

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Members of the Ras-related Rho family of GTPases act as key signalling nodes that regulate a variety of crucial cellular activities including morphology, motility, and proliferation. Their aberrant expression causes uncontrolled growth and transformation, and they are required for the transforming actions of Ras and other oncoproteins. However, whereas the identification of mutated Ras proteins in human cancers prompted immediate and intense interest in investigating their role in tumor formation, the evidence for the involvement of Rho GTPases in oncogenesis has come mainly from experimental cell culture and animal studies. Nevertheless, novel evidence for a direct link between Rho proteins and cancer development is beginning to accumulate, rendering them potential targets for drug discovery and cancer intervention.
Introduction
Malignant transformation is a complex multifaceted process involving systematic alterations in a variety of cellular properties. Cancer cells are characterized by deregulated cell cycle control, reduced contact inhibition, loss of matrix-dependent growth regulation, increased cell survival, morphologic alteration, increased motility, and acquisition of invasive and metastatic properties. Rho family GTPases act as key regulators of all of these cellular processes and mounting evidence implicates aberrant Rho GTPase function in human oncogenesis. Like the Ras oncoproteins, Rho GTPases function as regulated GDP/GTP switches that relay extracellular cues to cytoplasmic signalling cascades. In contrast to Ras proteins however, where direct mutational activation of Ras is prevalent in human cancers, much of the evidence linking Rho GTPases to oncogenesis is indirect, and implicates Rho proteins as critical downstream signalling components of a diverse array of oncoproteins. Recent reviews have provided excellent overviews on various facets of normal Rho GTPase function.1-5 Therefore, in this review we have especially focused on a summary of the evidence linking Rho GTPases to oncogenesis.
The Rho Family of GTPases Is a Major Branch of the Ras Superfamily
The Rho family constitutes one of the major branches of the Ras superfamily of small GTPases. Presently, 18 mammalian members have been described that share significant sequence identity ranging from 50 to 90%, with an overall 25% identity with Ras. This family is further subdivided into several groups and include the Rho (isoforms A, B, and C), Rac (isoforms 1, 2, 3), Cdc42 (and Wrch-1), TC10 (and TCL), Rnd (isoforms 1, 2, and 3), RhoD, RhoG, and TTF subfamilies. The most heavily studied and best characterized members are Rac1, RhoA, and Cdc42.6-8 Rho GTPase function is conserved in evolution, with members identified in yeast, plants, slime mold, worms, and flies.
Rho GTPases Function As Membrane-Associated GDP/GTP-Regulated Molecular Switches
In addition to sharing strong primary amino acid sequence identity, Ras and Rho GTPases share substantial structural, biochemical, and functional similarities. In the sections below, we have summarized these similarities (as well as differences) with regards to their function as regulated GDP/GTP signal transducers.
The Rho GDP/GTP Cycle Is Regulated by GEFs and GAPs
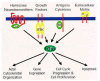
Like Ras, Rho GTPases act as binary switches that cycle between active GTP-bound and inactive GDP-bound states (fig. 1). The GTP-bound forms complex with and activate multiple downstream effector target molecules to stimulate cytoplasmic signalling cascades. Subsequently, an intrinsic GTP hydrolysis activity regenerates the GDP-bound form terminating the active state.
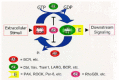
The intrinsic GTP hydrolysis and the GDP/GTP nucleotide exchange reactions of Rho and other Ras superfamily GTPases are both very slow, and require the enzymatic assistance of two families of regulators: GTPase-activating proteins (GAPs) and guanine nucleotide exchange factors (GEFs) (fig. 1). GAPs enhance the intrinsic GTP hydrolysis rate and therefore attenuate Rho protein activity by promoting the GDP-bound state.9 More than 20 Rho GAPs have been identified to date.10 Among these is BCR, which was identified originally as the translocation partner for the Abl tyrosine kinase in the Philadelphia Chromosome translocation found in a majority of human chronic myelogenous and acute lymphocytic leukemias.11
GEFs act as positive modulators by catalyzing the exchange of GDP for GTP, thereby increasing the GTP-bound protein levels in the cell (fig. 1). Rho GEFs, like Ras GEFs, eject the bound nucleotide (GDP or GTP) to promote the formation of the nucleotide-free form of the GTPase. However, since there is about 15-fold excess of GTP versus GDP in the cell, the net result of GEF-mediated exchange is to enhance formation of the GTP-bound protein.12 Rho GEFs are also referred to as Dbl family proteins and are named after the founding member characterized originally as a transforming protein from a human diffuse B-cell lymphoma (Dbl) cell line.13,14 To date, over 60 Dbl family proteins have been identified, and like Dbl, many were identified initially in screens for novel transforming oncoproteins. All Dbl family proteins contain a Dbl homology (DH) domain that catalyses GDP/GTP exchange, as well as a pleckstrin homology (PH) domain that regulates DH domain function.15,16 The tandem DH/PH domains are typically flanked by different modules (SH2, SH3, calponin homology (CH) domains, kinase domains, etc.) that contribute to the DH domain activation through diverse signalling activities.
