

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Although the primary deficiency in dystrophin and the concomitant reduction in surface glycoproteins are well established factors in the molecular pathogenesis of Duchenne muscular dystrophy, the pathophysiological events that render a muscle fibre more susceptible to necrosis are not well understood. One proposed mechanism involves abnormal calcium homeostasis in mechanically stressed fibres that eventually leads to skeletal muscle weakness. This chapter examines the calcium hypothesis of muscular dystrophy and outlines how unbalanced ion cycling through the sarcolemma and the sarcoplasmic reticulum may contribute to enhanced degradation of muscle proteins. Studying calcium handling in muscular dystrophy is not only important for increasing our knowledge on the multifaceted process of muscle degeneration, but might also have implications for the future design of therapeutic approaches to treating x-linked muscular dystrophy. In this respect, it is encouraging that the pharmacological elimination of Ca2+-dependent proteolysis counteracts dystrophic changes and that the removal of excess cytosolic Ca2+ appears to convey natural protection to dystrophin-deficient fibres.
Calcium Handling Proteins
Calcium ions represent an ubiquitous second messenger molecule involved in the regulation of various metabolic and physiological processes.1 In skeletal muscle, cytosolic Ca2+-levels dictate the overall contractile status, and Ca2+-cycling through intracellular compartments is maintained under precise spatial and temporal control.2 Signal transduction during excitation-contraction coupling and fibre relaxation is mediated by complex interactions between voltage sensors, Ca2+-channels, Ca2+-binding proteins, Ca2+-transporters, ion exchangers and ion pumps.3-5 Table 1 lists established elements involved in ion handling that are potentially impaired in muscular dystrophy. Almost all Ca2+-regulatory elements exist as several isoforms6 reflecting the heterogeneity of skeletal muscle fibres.7 Due to the enormous complexity of the Ca2+-handling apparatus, small changes in individual steps involved in modulating Ca2+-signals might result in major pathophysiological consequences. Although it is not yet established how many auxiliary proteins act as intrinsic regulators of Ca2+-release complexes, Ca2+-uptake units and the luminal Ca2+-reservoir complex, abnormal expression patterns of ion-regulatory components are clearly involved in muscular dystrophy.8-12 The exact degree of abnormal Ca2+-handling is difficult to judge,13 since it is not fully understood whether key Ca2+-handling proteins operate at their full capacity under normal conditions.3,14-16
Table 1
Ion-regulatory proteins of the excitation-contraction-relaxation cycle.
Calcium Hypothesis of Muscular Dystrophy
Historically, the formulation of the calcium hypothesis of muscular dystrophy17 preceded the pioneering discovery of the genetic defect responsible for Duchenne muscular dystrophy.18 A decade before Kunkel and coworkers described the primary abnormality in x-linked muscular dystrophy using a positional cloning strategy,18,19 anomalous intracellular Ca2+-accumulation was described in fibres from patients afflicted with x-linked muscular dystrophy.20 In conjunction with the electron microscopical demonstration of membrane damage in dystrophic fibres,21 this finding led to the idea that chronic Ca2+-overload might play a central role in fibre degeneration.17 Since dystrophinopathies are primarily diseases of the membrane cytoskeleton,22,23 it is important to consider that disruption of the cytoskeletal network has a direct effect on Ca2+-fluxes.24,25 This suggests a direct involvement of the membrane cytoskeleton in the maintenance of subsarcolemmal Ca2+-levels and Ca2+-mediated signal transduction pathways.
