

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

The sarcoglycans are transmembrane proteins found as a plasma membrane-associated complex. First characterized as a subunit of the dystrophin glycoprotein complex in skeletal muscle, the sarcoglycan complex is secondary disrupted and destabilized from the plasma membrane when dystrophin is mutated, as in Duchenne Muscular Dystrophy. Autosomal recessive mutations in several sarcoglycan genes, α, β, γ and δ, also lead to disruption of the sarcoglycan complex yielding a similar phenotype to what is seen in Duchenne Muscular Dystrophy and referred to as the type 2 Limb Girdle Muscular Dystrophies. Mutations in d-sarcoglycan have been reported in human cardiomyopathy patients. The exact function of the sarcoglycan complex is not known, but it appears to have both mechanical and nonmechanical roles in stabilizing the plasma membrane in cardiac and skeletal muscle. There is a variant of the sarcoglycan complex in vascular smooth muscle and in some nonmuscle cell and tissue types. Specifically, ε-sarcoglycan is highly expressed in the central and peripheral nervous systems. Dominant mutations in e-sarcoglycan lead to an inherited movement disorder. Thus, the sarcoglycan complex has important roles in both muscle and nonmuscle tissues.
Historical Perspective
The dystrophin glycoprotein complex (DGC) is a multimeric, multifaceted collection of proteins found at the plasma membrane of muscle cells (fig. 1). The DGC has cytoplasmic elements, dystrophin, dystrobrevins and syntrophins, and nitric oxide synthase, and transmembrane elements, dystroglycan, the sarcoglycans and sarcospan. The entire DGC can be purified as a macromolecular complex from detergent solubilized membranes.1-3 Using an additional detergent solubilization technique, the sarcoglycan complex can be separated from the remainder of the DGC.4 Mutations in dystrophin lead to Duchenne and Becker muscular dystrophy in humans and in the mdx mouse.5,6 Mutations that disrupt dystrophin cause instability of the remainder of the DGC including the sarcoglycans.1,3 Mutations that target the carboxyl-terminus of dystrophin lead to a severe muscular dystrophy phenotype; it is in this region that dystrophin interacts with dystroglycan and anchors the DGC.

Genetics
The characteristic phenotype in DMD, including early onset muscle degeneration and regeneration, calf hypertrophy and elevated creatine kinase (CK) is linked to the X chromosome and is present only in males. In regions of the world with higher rates of consanguinity, a phenotype similar to DMD was noted in both males and females and was termed Severe Childhood Autosomal Recessive Muscular Dystrophy (SCARMD).7 Genetic linkage analysis on families with SCARMD demonstrated that the genetic defect was located on chromosome 13q12, although some SCARMD families excluded involvement of the chromosome 13 locus, indicating genetic heterogeneity of this disorder.8-11
Biochemistry and Cell Biology
The first biochemical analyses to identify the sarcoglycan complex underestimated the full complexity of this subcomplex. Only through human genetic studies, biochemical analyses and the additional analysis of the human genome sequence has it become clear that there are six sarcoglycan sequences, α, β, γ, δ, ε and ζ. Antibodies were generated to a number of sarcoglycan proteins, initially referred to as 50 kDa dystrophin associated glycoprotein (DAG), 43 kDa DAG and the 35 kDa DAG.2,3 It was these three elements that were purified from the main DGC and, together were termed the sarcoglycan complex.4 It was subsequently shown that both the 50 kDa and 35 kDa proteins are heterogeneous in that they are produced from multiple genes (see below). Antibodies directed to the sarcoglycan components and the DGC were used to demonstrate the secondary reduction and degeneration of the DGC that occurs in the muscle of DMD subjects and the mdx mouse.1,12 Interestingly, SCARMD subjects also showed a reduction of sarcoglycan proteins as did the well-studied small animal model of cardiomyopathy and muscular dystrophy, the BIO 14.6 Syrian hamster model (see below).13-16 Genetic and mutational analyses in Duchenne-like muscular dystrophy patients revealed that mutations in the genes encoding α-sarcoglycan, β-sarcoglycan, γ-sarcoglycan and δ-sarcoglycan lead to human forms of muscular dystrophy.16-20
α-sarcoglycan
α-sarcoglycan is a type I transmembrane protein that migrates as a 50 kDa protein (fig. 2). The primary amino acid sequence predicts a protein of approximately 43 kDa with the difference in size related to N-linked glycosylation.21 The gene encoding human α-sarcoglycan maps to chromosome 17p21, and autosomal recessive mutations in the α-sarcoglycan have now been described from muscular dystrophy patients from all over the world.22,23 The majority of mutations in the α-sarcoglycan gene are point mutations that encode missense changes. Compound heterozygous mutations have been noted. These deleterious polymorphisms may affect cysteine residues that are important for secondary structure and potentially, interactions within the sarcoglycan complex. Like the other sarcoglycan gene mutations, in most carriers, heterozygous α-sarcoglycan mutations typically are not associated with any muscle phenotype. Mice engineered to lack α-sarcoglycan develop a progressive muscular dystrophy similar to what has been noted in the mdx mouse that lacks dystrophin.24,25 Mice lacking α-sarcoglycan do not develop cardiomyopathy, although cardiomyopathy has been noted rarely in human subjects with α-sarcoglycan mutations.24,26 The α-sarcoglycan protein has homology to ecto ATPases and biochemical studies have suggested that this may be a feature of α-sarcoglycan.27

β-sarcoglycan
β-sarcoglycan is a 43 kDa type II transmembrane protein (fig.2).16,19 There are three potential N-linked glycosylation sites in the extracellular domain and potential phosphorylation sites in the cytoplasmic portion. The gene encoding human β-sarcoglycan is on chromosome 4q12, and recessive mutations in the β-sarcoglycan gene lead to muscular dystrophy and cardiomyopathy. Mice lacking β-sarcoglycan have been generated and develop cardiomyopathy and muscular dystrophy similar to their human counterparts.28,29 Notably, the smooth muscle sarcoglycan complex is disrupted in these mice since β-sarcoglycan is expressed normally in smooth muscle including vascular smooth muscle.28
γ-sarcoglycan
The gene encoding γ-sarcoglycan maps to human chromosome 13q12, and recessive mutations in the γ-sarcoglycan gene lead to cardiomyopathy and muscular dystrophy.18 There is a single common mutation, Δ521-T, that is relatively prevalent. This frameshift mutation encodes a series of novel amino acids leading to a stop codon and is thought to produce an unstable, truncated protein. Generally, as a result of this Δ521-T mutation, there is little sarcoglycan expressed at the surface of muscle, although exceptions have been described.30,31 γ-sarcoglycan is a 35 kDa type II transmembrane protein that has a single N-linked glycosylation site and predicted phosphorylation sites in the cytoplasmic domain (fig.2). Mouse models have been engineered that harbor null alleles for ?-sarcoglycan.32,33 These mice display a phenotype similar to what is seen in β-sarcoglycan null mice (above) and δ-sarcoglycan null mice (see below).
