sokolov S. et al. Amino acids in segment IVS6 and β-subunit interaction support distinct conformational changes during Cav2.1 inactivation. J Biol Chem. 2001;276:17076–17082. [PubMed: 11350979]
- Introduction
- Amino Acid Residues Located in the Putative Drug-Binding Region Affect Drug-Sensitivity and Channel Inactivation
- Drug-Sensitivity Is Affected by Inactivation Determinants Located Outside the Putative Drug-Binding Region
- β-Subunits Modulate Inactivation and Channel Inhibition
- Inactivation Determinants and DHP Sensitivity
- On the Role of Ca2+-Dependent Inactivation in Drug Sensitivity
- Simulation of the Drug-Channel Interaction
- Summary and Outlook
- Acknowledgements
- References
- Calcium Channel Block and Inactivation: Insights from Structure-Activity Studies...Calcium Channel Block and Inactivation: Insights from Structure-Activity Studies - Madame Curie Bioscience Database
- Scaling Laws in the Functional Content of Genomes: Fundamental Constants of Evol...Scaling Laws in the Functional Content of Genomes: Fundamental Constants of Evolution? - Madame Curie Bioscience Database
- PDGF Pathways and Growth of Basal Cell and Squamous Cell Carcinomas - Madame Cur...PDGF Pathways and Growth of Basal Cell and Squamous Cell Carcinomas - Madame Curie Bioscience Database
- Phylogenetic, Structural and Functional Relationships between WD-and Kelch-Repea...Phylogenetic, Structural and Functional Relationships between WD-and Kelch-Repeat Proteins - Madame Curie Bioscience Database
- PROMOTER-ASSOCIATED LONG NONCODING RNAs REPRESS TRANSCRIPTION THROUGH AN RNA BIN...PROMOTER-ASSOCIATED LONG NONCODING RNAs REPRESS TRANSCRIPTION THROUGH AN RNA BINDING PROTEIN TLS - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...