Molecular Defects in Calcium Homeostasis
The current mechanical calcium hypothesis of muscular dystrophy assumes that a causal connection exists between the disintegration of sarcolemmal integrity and Ca2+-dependent proteolysis of muscle proteins,8-12 as outlined in the flow chart of Figure 1. Various aspects of individual steps involved in this destructive process have been discussed in recent reviews.8-12,26,27 Deficiency in the dystrophin isoform Dp42719 results in a drastic reduction in various sarcolemma-associated proteins,28 such as the sarcoglycans29 and dystroglycans.30 The resulting destabilisation of the linkage between the actin cytoskeleton and the extracellular matrix component laminin renders the muscle periphery more susceptible to micro-rupturing.31 This in turn, triggers a Ca2+-dependent membrane resealing process at the sites of sarcolemmal disintegration. The influx of extracellular Ca2+-ions initiates an exocytotic event that adds plasma membrane patches to seal leaky micro domains of the disrupted muscle periphery.8 Ca2+-leak channels present in the newly introduced sarcolemmal patches cause an increased influx of Ca2+-ions into the cytoplasm of dystrophic fibres. The exact extent of perturbation of Ca2+-handling in dystrophin-deficient fibres is still controversial.8-12 Contradictory findings may be partially due to the usage of different physiological techniques and standardisation in the measurement of intracellular Ca2+-levels. However, it is generally accepted that the Ca2+-overload is not global but restricted to cytosolic domains adjacent to the sarcolemma.32,33 The recent characterisation of dysferlin-null mice has highlighted the importance of proper membrane resealing in muscular disorders. Although dysferlin-deficient fibres maintain a functional dystrophin-glycoprotein complex, they develop a progressive muscular dystrophy.34 In skeletal muscle, surface membrane repair is an active process involving dysferlin whereby disruption of the sarcolemma repair apparatus triggers muscle degeneration.34 This clearly demonstrates the importance of a fast Ca2+-dependent sarcolemma resealing process to efficiently counter-act injuries to the muscle plasma membrane.
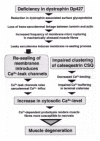
The pathological consequence of increased cytosolic Ca2+-levels is an activation of Ca2+-dependent proteases, such as skeletal muscle-specific calpains, resulting in the net degradation of muscle proteins in dystrophic muscle cells.35-38 Localised Ca2+-elevations are believed to contribute to a destructive cycle of enhanced protease activity and Ca2+-leak channel activation. 36 Because increased proteolytic activity renders Ca2+-leak channels constitutively active, insertion of membrane vesicles for resealing, following exercise-induced membrane rupturing, results in persistent Ca2+ influx at affected sarcolemma domains. Hence, more Ca2+ influx causes increased proteolysis, and more proteolysis causes increased Ca2+ influx, giving rise to a pathophysiological cycle.8 The theory of the initiating role of mechano-sensitive Ca2+-channels in triggering fibre degradation has recently been challenged by the study of transgenic mdx muscle that over-expresses utrophin.39 It therefore can not be excluded that the Ca2+-leak channel activation in mechanically stressed fibres is preceded by modifications in other Ca2+-handling proteins. For example, surface membrane leakage of Ca2+ into the cytoplasm may also be due to abnormalities in the nicotinic acetylcholine receptor.40,41 This fact, however, does not challenge the overall concept of deranged intracellular Ca2+-handling being involved in muscular dystrophy. Both, Ca2+-permeable growth factor-regulated channels42 (GRC) and transient receptor potential channels33 (TRPC) may facilitate abnormal Ca2+-influxes through the dystrophin-deficient muscle periphery. The expression of Ca2+-permeable nonselective cation channels of the GRC type were shown to be elevated in the sarcolemma of mdx fibres42 and isoforms TRPC-1, 4 and 6 of the transient receptor potential channels are present in mdx membrane.33 In contrast, surface L-type Ca2+-channels of the dihydropyridine receptor class do not appear to contribute to elevated cytosolic Ca2+-levels in muscular dystrophy.43
Impaired Excitation-Contraction Coupling