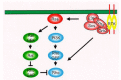
Unlike Ras, Rho GTPases are under the control of a third functional class of regulators called guanine nucleotide dissociation inhibitors (GDIs) comprising three members: GDI1 (RhoGDI or GDIα), GDI2 (D4/Ly-GDI or GDIβ), and GDI3 (GDIγ).17 GDIs inhibit Rho GTPase signalling by exerting three concerted functions (fig.1 and fig.2). First, they inhibit the nucleotide dissociation and block the GDP/GTP exchange, therefore antagonizing GEF activity.18-20 Second, GDIs interfere with GTP hydrolysis and hinder GAP activity.21 Finally, perhaps the most important function of GDIs involves their stimulation of the release of Rho GTPases from cellular membranes (fig. 2). The net effect is the transfer of the Rho protein from membrane compartments, where it is active, to the cytosol, shutting down the GTPase activation altogether.
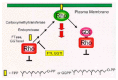
Membrane Asscociation Is Critical for Rho GTPase Function
Like Ras oncoproteins, Rho proteins also undergo COOH-terminal posttranslational modification by isoprenoid lipids, a process critical for their biological activity. Both Ras and Rho GTPases terminate in a carboxyl-terminal CAAX tetrapeptide motif (where C = cysteine, A = aliphatic amino acid, and X = terminal amino acid) that is necessary and sufficient for posttranslational modification. In the first step, a farnesyltransferase (FTase; when X = serine, methionine) or the related geranylgeranyltransferaseI (GGTaseI; when X = leucine), catalyze the covalent addition of a C15 farnesyl or a C20 geranylgeranyl isoprenoid moiety, respectively (fig. 2). Although the majority of Rho GTPases are modified by GGTaseI (e.g., RhoA, Rac1, and Cdc42), others are processed solely by FTase (e.g., RhoD and RhoE/Rnd3), or by both enzymes (such as RhoB). Prenylation is followed by a proteolytic activity that cleaves the terminal AAX residues prior to carboxymethylation of the terminal prenylated cysteine. The CAAX-signaled modifications, together with an additional membrane-targeting motif directly upstream of the CAAX motif, anchor Rho GTPases to membranes, a necessary step for GTPase function.22
In light of the critical importance of prenylation for the function of Ras and Rho GTPases, pharmacologic inhibitors of FTase (FTIs) or GGTaseI (GGTIs) have been developed as novel anti-cancer therapeutics (fig. 2). Initially developed as anti-Ras drugs, the anti-tumor activity of FTIs is speculated to be mediated, in part, by targeting other GTPases such as RhoB.23 Nevertheless, FTIs have demonstrated potent anti-oncogenic activity in cell culture and animal models, and are now under evaluation in phase III clinical trials.24 Similarly, inhibitors of GGTaseI (GGTIs) have been developed as inhibitors of Rho GTPases and have also shown anti-tumor activity in experimental models and tumor xenograft studies.25
Rho GTPase Mutants Are Molecular Tools for Dissection of Function
Alterations of specific residues in Rho GTPases can genetically modify their ability to interact with their regulators, effectors, or with membranes.26 Depending on the residue, these mutations can either generate dominant activated, inactive, or dominant negative GTPase. In particular, two classes of mutants have provided powerful reagents to evaluate the signalling and biological functions of Rho GTPases (fig. 3).