δ-sarcoglycan
δ-sarcoglycan is highly related to γ-sarcoglycan in that they share nearly 70 percent amino acid similarity (fig. 2).34 Moreover, the gene structure for δ-sarcoglycan on human chromosome 5q31 is similar to the gene structure for γ-sarcoglycan. The identical placement of intron and exons in these genes strongly suggest that they arose from a gene duplication event. Recessive mutations in the δ-sarcoglycan gene produce LGMD type 2F, a disorder similar to DMD and BMD.20 Rare cases of familial dilated cardiomyopathy have also been attributed in mutations in the δ-sarcoglycan gene, although in this case the pattern of inheritance of dilated cardiomyopathy was thought to be autosomal dominant.35 Like γ-sarcoglycan, δ-sarcoglycan is a type II transmembrane 35 kDa protein with a single N-linked glycosylation site. There is an alternative splice form that alters the very carboxyl-terminus of the protein.36 This region contains a cluster of conserved cysteine residues that have weak homology to the epidermal growth factor like cysteine residues.37 The cysteine cluster is found in β-, γ-, δ- and ζ-(see below) sarcoglycan, and may serve as a receptor site for an as yet unknown ligand. The alternative splice form of δ-sarcoglycan lacks these cysteine residues and is widely expressed by mRNA expression analysis. Like β-sarcoglycan, δ-sarcoglycan is expressed in both striated and smooth muscle, including vascular smooth muscle.
There are several different animal models of δ-sarcoglycan mutations. The first, the BIO4.6 Syrian Hamster model is a recessive model of cardiomyopathy and muscular dystrophy that was first described 40 years ago.38,39 This genetic model of cardiomyopathy displays pathology very similar to what is seen in humans with dystrophin or sarcoglycan gene mutations. That is, focal degeneration occurs in both heart and skeletal in response to dystrophin and sarcoglycan gene mutations. In the BIO 14.6 hamster, the focal nature of tissue damage was initially suggested to relate to vascular spasm since, in the heart, the appearance resembled focal micro-infarcts.40,41 A mouse model was generated with a null allele of δ-sarcoglycan, and these mice displayed the same focal nature of tissue damage.42 In these mice, it was noted that the sarcoglycan complex was disrupted in vascular smooth muscle, and it was reasoned that disruption of the vascular smooth muscle sarcoglycan complex was responsible for vascular spasm and tissue damage. This concept was further supported by studies of α-sarcoglycan mutant mice that displayed little cardiomyopathy and had an intact vascular smooth muscle sarcoglycan complex.24
An alternative explanation is that the defect is one that arises in striated muscle and that vascular defects arise as a nonvascular smooth muscle cell-autonomous defect. Supporting this idea is the cardiomyopathic findings in mice lacking γ-sarcoglycan. Mice lacking γ-sarcoglycan and δ-sarcoglycan have an identical phenotype with focal degeneration, but the vascular smooth muscle sarcoglycan complex is intact in these γ-sarcoglycan null animals.43 Abnormal vascular reactivity arises from degeneration in cardiomyocytes and furthers the course of cardiomyopathy progression. Vascular spasm mediated through this vascular smooth muscle-cell xtrinsic process is a target for therapeutic intervention.
ε-sarcoglycan
ε-sarcoglycan is highly related to α-sarcoglycan and is encoded by a gene on human chromosome 7p21.44,45 Like α-sarcoglycan, ε-sarcoglycan is type I transmembrane protein (fig. 2), but unlike α-sarcoglycan, ε-sarcoglycan is expressed in tissues outside of muscle.44,45 ε-sarcoglycan is expressed highly in the developing nervous system (EMM, unpublished results), and ε-sarcoglycan is expressed in striated and smooth muscle. Interestingly, mutations in the ε-sarcoglycan gene lead to an unusual phenotype of myoclonus dystonia.46 In this syndrome, ε-sarcoglycan gene mutations lead to a dominant, nonprogressive movement disorder. The genetics of myoclonus dystonia are complicated by variable penetrance related to parent-of-origin effects. This phenomenon is explained by imprinting of the maternal ε-sarcoglycan allele, first noted in mice.47 Thus, as the maternal allele is silenced, the gene defect, in effect is inherited from the parental allele.48,49
The high degree of sequence similarity between α-sarcoglycan and ε-sarcoglycan has been noted.44,45 It has been suggested that the mild to absent cardiac phenotype associated with a-sarcoglycan mutations may relate to upregulation and compensation by ε-sarcoglycan.25 In vascular smooth muscle, it is clear that ε-sarcoglycan can substitute for α-sarcoglycan.50
ζ-sarcoglycan
The most recently described sarcoglycan sequence is ζ-sarcoglycan.51 ζ-sarcoglycan is highly related to both γ-sarcoglycan and δ-sarcoglycan and is similarly a type II 35 kDa protein (fig. 2). The intron and exon structure of the ζ-sarcoglycan gene suggests a gene triplication event leading to the relationship between γ-, δ- and ζ-sarcoglycan. ζ-sarcoglycan is encoded by a gene on human chromosome 8p22. ζ-sarcoglycan can be purified from muscle microsomal membranes and through immunoprecipitation experiments, was shown to interact with dystrophin and β-sarcoglycans.51 Moreover, ζ-sarcoglycan expression is reduced in microsomal membranes from muscle with sarcoglycan gene mutations. Genetic mutations have not been described in ζ-sarcoglycan gene so it is unclear whether genetic defects in this gene can lead to muscular dystrophy. In addition to being expressed in striated muscle, ζ-sarcoglycan is highly expressed in smooth muscle sources including vascular smooth muscle (fig. 3).