In addition to abnormal sarcolemma physiology, changes in the Ca2+-cycling patterns through the lumen of the sarcoplasmic reticulum44 and mitochondria45 have also been shown to occur in dystrophin-deficient skeletal muscle. The excitation-contraction-relaxation cycle is regulated by the physiological interplay between the voltage-sensing dihydropyridine receptor of the transverse tubules, the ryanodine receptor Ca2+-release channel of the junctional sarcoplasmic reticulum and the Ca2+-ATPases of the longitudinal tubules.3-5 It would not be surprising if these ion-regulatory elements were secondarily involved in muscular dystrophy, since numerous inherited muscle diseases exhibit defects in excitation-contraction coupling or muscle relaxation. This includes malignant hyperthermia, central core disease, hypokalemic periodic paralysis and Brody's disease.46 In normal muscle fibres, the release of intracellular Ca2+ in response to sarcolemmal depolarization is one of the key steps of excitation-contraction coupling. 3 Following voltage-sensing by the α1S-subunit of the dihydropyridine receptor, physical coupling between the II-III loop domain of the transverse-tubular tetrad complex and the cytosolic foot region of the junctional ryanodine receptor tetramer triggers transient opening of the Ca2+-release channel.5,16 Thus, at least initially, a direct physical receptor interaction process couples membrane depolarization to the release of Ca2+-ions into the cytosol. In a subsequent step, comparable to the cardiac Ca2+-induced Ca2+ -release mechanism, sarcoplasmic reticulum Ca2+-channels are also locally activated by excess Ca2+ that greatly amplifies the overall Ca2+-signal.3-5,16 Fibre relaxation is initiated by the energy-dependent reversal of the cytosolic Ca2+-signal. Reuptake of Ca2+-ions in the lumen of the sarcoplasmic reticulum is achieved by the SERCA type Ca2+-pumps.14 In contrast to heart muscle, Ca2+-removal via the surface Na+/Ca2+-exchanger does not play a major role in skeletal muscle fibres. An important physiological regulator that mediates between Ca2+-release and Ca2+-uptake is the luminal Ca2+-reservoir complex of the sarcoplasmic reticulum.15 Besides the histidine-rich Ca2+-binding protein, calreticulin, sarcalumenin and calsequestrin-like proteins, the most abundant Ca2+-binding element of the terminal cisternae are clusters of 63 kDa calsequestrin molecules.4
It has not been determined how many auxiliary proteins are involved in the fine regulation of receptor coupling, Ca2+-release, Ca2+-buffering and Ca2+-uptake and remains to be elucidated which triad-associated components prevent passive disintegration of supramolecular membrane assemblies. Recently identified auxiliary proteins such as triadin, junctin, JP-45 and JP-90 only represent part of the total protein complement of the junctional couplings involved in excitation-contraction coupling.47-50 Current muscle proteome projects are addressing this question and aim at the identification of all sarcoplamic reticulum proteins involved in Ca2+-cycling. Until these results become available, we lack an overall understanding of the complexity of the Ca2+-handling apparatus. Nevertheless, the initial analysis of the main players involved in excitation-contraction coupling revealed that calsequestrin-like proteins (CLP) are greatly reduced in muscular dystrophy,44 as illustrated in the immunoblot analysis shown in Figure 2. While the expression of the junctional ryanodine receptor Ca2+-release channel, the voltage-sensing dihydropyridine receptor, the Ca2+-ATPase and calsequestrin was not affected, a drastic decline in CLP-150, CLP-170 and CLP-220 was observed in dystrophic microsomes.44 Although immunoblotting could clearly demonstrate the reduction in these terminal cisternae markers, this technique could not address the question of whether the reduction in high-molecular-mass isoforms of calsequestrin is due to decreased expression levels or because of an impaired oligomersiation pattern.
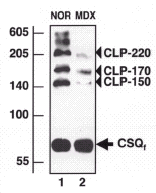
Comparative chemical crosslinking analysis of normal and dystrophic microsomal membranes revealed a crosslinker-induced reappearance of CLP species in dystrophin-deficient preparations. 51 Hence, CLP-150, CLP-170 and CLP-220 appear to represent clusters of the calsequestrin monomers and not distinct high-molecular-mass isoforms. Since the introduction of short crosslinker probes could restore the appearance of CLPs in dystrophic membranes, their drastically reduced expression in muscular dystrophy is due to impaired oligomerisation of calsequestrin monomers, and not based on a loss in individual isoforms of this Ca2+-binding protein. Equilibrium dialysis experiments have shown that the overall Ca2+-binding capacity was reduced by approximately 20% in dystrophic sarcoplasmic reticulum. 44 Thus, impaired protein coupling between calsequestrin units in the terminal cisternae region might be directly involved in triggering impaired Ca2+-sequestration within the lumen of the sarcoplasmic reticulum. In muscular dystrophy, disturbed sarcolemmal Ca2+-fluxes appear to influence the complex Ca2+-cycling apparatus causing distinct changes in the oligomerisation pattern of a subset of Ca2+-handling proteins (fig. 3).