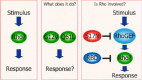
Mutations analogous to those found in tumor-associated mutants of Ras (e.g., G12V and Q61L) interfere with the intrinsic GTP hydrolysis reaction, and de-sensitize the G-protein to GAP regulation, resulting in constitutively active GTP-bound proteins that are activated in a ligand-independent fashion27-29 (fig. 4). A second type of activated mutants is based on the Ras(F28L) variant, which shows decreased affinity for guanine nucleotides and results in a “fast-cycling” protein.30 Their increased intrinsic nucleotide exchange rate, coupled with excess in cellular GTP, favor the formation of GTP-bound proteins. Like the GAP-deficient mutants, F28 versions of Ras, Cdc42, RhoA, and Rac1 are constitutively activated and transforming.31
Another important class of GTPase mutants is those that display a dominant-negative phenotype.32 These mutants have been essential reagents that implicated Rho GTPases as mediators of oncogenesis.33 In particular, the S17N mutant of Ras has been the prototype for analogous versions in Rho proteins. Introducing an aspargine at position 17 (17N) favors formation of nucleotide-free GTPases that bind GEFs with higher affinity than their wild type counterparts. Hence, they prevent GTPase activation via inhibition of GEF function. When introduced in cells, Rac1(17N) for instance, would compete with the endogenous Rac for GEF binding, forming tight complexes unable to relay effector-mediated downstream signalling. As described in the following sections, both dominant activating and dominant negative mutants of Rho GTPases have been instrumental in linking Rho GTPases to oncogenesis.
A third functional class of mutants are those that are impaired in downstream effector interaction26 (fig. 4). As described below, Rho GTPase function is mediated through multiple downstream effector targets. Although all effector interactions are dependent on a core effector domain (corresponding to Ras residues 32-40), select missense mutations in the Rho protein core effector domain can result in differential impairment in effector binding (e.g., see refs. 34 and 35). Such mutants have provided powerful reagents to assess the contribution of a particular effector to a specific GTPase signalling and/or function.
In contrast to the Ras superfamily of proteins, Rho GTPases possess an additional ∼13 amino acid “insert region” positioned between amino acids corresponding to Ras residues 122 and 123.36 Although the insert region is distant some 18 _om the switch regions and the effector domain, it still participates in effector-mediated functions, such as cytoskeletal remodeling.37 The sequence and structural divergence of the insert domain between different Rho GTPases suggests it may serve very distinct roles, particularly in transformation.37-39
Rho Family GTPases Are Critical Components of Diverse Signalling Pathways
A large and diverse variety of cell surface agonists utilize Rho GTPases to modify cellular behavior.40 An expanding body of evidence has implicated Rho proteins in growth factor, cytokine, cell-cell as well as cell-extracellular matrix signalling. The ligand-activated receptors are thought to regulate Rho GTPase activation mainly via activation of Rho GEFs, although in some situations regulation of Rho GAP activity has been described. Once activated, the GTP-bound Rho GTPase interacts with a wide spectrum of downstream effectors, initiating signalling cascades that regulate events in the cytoplasm and the nucleus (fig. 5). In the following sections, we have described the cellular responses regulated by signalling cascades that may contribute to the aberrant growth properties of cancer cells.
Rho GTPases Regulate Actin Cytoskeletal Organization
The best-characterized cytoplasmic function of Rho family proteins is their role as regulators of actin cytoskeletal organization.3,6,8 For example, Rac1 promotes the reorganization of filamentous actin into membrane structures called lamellipodia which serve as new adhesive contacts typically found at the leading edge of migrating fibroblasts.41 On the other hand, RhoA promotes the formation of actin stress fibers and focal adhesions, promoting cell attachment, whereas RhoE causes a disruption of these structures and causes cell rounding.42-44 Other GTPases such as Cdc42 promote the protrusion of finger-like actin-rich projections that act as sensors of the extracellular environment.45
Additional processes regulated by Rho proteins and requiring cytoskeletal changes extend to membrane transport, organization of tight junctions, chemotaxis, cytokinesis, and cell polarity.46-54 Collectively, these dynamic RhoGTPase-controlled cytoskeletal rearrangements promote shape adjustment and regulate cell-cell and cell-matrix contacts, processes that may influence invasion and metastasis.