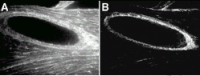
Sarcospan
Sarcospan is a member of the tetraspanin family and, as it name implies, contains four transmembrane domains. Initially characterized as the 25 kDa component of the DGC, the identity of sarcospan was clarified by microsequencing.52 Mice deficient for sarcospan have been generated and display no obvious phenotype.53 This apparent lack of phenotype may arise from the expression of additional tetraspanins in muscle and potential compensation by these alternative tetraspanins. Biochemical and genetic studies have confirmed that the sarcoglycan complex is required for proper targeting of sarcospan.54 Sarcospan is intimately associated with the sarcoglycan complex, but the detergent approach used to define the sarcoglycan complex does not include sarcospan.55 Thus, it appears that sarcospan is not, by strict criteria, a member of the sarcoglycan complex although its close association with the sarcoglycan complex is important. In other tissues, tetraspanin proteins interact with integrins to mediate interaction with other membrane complexes.56
Heterogeneity of the Sarcoglycan Complex
In mammals, the six sarcoglycan sequences, α, β, γ, δ, ε, and ζ, appear to constitute the complete sarcoglycan family. The major sarcoglycan complex found in striated muscle includes α-sarcoglycan, β-sarcoglycan, γ-sarcoglycan and δ-sarcoglycan. As originally characterized through biochemical purification, the sarcoglycan complex contained a 50 kDa subunit (α-sarcoglycan), a 43 kDa subunit (β-sarcoglycan) and a 35 kDa subunit with a ratio of 1:1:2.2,57 The heterogeneity of the 35 kDa component was not originally appreciated, and it was through genomic and subsequent biochemical analysis that it became clear that the 35 kDa sarcoglycan subunit in striated muscle contains both γ-sarcoglycan and δ-sarcoglycan in a 1:1 ratio.2,57 Further complicating this issue is the recent identification of ζ-sarcoglycan.51 γ-sarcoglycan, δ-sarcoglycan and ζ-sarcoglycan are very similar in amino acid composition, so the specificity of antibodies should be considered when reexamining earlier studies. Cross reactivity of antibodies between γ-sarcoglycan, δ-sarcoglycan and ζ-sarcoglycan may confound some of the earlier analyses examining expression patterns and tissue specificity.
ε-sarcoglycan can be detected at the plasma membrane of skeletal muscle, but by immunoblotting and immunostaining, the amount of this protein is less that the related protein α-sarcoglycan.32 However, the apparent lower expression of ε-sarcoglycan may arise from a lower affinity anti-ε-sarcoglycan antibody.32 That said, the original biochemical characterization of the sarcoglycan complex from skeletal muscle supports that the 50 kDa component is mainly comprised by α-sarcoglycan. The pI of each of α-sarcoglycan and ε-sarcoglycan is similar (5.44 for α-sarcoglycan versus 6.10 for ε-sarcoglycan) but the molecular mass of these two species differs slightly with ε-sarcoglycan migrating slightly slower on SDS-PAGE analysis. Furthermore, using an anti-ε-sarcoglycan antibody, ε-sarcoglycan appears to be more highly expressed in vascular smooth muscle tissue when examining tissue sections where both striated and vascular smooth muscle are found in the same sections.50 Like e-sarcoglycan, ζ-sarcoglycan appears to be more highly expressed in vascular smooth muscle than in striated muscle, although expression can also be detected in striated muscle.51 In vascular smooth muscle, a complex of ζ-sarcoglycan, δ-sarcoglycan and β-sarcoglycan constitutes the vascular smooth muscle sarcoglycan complex (fig. 4).50 Whether this same complex is also present in striated muscle is not known. The major sarcoglycan complex of striated muscle is α-sarcoglycan, β-sarcoglycan, γ-sarcoglycan and δ-sarcoglycan.