With respect to cytosolic Ca2+-binding elements, parvalbumin plays an ion-buffering role in fast muscle fibres.52 Chronic low-frequency stimulation suppresses parvalbumin expression in fast muscles demonstrating that a slow motoneuron-like impulse pattern rapidly silences the parvalbumin gene.53 An increased turnover of parvalbumin has been described in dystrophic fast-twitching mdx fibres.54 Elevated serum parvalbumin levels appear to be indicative of the severity of the disease status in dystrophic mouse muscle.54 Compared to the severe dystrophic phenotype of Duchenne patients, the mdx mouse model exhibits a much milder dystrophy. Since parvalbumin is expressed at only very low levels in human fibres, but is abundant in rodent IIB fibres,55 the presence of parvalbumin might be responsible for this difference in the severity of the dystrophinopathy. This relatively species-specific Ca2+-binding element could buffer the increased Ca2+-influx through the mdx sarcolemma thereby maintaining a lower cytosolic Ca2+-level as compared to dystrophic human muscle fibres. However, double mutant mice that are deficient in both dystrophin and parvalbumin exhibit a cytosolic Ca2+-content similar to that of mdx fibres.56 Thus, parvalbumin does not appear to play a central role in eliminating abnormal Ca2+-cycling through the cytosol of mdx muscle.
Therapeutic Implications
In addition to helping in understanding the molecular pathways underlying muscular dystrophy, studying Ca2+-regulatory proteins might also lead to the discovery of new therapeutic targets. One potential way forward is the analysis of naturally protected muscle phenotypes that lack dystrophin but do not exhibit severe symptoms. The sparing of dystrophin-deficient extraocular fibres is attributed to the special protective properties of small-diameter fibres.57 Recently, it was shown that in contrast to leg muscles, toe fibres from the dystrophic animal model mdx also show a very low degree of muscle degeneration.58,59 This is histochemically evident by a low percentage of centrally located nuclei. Toe fibres from the mdx mouse exhibit a preserved expression of the critical trans-sarcolemmal linker α-dystroglycan58 and an up-regulation and extra-junctional localization of the autosomal dystrophin homologue utrophin.58,59 Possibly utrophin acts as a substitute for the deficient Dp427 isoform and thereby prevents the disintegration of the surface-associated glycoprotein complex.60 Interestingly, an increase in both the expression of the fast SERCA1 isoform of the sarcoplasmic reticulum Ca2+-pump and the total Ca2+-ATPase activity was described in protected mdx toe fibres.58 Possibly, the up-regulation of Ca2+-ATPase units causes an increased removal of cytosolic Ca2+-ions from mdx toe fibres thereby significantly reducing the degree of Ca2+-induced myonecrosis in dystrophin-deficient fibres.58 That a decrease in cytosolic Ca2+-levels has a protective effect was shown by experiments with carnitine-linked leupeptin.61,62 The targeted introduction of protease inhibitors appears to specifically inactivate Ca2+-dependent calpain activity. 62 Calpain inhibition could be correlated with rentention of myofibre size.61 Overexpression of the calpain inhibitor calpastatin in dystrophin-deficient fibres reduced the dystrophic phenotype63 suggesting that specific calpains are primarily involved in muscle necrosis. Based on the pathophysiological role of abnormal Ca2+-cycling in dystrophic fibres, potential sites of pharmacological interventions present themselves at various Ca2+ regulatory processes including the sarcolemma and sarcoplasmic reticulum.64-66 Especially calpain inhibition promises to be an excellent therapeutic target to prevent muscular degeneration in Duchennne muscular dystrophy.61-63 Hence, besides myoblast transfer,67 stem cell treatment68 or gene therapy69 for the treatment of x-linked muscular dystrophy (see Chapters 17 and 18), a more traditional pharmacological approach might also be a promising option.70
Conclusions
In conclusion, abnormal Ca2+-handling might be an important factor in the progressive functional decline of dystrophic muscle fibres. Although the total extent and exact micro-domain localisation of the initial Ca2+-disturbance has not yet been fully established, it is understood that even small changes in Ca2+-cycling trigger a cascade of modifications in ion-regulatory muscle proteins. The deficiency in dystrophin leads primarily to a reduction in dystrophin-associated proteins which in turn destroys sarcolemmal integrity by weakening the linkage between the extracellular matrix and the membrane cytoskeleton. The introduction of Ca2+-leak channels during membrane resealing appears to be the central pathophysiological incident that causes an increase in cytosolic Ca2+-levels and consequently Ca2+-induced myonecrosis. In addition, impaired calsequestrin oligomerisation causes decreased Ca2+-sequestration within the lumen of the sarcoplasmic reticulum. Thus, the disintegration of the surface dystrophin-glycoprotein complex triggers disturbed sarcolemmal Ca2+-fluxes which in turn impairs other Ca2+-handling steps thereby resulting in distinct changes in the oligomerisation pattern of Ca2+-binding elements. This pathophysiological scenario might explain one of the key routes leading to cellular degeneration in muscular dystrophy. Most importantly, a more detailed knowledge of impaired Ca2+ handling in muscular dystrophy might lead to the design of novel approaches to counter-act fibre degeneration in genetic muscle diseases.