Rho GTPases Regulate Transcription Factor Function and Gene Expression
A second major function of Rho family GTPases involves their regulation of the activity of a variety of transcription factors.55 For instance, the three GTPases Rac1, RhoA, and Cdc42 activate the serum response factor (SRF).56 SRF is a transcription factor that cooperates with ternary complex factors (e.g., Elk-1) to modulate the activity of serum response element (SRE)-containing promoters of many early response genes such as c-fos.57 Rho proteins also stimulate the activation of various other transcription factors such as NF-κB, E2F-1, c-Jun, ATF-2, Elk-1, AP-1, and STAT3.4,58-67 The diverse multitude of transcription factors that are regulated by Rho GTPases emphasizes that changes in gene expression form an important outcome and may be a critical mediator of Rho GTPase-induced transformation.
Rho GTPases Regulate Cell Cycle Progression and Cell Proliferation
In light of their ability to promote uncontrolled growth, it is not surprising that Rho GTPases, like Ras, also regulate cell cycle progression.5 Dominant negative Rac1, RhoA, and Cdc42 block, whereas activated forms of these GTPases stimulate, progression through the G1 phase of the cell cycle.68,69 Rho GTPases promote cell cycle progression by modulating the activity of both positive as well as negative regulators of the Rb tumor suppressor, which functions in regulating G1 progression. For example, RhoA and Rac1 up-regulate cyclin D1 gene expression34 and the transforming activity of various Dbl family oncoproteins correlated with their ability in promote cyclin D1 expression.70 Conversely, RhoA has been found to antagonize the expression of two negative regulators of G1 progression, the cyclin-dependent kinase inhibitors p21CIP1 and p27KIP1.71 For example, RhoA inhibition of p21CIP1 has been described as one mechanism by which Rho GTPases may facilitate the growth promoting actions of Ras.72
Rho GTPases and Regulation of Apoptosis
Rho GTPases have also been shown to regulate cell survival by either promoting or antagonizing apoptosis depending on the cell type and signalling contexts. Several reports have indicated that RhoA, RhoC, Rac1, Rac2 and Cdc42 proteins were involved in apoptotic stimulation in cells such as fibroblasts, thymocytes, neurons, and epithelial cells. NIH 3T3 cells expressing Rac1 seem to be sensitized to serum withdrawal through a mechanism involving up-regulation of Fas ligand transcription.73 Overexpression of RhoA also correlates with an increased ceramide production, which is a second messenger for apoptosis.74,75 In addition, RhoG is thought to mediate the toxic effects of FTI treatment and Taxol.77,76 On the other hand, there is also considerable evidence for the anti-apoptotic activities of Rho GTPases. For example, Rac1 appears to protect Rat-1 fibroblasts from serum deprivation-induced apoptosis, or MDCK epithelial cells from matrix deprivation-induced apoptosis.78,79 Inhibition of RhoA, but not of Rac1, caused apoptosis of RET/PTC1-expressing thyroid cells.80 Similarly, Cdc42 inhibition through caspase-mediated degradation of Cdc42 was suggested to facilitate Fas ligand-induced apoptosis in T47D breast cancer cell line.81 While oncogenesis clearly involves increased cell survival, how Rho GTPase regulation of apoptosis factors into Rho GTPase-mediated oncogenesis is presently not clear.
Rho GTPases Promote Oncogenic Transformation
Several lines of evidence have implicated Rho GTPases in cellular transformation. First, Rho GTPases are required for Ras-mediated oncogenesis.29,82-88 Rho proteins are also required for the functions of other oncoproteins such as polyoma virus middle T antigen, tyrosine kinases such as Abl, Met, Fps, BCR-Abl, RET, epidermal growth factor receptor, insulin-like growth factor receptor, G protein-coupled receptors (Mas, G2A, M1-muscarinic receptor, and Par-1), and Dbl family oncoproteins (see below).80,89-101 Finally, Rho GTPases are associated with tumor suppressor function. For example, the NF2 tumor suppressor merlin may mediate its actions, in part, by inhibiting the functions of Rac.102 Moreover, the hematopoietic-specific Rho GTPase Rac2 was shown to cooperate with Ras to promote the proliferation of mast cells heterozygous for the tumor suppressor neurofibromin (NF1).103
Second, ectopic expression of dominant active forms of Rho GTPases, analogous to the oncogenic mutants of Ras (e.g., G12V and Q61L), cause growth and morphologic transformation, and tumorigenic and metastatic growth of rodent fibroblasts.29,82,83,85,104,105 Similarly, activated Rac1 was shown to induce anchorage-independent growth29,82,83 and to promote motility, invasion, and metastasis in a variety of cell types, including fibroblasts, T lymphoma, epithelial, and melanoma cells.106-109 In the following sections below, we provide a more detailed discussion of the evidence that implicates Rho GTPases in the transforming functions of Ras, Dbl family oncoproteins, and GPCRs, as well as the emerging evidence that Rho GTPases themselves are aberrantly expressed in human cancers (see below).