Sarcoglycans Are Conserved in Invertebrates
Genomic information from both C. elegans and Drosophila melanogaster support the presence of at least three distinct sarcoglycan genes in each of these genetic model systems.58,59 In both species, there is a single α/ε-sarcoglycan-like sequence, a β-sarcoglycan-like sequence and a single γ/δ/ζ-sarcoglycan-like sequence. In mammals, gene duplication produced α-sarcoglycan and ε-sarcoglycan while there is even greater heterogeneity for γ-, δ- and ζ-sarcoglycan. Analysis of mice with null mutations in sarcoglycan genes suggests that each sarcoglycan protein possesses unique function and that sarcoglycan subunits cannot substitute for one and other, except potentially in the case of α-sarcoglycan.25 There is upregulation of ε-sarcoglycan in α-sarcoglycan null hearts and little to no cardiomyopathy in these mice, suggesting that ε-sarcoglycan can potentially assume some of the function of α-sarcoglycan.25
Sequences identified as sarcoglycans are noted in the Danio rerio and Fugu databases (unpublished results). Antibodies raised against mammalian sarcoglycan epitopes demonstrate crossreactivity to muscle membrane proteins from lower vertebrates confirming the presence of a sarcoglycan complex in leech and torpedo.60-62 Genetic studies have been initiated in C. elegans where genetic mutants were studied using an RNA interference technique.58 Worms with a combination of dystrophin, dystroglycan and γ/δ/ζ “knock-downs” displayed a head-bending phenotype, and the significance of this finding is still under investigation. Whether an invertebrate model of sarcoglycan gene mutations may be useful for larger scale genetic screens, including suppressor screens, remains to be established. Of note, in examining the worm and fly databases, orthologues for sarcospan were not identified.58,59
Assembly of the Sarcoglycan Complex
The assembly of the sarcoglycan complex is of relevance since so many sarcoglycan mutations result in the dissolution and/or aberrant assembly of the sarcoglycan complex (fig. 5). Sarcoglycan assembly has been studied in cultured cells, including the C2C12 mouse muscle cell line and primary myoblasts cultures, in mouse models of sarcoglycan mutations and inferred from staining patterns of residual sarcoglycans in muscle biopsies from human patients with sarcoglycan mutations.63-65 Taking these data together, in striated muscle, δ-sarcoglycan and β-sarcoglycan form a core unit to which α-sarcoglycan or ε-sarcoglycan can bind. γ-sarcoglycan is added last to this complex. The complex assembles in the endoplasmic reticulum where it is also coassembles with β-dystroglycan, although this interaction is less well studied.64
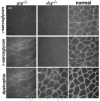
Interactions with the Remainder of the DGC and Other Cytoskeletal Proteins
Recent studies using a differential ionic strength to dissolve interactions within the DGC have suggested that the sarcoglycan complex associates with dystrophin by way of an interaction with dystrobrevin.55 a-dystrobrevin is a splice form highly expressed in muscle that has homology to the carboxyl-terminus of dystrophin and, like dystrophin, can bind to the syntrophins.66,67 The syntrophin-dystrobrevin interaction is tight and there is evidence that this complex preferentially interacts with the sarcoglycan complex.67
It has also been suggested that the role of the sarcoglycan complex is to stabilize the interaction between α-dystroglycan and β-dystroglycan.68 The two subunits, α and β, of dystroglycan are produced from a single gene and result from proteolytic cleavage from a single polypeptide precursor (see chapter 16). The α subunit of dystroglycan is differentially glycosylated in many different tissues types and was originally identified as cranin in the nervous system.69,70 In muscle, the predominant form of α-dystroglycan is a 156 kDa protein. In the BIO 14.6 Syrian hamster that is mutant for δ-sarcoglycan, biochemical analysis of the residual dystroglycan complex reveals that it no longer associates with the sarcoglycan complex.68
There is a muscle specific form of the protein filamin, called filamin C, is a cytoskeletal protein that is found at both the Z band and plasma membrane of muscle.71,72 Filamins are actin bindings proteins that play a role in reorganizing the cytoskeleton in nonmuscle cells. The precise function of filamin C in striated muscle is not known. It was shown that filamin C can bind directly to the cytoplasmic domains of both γ-sarcoglycan and δ-sarcoglycan (fig. 1).71 In addition, the cellular distribution of filamin C changes in response to the loss of the sarcoglycan complex where it redistributes from overlying the Z band to the plasma membrane. The genetic defect in human subjects is a dominant form of LGMD that maps to chromosome 7 where the locus for filamin C is. However, mutations in filamin C gene have not been identified.73,74
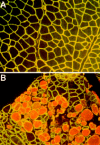
Where sarcoglycan is absent, whether by a primary mutation in sarcoglycan genes or through secondary destabilization in the case of dystrophin gene mutations, the plasma membrane becomes abnormally leaky. This has been best demonstrated with the vital tracer Evans blue dye (EBD) as normal muscle is impermeable to EBD. EBD uptake is a feature of dystrophin deficient muscle but not of muscle with a mutation in a2 chain of laminin-2 (merosin).75,76 This laminin is the extracellular ligand for dystroglycan and mutations in this gene lead to congenital muscular dystrophy (see chapter 6).69 Disruption of the sarcoglycan complex, where the dystrophin protein is still normally found at the plasma membrane, is also associated with EBD uptake.32 Therefore, aspects of the membrane permeability defects can be attributed specifically to the loss of the sarcoglycan complex (fig. 6).
The function of the sarcoglycan complex is not known, but like the DGC itself, the complex appears to have both mechanical and signaling roles. The only domain found within a sarcoglycan sequence to date is the ectoATPase domain in α-sarcoglycan.27 As for physiological function, the function of the DGC appears to be to stabilize the plasma membrane against some of the forces associated with muscle contraction.77,78 Mice lacking dystrophin display enhanced damage in response to repeated contraction, especially eccentric contraction that is thought to mimic the forces associated with physiologic exercise. Muscle from mice lacking δ-sarcoglycan display an intermediate phenotype with an intermediate degree of increased damage in response to eccentric contraction.65 Muscle from mice lacking γ-sarcoglycan do not show enhanced damage in response to eccentric contraction highlighting that the sarcoglycan complex, and the DGC itself, play more than just a simple mechanical stabilizing role.79 The sarcoglycan complex, like the DGC, is concentrated over costameres (fig. 7). Costameres are specialized structures of striated muscle that overlie the Z band.80,81 It is clear that the Z band had additional roles beyond simply anchoring thin filaments; in cardiac muscle, titin, MLP and telethonin play a role in elastic recoil through Z band anchoring.82,83 As the DGC and sarcoglycan form the specific attachment of the Z band to the membrane, they are also likely to be multifunctional.