Acknowledgments
Research in the author's laboratory on calcium handling in neuromuscular disorders was supported by project grants from the Irish Health Research Board (HRB-95/95, HRB-01/98, HRB-01/99, HRB-01/01), Enterprise Ireland (SC/2000/386) and Muscular Dystrophy Ireland (MDI-95, MDI-02), as well as network grants from the European Commission (FMRX-CT960032, QLRT-1999-02034, RTN2-2001-00337). I would like to thank Dr. Kevin Culligan for the immunoblot analysis of dystrophic fibres.
References
- 1.
- Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: Dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4:517–529. [PubMed: 12838335]
- 2.
- Berchtold MW, Brinkmeier H, Muntener M. Calcium ion in skeletal muscle: Its crucial role for muscle function, plasticity, and disease. Physiol Rev. 2000;80:1215–1265. [PubMed: 10893434]
- 3.
- Melzer W, Herrmann-Frank A, Luttgau HC. The role of Ca2+ ions in excitation-contraction coupling of skeletal muscle fibres. Biochim Biophys Acta. 1995;1241:59–116. [PubMed: 7742348]
- 4.
- Murray BE, Froemming GR, Maguire PB. et al. Excitation-contraction-relaxation cycle: Role of Ca2+-regulatory membrane proteins in normal, stimulated and pathological skeletal muscle fibres (review). Int J Mol Med. 1998;1:677–697. [PubMed: 9852282]
- 5.
- Leong P, MacLennan DH. Complex interactions between skeletal muscle ryanodine receptor and dihydropyridine receptor proteins. Biochem Cell Biol. 1998;76:681–694. [PubMed: 10353700]
- 6.
- Ohlendieck K. Changes in Ca2+-regulatory membrane proteins during the stimulation-induced fast-to-slow transition process. Bas Appl Myol. 2000;10:99–106.
- 7.
- Pette D, Staron RS. Transitions of muscle fiber phenotypic profiles. Histochem Cell Biol. 2001;115:359–372. [PubMed: 11449884]
- 8.
- Alderton JM, Steinhardt RA. How calcium influx through calcium leak channels is responsible for the elevated levels of calcium-dependent proteolysis in dystrophic myotubes. Trends Cardiovasc Med. 2000;10:268–272. [PubMed: 11282306]
- 9.
- Gailly P. New aspects of calcium signaling in skeletal muscle cells: Implications in Duchenne muscular dystrophy. Biochim Biophys Acta. 2002;1600:38–44. [PubMed: 12445457]
- 10.
- Gillis JM. Membrane abnormalities and Ca homeostasis in muscles of the mdx mouse, an animal model of the Duchenne muscular dystrophy: A review. Acta Physiol Scand. 1996;156:397–406. [PubMed: 8729700]
- 11.
- Ruegg UT, Nicolas-Metral V, Challet C. et al. Pharmacological control of cellular calcium handling in dystrophic skeletal muscle. Neuromuscul Disord. 2002;12:S155–S161. [PubMed: 12206810]
- 12.
- Culligan K, Ohlendieck K. Abnormal calcium handling in muscular dystrophy. Bas Appl Myol. 2002;12:147–157.
- 13.
- Divet A, Huchet-Cadiou C. Sarcoplasmic reticulum function in slow- and fast-twitch skeletal muscles from mdx mice. Pflugers Arch. 2002;444:634–643. [PubMed: 12194017]
- 14.
- Stokes DL, Wagenknecht T. Calcium transport across the sarcoplasmic reticulum: Structure and function of Ca2+-ATPase and the ryanodine receptor. Eur J Biochem. 2000;267:5274–9. [PubMed: 10951184]
- 15.
- MacLennan DH, Reithmeier RA. Ion tamers. Nat Struct Biol. 1998;5:409–411. [PubMed: 9628471]
- 16.
- Franzini-Armstrong C, Protasi F. Ryanodine receptors of striated muscles: A complex channel capable of multiple interactions. Physiol Rev. 1997;77:699–729. [PubMed: 9234963]
- 17.
- Duncan CJ. Role of intracellular calcium in promoting muscle damage: A strategy for controlling the dystrophic condition. Experientia. 1978;34:1531–1535. [PubMed: 365566]
- 18.