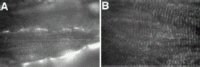
Future Directions
Future studies of the sarcoglycan and the DGC complex center on understanding the nonmechanical function of the DGC and identifying the ligand that sarcoglycan binds to in the extracellular matrix. Understanding better the downstream signaling defects that arise from dissolution of the sarcoglycan complex have bearing, in that these pathways may be targets for therapy in treating both the LGMDs and DMD/BMD. Finally, signalling defects that arise form the loss of this complex may be relevant to understand better muscle wasting and degeneration as it occurs in other muscle diseases and with the muscle loss that occurs in aging.
Ackowledgments
EMM is supported by the NIH, the American Heart Association, the Muscular Dystrophy Association and the Burroughs Wellcome Fund.
References
- 1.
- Ervasti JM, Ohlendieck K, Kahl SD. et al. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature. 1990;345(6273):315–319. [PubMed: 2188135]
- 2.
- Ervasti JM, Campbell KP. Membrane organization of the dystrophin-glycoprotein complex. Cell. 1991;66(6):1121–1131. [PubMed: 1913804]
- 3.
- Yoshida M, Ozawa E. Glycoprotein complex anchoring dystrophin to sarcolemma. J Biochem (Tokyo). 1990;108(5):748–752. [PubMed: 2081733]
- 4.
- Yoshida M, Suzuki A, Yamamoto H. et al. Dissociation of the complex of dystrophin and its associated proteins into several unique groups by n-octyl beta-D-glucoside. Eur J Biochem. 1994;222(3):1055–1061. [PubMed: 8026484]
- 5.
- Hoffman EP, Brown JrRH, Kunkel LM. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51(6):919–928. [PubMed: 3319190]
- 6.
- Sicinski P, Geng Y, Ryder-Cook AS. et al. The molecular basis of muscular dystrophy in the mdx mouse: A point mutation. Science. 1989;244(4912):1578–1580. [PubMed: 2662404]
- 7.
- Ben HamidaM, Fardeau M, Attia N. Severe childhood muscular dystrophy affecting both sexes and frequent in Tunisia. Muscle Nerve. 1983;6(7):469–480. [PubMed: 6633560]
- 8.
- Ben OthmaneK, Ben HamidaM, Pericak-Vance MA. et al. Linkage of Tunisian autosomal recessive Duchenne-like muscular dystrophy to the pericentromeric region of chromosome 13q. Nat Genet. 1992;2(4):315–317. [PubMed: 1303286]
- 9.
- Romero NB, Tome FM, Leturcq F. et al. Genetic heterogeneity of severe childhood autosomal recessive muscular dystrophy with adhalin (50 kDa dystrophin-associated glycoprotein) deficiency. C R Acad Sci III. 1994;317(1):70–76. [PubMed: 7987694]
- 10.
- Passos-Bueno MR, Oliveira JR, Bakker E. et al. Genetic heterogeneity for Duchenne-like muscular dystrophy (DLMD) based on linkage and 50 DAG analysis. Hum Mol Genet. 1993;2(11):1945–1947. [PubMed: 8281158]
- 11.
- Azibi K, Bachner L, Beckmann JS. et al. Severe childhood autosomal recessive muscular dystrophy with the deficiency of the 50 kDa dystrophin-associated glycoprotein maps to chromosome 13q12. Hum Mol Genet. 1993;2(9):1423–1428. [PubMed: 8242065]
- 12.
- Ohlendieck K, Campbell KP. Dystrophin-associated proteins are greatly reduced in skeletal muscle from mdx mice. J Cell Biol. 1991;115(6):1685–1694. [PMC free article: PMC2289197] [PubMed: 1757468]
- 13.
- Matsumura K, Tome FM, Collin H. et al. Deficiency of the 50K dystrophin-associated glycoprotein in severe childhood autosomal recessive muscular dystrophy. Nature. 1992;359(6393):320–322. [PubMed: 1406935]
- 14.
- Yamanouchi Y, Mizuno Y, Yamamoto H. et al. Selective defect in dystrophin-associated glycoproteins 50DAG (A2) and 35DAG (A4) in the dystrophic hamster: An animal model for severe childhood autosomal recessive muscular dystrophy (SCARMD). Neuromuscul Disord. 1994;4(1):49–54. [PubMed: 8173351]
- 15.
- Roberds SL, Ervasti JM, Anderson RD. et al. Disruption of the dystrophin-glycoprotein complex in the cardiomyopathic hamster. J Biol Chem. 1993;268(16):11496–11499. [PubMed: 8505286]
- 16.
- Bonnemann CG, Modi R, Noguchi S. et al. Beta-sarcoglycan (A3b) mutations cause autosomal recessive muscular dystrophy with loss of the sarcoglycan complex. Nat Genet. 1995;11(3):266–273. [PubMed: 7581449]
- 17.
- Roberds SL, Leturcq F, Allamand V. et al. Missense mutations in the adhalin gene linked to autosomal recessive muscular dystrophy. Cell. 1994;78(4):625–633. [PubMed: 8069911]
- 18.
- Noguchi S, McNally EM, Ben Othmane K. et al. Mutations in the dystrophin-associated protein gamma-sarcoglycan in chromosome 13 muscular dystrophy. Science. 1995;270(5237):819–822. [PubMed: 7481775]
- 19.
- Lim LE, Duclos F, Broux O. et al. Beta-sarcoglycan: Characterization and role in limb-girdle muscular dystrophy linked to 4q12. Nat Genet. 1995;11(3):257–265. [PubMed: 7581448]
- 20.
- Nigro V, de Sa Moreira E, Piluso G. et al. Autosomal recessive limb-girdle muscular dystrophy, LGMD2F, is caused by a mutation in the delta-sarcoglycan gene. Nat Genet. 1996;14(2):195–198. [PubMed: 8841194]
- 21.