- Monaco AP, Neve RL, Colletti-Feener C. et al. Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature. 1986;323:646–650. [PubMed: 3773991]
- 19.
- Ahn AH, Kunkel LM. The structural and functional diversity of dystrophin. Nat Genet. 1993;3:283–291. [PubMed: 7981747]
- 20.
- Bodensteiner JB, Engel AG. Intracellular calcium accumulation in Duchenne dystrophy and other myopathies: A study of 567,000 muscle fibers in 114 biopsies. Neurology. 1978;28:439–446. [PubMed: 76996]
- 21.
- Cullen MJ, Fulthorpe JJ. Stages in fibre breakdown in Duchenne muscular dystrophy. An electron-microscopic study. J Neurol Sci. 1975;24:179–200. [PubMed: 163299]
- 22.
- Campbell KP. Three muscular dystrophies: Loss of cytoskeleton-extracellular matrix linkage. Cell. 1995;80:675–679. [PubMed: 7889563]
- 23.
- Ohlendieck K. Towards an understanding of the dystrophin-glycoprotein complex: Linkage between the extracellular matrix and the subsarcolemmal membrane cytoskeleton. Eur J Cell Biol. 1996;69:1–10. [PubMed: 8825019]
- 24.
- Menke A, Jockusch H. Extent of shock-induced membrane leakage in human and mouse myotubes depends on dystrophin. J Cell Sci. 1995;108:727–33. [PubMed: 7769014]
- 25.
- Hajnoczky G, Lin C, Thomas AP. Luminal communication between intracellular calcium stores modulated by GTP and the cytoskeleton. J Biol Chem. 1994;269:10280–10287. [PubMed: 8144609]
- 26.
- Petrof BJ. The molecular basis of activity-induced muscle injury in Duchenne muscular dystrophy. Mol Cell Biochem. 1998;179:111–23. [PubMed: 9543354]
- 27.
- Blake DJ, Weir A, Newey SE. et al. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. 2002;82:291–329. [PubMed: 11917091]
- 28.
- Culligan K, Mackey A, Finn D. et al. Role of dystrophin isoforms and associated glycoproteins in muscular dystrophy (review). Int J Mol Med. 1998;2:639–648. [PubMed: 9850730]
- 29.
- Hack AA, Groh ME, McNally EM. Sarcoglycans in muscular dystrophy. Microsc Res Tech. 2000;48:167–80. [PubMed: 10679964]
- 30.
- Winder SJ. The complexities of dystroglycan. Trends Biochem Sci. 2001;26:118–124. [PubMed: 11166570]
- 31.
- Clarke MS, Khakee R, McNeil PL. Loss of cytoplasmic basic fibroblast growth factor from physiologically wounded myofibers of normal and dystrophic muscle. J Cell Sci. 1993;106:121–133. [PubMed: 8270618]
- 32.
- Mallouk N, Jacquemond V, Allard B. Elevated subsarcolemmal Ca2+ in mdx mouse skeletal muscle fibres detected with Ca2+-activated K+ channels. Proc Natl Acad Sci USA. 2000;97:4950–4955. [PMC free article: PMC18338] [PubMed: 10781103]
- 33.
- Vandebrouck C, Martin D, Colson-Van Schoor M. et al. Involvement of TRPC in the abnormal calcium influx observed in dystrophic (mdx) mouse skeletal muscle fibers. J Cell Biol. 2002;158:1089–96. [PMC free article: PMC2173225] [PubMed: 12235126]
- 34.
- Bansal D, Miyake K, Vogel SS. et al. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature. 2003;423:168–172. [PubMed: 12736685]
- 35.
- MacLennan PA, McArdle A, Edwards RH. Effects of calcium on protein turnover of incubated muscles from mdx mice. Am J Physiol. 1991;260:E594–E598. [PubMed: 2018123]
- 36.
- Turner PR, Fong PY, Denetclaw WF. et al. Increased calcium influx in dystrophic muscle. J Cell Biol. 1991;115:1701–1712. [PMC free article: PMC2289194] [PubMed: 1661733]
- 37.
- Turner PR, Schultz R, Ganguly B. et al. Proteolysis results in altered leak channel kinetics and elevated free calcium in mdx muscle. J Membr Biol. 1993;133:243–251. [PubMed: 8392585]
- 38.
- Alderton JM, Steinhardt RA. Calcium influx through calcium leak channels is responsible for the elevated levels of calcium-dependent proteolysis in dystrophic myotubes. J Biol Chem. 2000;275:9452–9460. [PubMed: 10734092]
- 39.