- Roberds SL, Anderson RD, Ibraghimov-Beskrovnaya O. et al. Primary structure and muscle-specific expression of the 50-kDa dystrophin-associated glycoprotein (adhalin). J Biol Chem. 1993;268(32):23739–23742. [PubMed: 8226900]
- 22.
- McNally EM, Yoshida M, Mizuno Y. et al. Human adhalin is alternatively spliced and the gene is located on chromosome 17q21. Proc Natl Acad Sci USA. 1994;91(21):9690–9694. [PMC free article: PMC44882] [PubMed: 7937874]
- 23.
- Piccolo F, Roberds SL, Jeanpierre M. et al. Primary adhalinopathy: A common cause of autosomal recessive muscular dystrophy of variable severity. Nat Genet. 1995;10(2):243–245. [PubMed: 7663524]
- 24.
- Duclos F, Straub V, Moore SA. et al. Progressive muscular dystrophy in alpha-sarcoglycan-deficient mice. J Cell Biol. 1998;142(6):1461–1471. [PMC free article: PMC2141773] [PubMed: 9744877]
- 25.
- Liu LA, Engvall E. Sarcoglycan isoforms in skeletal muscle. J Biol Chem. 1999;274(53):38171–38176. [PubMed: 10608889]
- 26.
- van der Kooi AJ, de VoogtWG, Barth PG. et al. The heart in limb girdle muscular dystrophy. Heart. 1998;79(1):73–77. [PMC free article: PMC1728583] [PubMed: 9505924]
- 27.
- Betto R, Senter L, Ceoldo S. et al. Ecto-ATPase activity of alpha-sarcoglycan (adhalin). J Biol Chem. 1999;274(12):7907–7912. [PubMed: 10075685]
- 28.
- Durbeej M, Cohn RD, Hrstka RF. et al. Disruption of the beta-sarcoglycan gene reveals pathogenetic complexity of limb-girdle muscular dystrophy type 2E. Mol Cell. 2000;5(1):141–151. [PubMed: 10678176]
- 29.
- Araishi K, Sasaoka T, Imamura M. et al. Loss of the sarcoglycan complex and sarcospan leads to muscular dystrophy in beta-sarcoglycan-deficient mice. Hum Mol Genet. 1999;8(9):1589–1598. [PubMed: 10441321]
- 30.
- Bonnemann CG, Wong J, Jones KJ. et al. Primary gamma-sarcoglycanopathy (LGMD 2C): Broadening of the mutational spectrum guided by the immunohistochemical profile. Neuromuscul Disord. 2002;12(3):273–280. [PubMed: 11801399]
- 31.
- Crosbie RH, Lim LE, Moore SA. et al. Molecular and genetic characterization of sarcospan: Insights into sarcoglycan-sarcospan interactions. Hum Mol Genet. 2000;9(13):2019–2027. [PubMed: 10942431]
- 32.
- Hack AA, Ly CT, Jiang F. et al. Gamma-sarcoglycan deficiency leads to muscle membrane defects and apoptosis independent of dystrophin. J Cell Biol. 1998;142(5):1279–1287. [PMC free article: PMC2149352] [PubMed: 9732288]
- 33.
- Sasaoka T, Imamura M, Araishi K. et al. Pathological analysis of muscle hypertrophy and degeneration in muscular dystrophy in gamma-sarcoglycan-deficient mice. Neuromuscul Disord. 2003;13(3):193–206. [PubMed: 12609501]
- 34.
- Nigro V, Piluso G, Belsito A. et al. Identification of a novel sarcoglycan gene at 5q33 encoding a sarcolemmal 35 kDa glycoprotein. Hum Mol Genet. 1996;5(8):1179–1186. [PubMed: 8842738]
- 35.
- Tsubata S, Bowles KR, Vatta M. et al. Mutations in the human delta-sarcoglycan gene in familial and sporadic dilated cardiomyopathy. J Clin Invest. 2000;106(5):655–662. [PMC free article: PMC381284] [PubMed: 10974018]
- 36.
- Jung D, Duclos F, Apostol B. et al. Characterization of delta-sarcoglycan, a novel component of the oligomeric sarcoglycan complex involved in limb-girdle muscular dystrophy. J Biol Chem. 1996;271(50):32321–32329. [PubMed: 8943294]
- 37.
- McNally EM, Duggan D, Gorospe JR. et al. Mutations that disrupt the carboxyl-terminus of gamma-sarcoglycan cause muscular dystrophy. Hum Mol Genet. 1996;5(11):1841–1847. [PubMed: 8923014]
- 38.
- Gertz EW. Cardiomyopathic Syrian hamster: A possible model of human disease. Prog Exp Tumor Res. 1972;16:242–260. [PubMed: 4261129]
- 39.
- Bajusz E. Hereditary cardiomyopathy: A new disease model. Am Heart J. 1969;77(5):686–696. [PubMed: 4888117]
- 40.
- Factor SM, Sonnenblick EH. Hypothesis: Is congestive cardiomyopathy caused by a hyperreactive myocardial microcirculation (microvascular spasm)? Am J Cardiol. 1982;50(5):1149–1152. [PubMed: 6215853]
- 41.
- Factor SM, Minase T, Cho S. et al. Microvascular spasm in the cardiomyopathic Syrian hamster: A preventable cause of focal myocardial necrosis. Circulation. 1982;66(2):342–354. [PubMed: 7094244]
- 42.
- Coral-Vazquez R, Cohn RD, Moore SA. et al. Disruption of the sarcoglycan-sarcospan complex in vascular smooth muscle: A novel mechanism for cardiomyopathy and muscular dystrophy. Cell. 1999;98(4):465–474. [PubMed: 10481911]
- 43.
- Wheeler MT, Allikian MJ, Heydeman A. et al. Smooth muscle cell extrinsic vascular spasm arises from cardiomyocyte degeneration in sarcoglycan mutant cardiomyopathy. J Clin Invest. 2004;113:668–675. [PMC free article: PMC351323] [PubMed: 14991064]
- 44.
- Ettinger AJ, Feng G, Sanes JR. Epsilon-Sarcoglycan, a broadly expressed homologue of the gene mutated in limb-girdle muscular dystrophy 2D. J Biol Chem. 1997;272(51):32534–32538. [PubMed: 9405466]
- 45.
- McNally EM, Ly CT, Kunkel LM. Human epsilon-sarcoglycan is highly related to alpha-sarcoglycan (adhalin), the limb girdle muscular dystrophy 2D gene. FEBS Lett. 1998;422(1):27–32. [PubMed: 9475163]
- 46.
- Zimprich A, Grabowski M, Asmus F. et al. Mutations in the gene encoding epsilon-sarcoglycan cause myoclonus-dystonia syndrome. Nat Genet. 2001;29(1):66–69. [PubMed: 11528394]
- 47.
- Piras G, El KharroubiA, Kozlov S. et al. Zac1 (Lot1), a potential tumor suppressor gene, and the gene for epsilon-sarcoglycan are maternally imprinted genes: Identification by a subtractive screen of novel uniparental fibroblast lines. Mol Cell Biol. 2000;20(9):3308–3315. [PMC free article: PMC85624] [PubMed: 10757814]
- 48.
- Grabowski M, Zimprich A, Lorenz-Depiereux B. et al. The epsilon-sarcoglycan gene (SGCE), mutated in myoclonus-dystonia syndrome, is maternally imprinted. Eur J Hum Genet. 2003;11(2):138–144. [PubMed: 12634861]
- 49.
- Muller B, Hedrich K, Kock N. et al. Evidence that paternal expression of the epsilon-sarcoglycan gene accounts for reduced penetrance in myoclonus-dystonia. Am J Hum Genet. 2002;71(6):1303–1311. [PMC free article: PMC378568] [PubMed: 12444570]
- 50.
- Straub V, Ettinger AJ, Durbeej M. et al. epsilon-sarcoglycan replaces alpha-sarcoglycan in smooth muscle to form a unique dystrophin-glycoprotein complex. J Biol Chem. 1999;274(39):27989–27996. [PubMed: 10488149]
- 51.
- Wheeler MT, Zarnegar S, McNally EM. zeta-Sarcoglycan, a novel component of the sarcoglycan complex, is reduced in muscular dystrophy. Hum Mol Genet. 2002;11(18):2147–2154. [PubMed: 12189167]
- 52.
- Crosbie RH, Heighway J, Venzke DP. et al. Sarcospan, the 25-kDa transmembrane component of the dystrophin-glycoprotein complex. J Biol Chem. 1997;272(50):31221–31224. [PubMed: 9395445]
- 53.
- Lebakken CS, Venzke DP, Hrstka RF. et al. Sarcospan-deficient mice maintain normal muscle function. Mol Cell Biol. 2000;20(5):1669–1677. [PMC free article: PMC85350] [PubMed: 10669744]
- 54.
- Crosbie RH, Lebakken CS, Holt KH. et al. Membrane targeting and stabilization of sarcospan is mediated by the sarcoglycan subcomplex. J Cell Biol. 1999;145(1):153–165. [PMC free article: PMC2148225] [PubMed: 10189375]
- 55.
- Yoshida M, Hama H, Ishikawa-Sakurai M. et al. Biochemical evidence for association of dystrobrevin with the sarcoglycan-sarcospan complex as a basis for understanding sarcoglycanopathy. Hum Mol Genet. 2000;9(7):1033–1040. [PubMed: 10767327]
- 56.
- Hemler ME. Specific tetraspanin functions. J Cell Biol. 2001;155(7):1103–1107. [PMC free article: PMC2199333] [PubMed: 11756464]
- 57.
- Yamamoto H, Hagiwara Y, Mizuno Y. et al. Heterogeneity of dystrophin-associated proteins. J Biochem (Tokyo). 1993;114(1):132–139. [PubMed: 8407865]
- 58.
- Grisoni K, Martin E, Gieseler K. et al. Genetic evidence for a dystrophin-glycoprotein complex (DGC) in Caenorhabditis elegans. Gene. 2002;294(1-2):77–86. [PubMed: 12234669]
- 59.
- Greener MJ, Roberts RG. Conservation of components of the dystrophin complex in Drosophila. FEBS Lett. 2000;482(1-2):13–18. [PubMed: 11018515]
- 60.
- Royuel M, Hugon G, Rivier Fl. et al. Dystrophin-associated proteins in obliquely striated muscle of the leech. Histochem J. 2001;33(3):135–139. [PubMed: 11508336]
- 61.
- Royuela M, Paniagua R, Rivier F. et al. Presence of invertebrate dystrophin-like products in obliquely striated muscle of the leech, Pontobdella muricata (Annelida, Hirudinea). Histochem J. 1999;31(9):603–608. [PubMed: 10579629]
- 62.
- Royuela M, Hugon G, Rivier F. et al. Variations in dystrophin complex in red and white caudal muscles from Torpedo marmorata. J Histochem Cytochem. 2001;49(7):857–865. [PubMed: 11410610]
- 63.
- Chan YM, Bonnemann CG, Lidov HG. et al. Molecular organization of sarcoglycan complex in mouse myotubes in culture. J Cell Biol. 1998;143(7):2033–2044. [PMC free article: PMC2175228] [PubMed: 9864373]
- 64.
- Noguchi S, Wakabayashi E, Imamura M. et al. Formation of sarcoglycan complex with differentiation in cultured myocytes. Eur J Biochem. 2000;267(3):640–648. [PubMed: 10651799]
- 65.