- Squire S, Raymackers JM, Vandebrouck C. et al. Prevention of pathology in mdx mice by expression of utrophin: Analysis using inducible transgenic expression system. Hum Mol Genet. 2002;11:3333–3344. [PubMed: 12471059]
- 40.
- Carlson CG. The dystrophinopathies: An alternative to the structural hypothesis. Neurobiol Dis. 1998;5:3–15. [PubMed: 9702783]
- 41.
- Carlson CG. Acetylcholine receptor and calcium leakage activity in nondystrophic and dystrophic myotubes (MDX). Muscle Nerve. 1996;19:1258–1267. [PubMed: 8808651]
- 42.
- Iwata Y, Katanosaka Y, Arai Y. et al. A novel mechanism of myocyte degeneration involving the Ca2+-permeable growth factor-regulated channel. J Cell Biol. 2003;161:957–967. [PMC free article: PMC2172975] [PubMed: 12796481]
- 43.
- Collet C, Csernoch L, Jacquemond V. Intramembrane charge movement and L-type calcium current in skeletal muscle fibers isolated from control and mdx mice. Biophys J. 2003;84:251–265. [PMC free article: PMC1302607] [PubMed: 12524279]
- 44.
- Culligan K, Banville N, Dowling P. et al. Drastic reduction of calsequestrin-like proteins and impaired calcium binding in dystrophic mdx muscle. J Appl Physiol. 2002;92:435–445. [PubMed: 11796649]
- 45.
- Robert V, Massimino ML, Tosello V. et al. Alteration in calcium handling at the subcellular level in mdx myotubes. J Biol Chem. 2001;276:4647–4651. [PubMed: 11029464]
- 46.
- Froemming GR, Ohlendieck K. The role of ion-regulatory membrane proteins of excitation-contraction coupling and relaxation in inherited muscle diseases. Front Biosci. 2001;6:D65–D74. [PubMed: 11145921]
- 47.
- Froemming GR, Murray BE, Ohlendieck K. Self-aggregation of triadin in the sarcoplasmic reticulum of rabbit skeletal muscle. Biochim Biophys Acta. 1999;1418:197–205. [PubMed: 10209224]
- 48.
- Froemming GR, Pette D, Ohlendieck K. The 90 kDa junctional sarcoplasmic reticulum protein forms an integral part of a supramolecular triad complex in skeletal muscle. Biochem Biophys Res Comm. 1999;261:603–609. [PubMed: 10441473]
- 49.
- Zorzato F, Anderson AA, Ohlendieck K. et al. Identification of a novel 45 kDa protein (JP-45) from rabbit sarcoplasmic-reticulum junctional-face membrane. Biochem J. 2000;351:537–543. [PMC free article: PMC1221391] [PubMed: 11023841]
- 50.
- Jones LR, Zhang L, Sanborn K. et al. Purification, primary structure, and immunological characterization of the 26-kDa calsequestrin binding protein (junctin) from cardiac junctional sarcoplasmic reticulum. J Biol Chem. 1995;270:30787–30796. [PubMed: 8530521]
- 51.
- Dowling P, Lohan J, Ohlendieck K. Comparative analysis of Dp427-deficient mdx tissues shows that the milder dystrophic phenotype of extraocular and toe muscle fibres is associated with a persistent expression of beta-dystroglycan. Eur J Cell Biol. 2003;82:222–230. [PubMed: 12800977]
- 52.
- Schleef M, Zuhlke C, Jockusch H. et al. The structure of the mouse parvalbumin gene. Mamm Genome. 1992;3:217–225. [PubMed: 1611216]
- 53.
- Huber B, Pette D. Dynamics of parvalbumin expression in low-frequency-stimulated fast-twitch rat muscle. Eur J Biochem. 1996;236:814–819. [PubMed: 8665899]
- 54.
- Jockusch H, Friedrich G, Zippel M. Serum parvalbumin, an indicator of muscle disease in murine dystrophy and myotonia. Muscle Nerve. 1990;13:551–555. [PubMed: 2366828]
- 55.
- Schmitt TL, Pette D. Fiber type-specific distribution of parvalbumin in rabbit skeletal muscle. A quantitative microbiochemical and immunohistochemical study. Histochemistry. 1991;96:459–465. [PubMed: 1837548]
- 56.