- Hack AA, Lam MY, Cordier L. et al. Differential requirement for individual sarcoglycans and dystrophin in the assembly and function of the dystrophin-glycoprotein complex. J Cell Sci. 2000;113(Pt 142):2535–2544. [PubMed: 10862711]
- 66.
- Newey SE, Benson MA, Ponting CP. et al. Alternative splicing of dystrobrevin regulates the stoichiometry of syntrophin binding to the dystrophin protein complex. Curr Biol. 2000;10(20):1295–1298. [PubMed: 11069112]
- 67.
- Sadoulet-Puccio HM, Rajala M, Kunkel LM. Dystrobrevin and dystrophin: An interaction through coiled-coil motifs. Proc Natl Acad Sci USA. 1997;94(23):12413–12418. [PMC free article: PMC24974] [PubMed: 9356463]
- 68.
- Straub V, Duclos F, Venzke DP. et al. Molecular pathogenesis of muscle degeneration in the delta-sarcoglycan-deficient hamster. Am J Pathol. 1998;153(5):1623–1630. [PMC free article: PMC1853419] [PubMed: 9811355]
- 69.
- Ibraghimov-Beskrovnaya O, Ervasti JM, Leveille CJ. et al. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature. 1992;355(6362):696–702. [PubMed: 1741056]
- 70.
- Smalheiser NR, Schwartz NB. Cranin: A laminin-binding protein of cell membranes. Proc Natl Acad Sci USA. 1987;84(18):6457–6461. [PMC free article: PMC299096] [PubMed: 2957695]
- 71.
- Thompson TG, Chan YM, Hack AA. et al. Filamin 2 (FLN2): A muscle-specific sarcoglycan interacting protein. J Cell Biol. 2000;148(1):115–126. [PMC free article: PMC3207142] [PubMed: 10629222]
- 72.
- Bonnemann CG, Thompson TG, van der Ven PF. et al. Filamin C accumulation is a strong but nonspecific immunohistochemical marker of core formation in muscle. J Neurol Sci. 2003;206(1):71–78. [PubMed: 12480088]
- 73.
- Speer MC, Vance JM, Grubber JM. et al. Identification of a new autosomal dominant limb-girdle muscular dystrophy locus on chromosome 7. Am J Hum Genet. 1999;64(2):556–562. [PMC free article: PMC1377765] [PubMed: 9973293]
- 74.
- Palenzuela L, Andreu AL, Gamez J. et al. A novel autosomal dominant limb-girdle muscular dystrophy (LGMD 1F) maps to 7q32.1-32.2. Neurology. 2003;61(3):404–406. [PubMed: 12913210]
- 75.
- Straub V, Rafael JA, Chamberlain JS. et al. Animal models for muscular dystrophy show different patterns of sarcolemmal disruption. J Cell Biol. 1997;139(2):375–385. [PMC free article: PMC2139791] [PubMed: 9334342]
- 76.
- Matsuda R, Nishikawa A, Tanaka H. Visualization of dystrophic muscle fibers in mdx mouse by vital staining with Evans blue: Evidence of apoptosis in dystrophin-deficient muscle. J Biochem (Tokyo). 1995;118(5):959–964. [PubMed: 8749313]
- 77.
- Petrof BJ, Shrager JB, Stedman HH. et al. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci USA. 1993;90(8):3710–3714. [PMC free article: PMC46371] [PubMed: 8475120]
- 78.
- Lynch GS, Rafael JA, Chamberlain JS. et al. Contraction-induced injury to single permeabilized muscle fibers from mdx, transgenic mdx, and control mice. Am J Physiol Cell Physiol. 2000;279(4):C1290–1294. [PubMed: 11003610]
- 79.
- Hack AA, Cordier L, Shoturma DI. et al. Muscle degeneration without mechanical injury in sarcoglycan deficiency. Proc Natl Acad Sci USA. 1999;96(19):10723–10728. [PMC free article: PMC17950] [PubMed: 10485893]
- 80.
- Ervasti JM. Costameres: The Achilles' heel of Herculean muscle. J Biol Chem. 2003;278(16):13591–13594. [PubMed: 12556452]
- 81.
- Bloch RJ, Capetanaki Y, O'Neill A. et al. Costameres: Repeating structures at the sarcolemma of skeletal muscle. Clin Orthop. 2002;(403Suppl):S203–210. [PubMed: 12394470]
- 82.
- Hoshijima M, Pashmforoush M, Knoll R. et al. The MLP family of cytoskeletal Z disc proteins and dilated cardiomyopathy: A stress pathway model for heart failure progression. Cold Spring Harb Symp Quant Biol. 2002;67:399–408. [PubMed: 12858565]
- 83.
- Knoll R, Hoshijima M, Hoffman HM. et al. The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell. 2002;111(7):943–955. [PubMed: 12507422]
- The Sarcoglycans - Madame Curie Bioscience DatabaseThe Sarcoglycans - Madame Curie Bioscience Database
- Nucleic Acids Are Not Boring Long Polymers of Only Four Types of Nucleotides: A ...Nucleic Acids Are Not Boring Long Polymers of Only Four Types of Nucleotides: A Guided Tour - Madame Curie Bioscience Database
- Surgical Stress Response and Cancer Metastasis: The Potential Benefit of Periope...Surgical Stress Response and Cancer Metastasis: The Potential Benefit of Perioperative Beta Blockade - Madame Curie Bioscience Database
- Branching Morphogenesis of the Prostate - Madame Curie Bioscience DatabaseBranching Morphogenesis of the Prostate - Madame Curie Bioscience Database
- From Artificial Antibodies to Nanosprings:The Biophysical Properties of Repeat P...From Artificial Antibodies to Nanosprings:The Biophysical Properties of Repeat Proteins - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...