- Raymackers JM, Debaix H, Colson-Van Schoor M. et al. Consequence of parvalbumin deficiency in the mdx mouse: Histological, biochemical and mechanical phenotype of a new double mutant. Neuromuscul Disord. 2003;13:376–387. [PubMed: 12798793]
- 57.
- Porter JD. Extraocular muscle sparing in muscular dystrophy: A critical evaluation of potential protective mechanisms. Neuromusc Disord. 1998;8:198–203. [PubMed: 9631402]
- 58.
- Dowling P, Culligan K, Ohlendieck K. Distal mdx muscle groups exhibiting up-regulation of utrophin and rescue of dystrophin-associated glycoproteins exemplify a protected phenotype in muscular dystrophy. Naturwiss. 2002;89:75–88. [PubMed: 12046625]
- 59.
- Matsumura K, Ervasti JM, Ohlendieck K. et al. Association of dystrophin-related protein with dystrophin-associated proteins in mdx mouse muscle. Nature. 1992;360:588–591. [PubMed: 1461282]
- 60.
- Rafael JA, Tinsley JM, Potter AC. et al. Skeletal muscle-specific expression of utrophin transgene rescues utrophin-dystrophin deficient mice. Nature Genet. 1998;19:79–82. [PubMed: 9590295]
- 61.
- Badalamente WA, Stracher A. Delay of muscle degeneration and necrosis in mdx mice by calpain inhibition. Muscle Nerve. 2000;23:106–111. [PubMed: 10590413]
- 62.
- Stracher A. Calpain inhibitors as therapeutic agents in nerve and muscle degeneration. Ann NY Acad Sci. 1999;884:52–59. [PubMed: 10842583]
- 63.
- Spencer MJ, Mellgren RL. Overexpression of a calpastatin transgene in mdx muscle reduces dystrophic pathology. Hum Mol Genet. 2002;11:2645–2655. [PubMed: 12354790]
- 64.
- Leijendekker WJ, Passaquin AC, Metzinger L. et al. Regulation of cytosolic calcium in skeletal muscle cells of the mdx mouse under conditions of stress. Br J Pharmacol. 1996;118:611–616. [PMC free article: PMC1909736] [PubMed: 8762085]
- 65.
- Tutdibi O, Brinkmeier H, Rudel R. et al. Increased calcium entry into dystrophin-deficient muscle fibres of MDX and ADR-MDX mice is reduced by ion channel blockers. J Physiol. 1999;515:859–68. [PMC free article: PMC2269189] [PubMed: 10066910]
- 66.
- DeLuca A, Pierno S, Liantonio A. et al. Alteration of excitation-contraction coupling mechanism in extensor digitorum longus muscle fibres of dystrophic mdx mouse and potential efficacy of taurine. Br J Pharmacol. 2001;132:1047–1054. [PMC free article: PMC1572646] [PubMed: 11226135]
- 67.
- Partridge TA. Myoblast transplantation. Neuromuscul Disord. 2002;12:S3–S6. [PubMed: 12206788]
- 68.
- Partridge TA. Stem cell route to neuromuscular therapies. Muscle Nerve. 2003;27:133–141. [PubMed: 12548520]
- 69.
- Chamberlain JS. Gene therapy of muscular dystrophy. Hum Mol Genet. 2002;11:2355–2362. [PubMed: 12351570]
- 70.
- Khurana TS, Davies KE. Pharmacological strategies for muscular dystrophy. Nat Rev Drug Discov. 2003;2:379–390. [PubMed: 12750741]
- The Pathophysiological Role of Impaired Calcium Handling in Muscular Dystrophy -...The Pathophysiological Role of Impaired Calcium Handling in Muscular Dystrophy - Madame Curie Bioscience Database
- ER Calcium and ER Chaperones: New Players in Apoptosis? - Madame Curie Bioscienc...ER Calcium and ER Chaperones: New Players in Apoptosis? - Madame Curie Bioscience Database
- IL-10 Gene Polymorphisms in Transplantation - Madame Curie Bioscience DatabaseIL-10 Gene Polymorphisms in Transplantation - Madame Curie Bioscience Database
- MDM2: RING Finger Protein and Regulator of p53 - Madame Curie Bioscience Databas...MDM2: RING Finger Protein and Regulator of p53 - Madame Curie Bioscience Database
- Cytogenetics of Human Sperm - Madame Curie Bioscience DatabaseCytogenetics of Human Sperm - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...