Rho GTPases Are Critical for Ras-Mediated Oncogenesis
The evidence implicating Rho GTPases as critical mediators of oncogenic Ras function has come mainly from studies of NIH 3T3 and Rat-1 rodent fibroblast cell lines. First, it was found that dominant negative versions of Rac1, RhoA, RhoB, RhoG, Cdc42, and TC10 blocked Ras-induced transformation of rodent fibroblasts.29,82-87,110 Second, constitutively activated mutants of Rho GTPases were shown to cooperate with activated Raf to cause synergistic transformation of rodent fibroblasts. Similarly, coordinate expression of activated Rac1 and MEK1 caused synergistic growth transformation of FRTL-5 rat thyroid epithelial cells.111 Finally, like Ras, Rac and Raf cooperated with E1A to cause transformation of primary BRK rat epithelial cells.112 These observations suggested a model where Ras activation of Rho GTPases via a Raf-independent effector(s) pathway can promote various aspects of Ras transformation of both fibroblasts and epithelial cells.
Studies with activated or dominant negative Rho GTPase mutants have delineated the contribution of specific Rho family GTPases to distinct aspects of the transformed phenotype caused by oncogenic Ras. Although activated Rho GTPases are not sufficient to reproduce the drastic morphologic transformation caused by activated Ras or Raf, NIH 3T3 cells stably-expressing activated RhoA or Rac1, or Rat-1 cells expressing activated Cdc42, grow in anchorage-independent conditions and form tumors in nude mice, indicating that each GTPase alone can contribute to the growth-promoting actions of oncogenic Ras.29,31,84 Furthermore, analyses of Ras transformation of Rat-1 fibroblasts indicated that while RhoA, Rac1 and Cdc42 all contribute to Ras induction of anchorage-independent growth, Rac1 contributed to the reduced growth factor requirements, whereas Cdc42 and RhoA were required for morphologic transformation.82-84
Interestingly, there is evidence that Rac1 may also contribute to Ras-mediated transformation by blocking apoptosis.113 Similarly, Symons and colleagues determined that Rac1 protected MDCK epithelial cells79 and Rat1 cells78 against apoptosis induced by matrix deprivation. Finally, activated Rac1 can promote the invasion and metastasis of a variety of cell types, suggesting that the invasive properties of Ras may be mediated, in part, by activation of Rac1.107,114
Whether Rho GTPases are found to be constitutively activated in Ras-transformed rodent fibroblasts, and whether Rho GTPases are important for Ras transformation of epithelial cells, are issues that have been addressed with conflicting results, and are yet to be fully resolved. Earlier microinjection analyses showed that transient expression of oncogenic Ras caused activation of Rac, which in turn caused activation of RhoA.41,42 However, Ras-transformed rodent fibroblasts lack well-developed actin stress fibers, an observation that is inconsistent with an up-regulated RhoA activity. In fact, activated RhoA restored stress fibers to Ras-transformed cells, arguing for a down-regulated Rho function.115 Similarly, inhibition of RhoA/B/C function in mouse T cells by the C3 toxin promoted the development of malignant thymic lymphoblastic lymphomas, consistent with the model that the loss of Rho function supports oncogenesis.116 On the other hand, Rho activation in Ras-transformed MDCK cells was found to contribute to their transformation from an epithelial to a mesenchymal morphology, an observation consistent with RhoA up-regulation in Ras-transformed cells.117 Finally, as described below, observations from RhoA-specific GEF studies as well as others using pharmacologic inhibition of RhoA signalling, provide further evidence that up-regulation, rather than down-regulation, of RhoA activity is consistent with promoting oncogenesis.
Similarly, Ras-transformed cells show increased membrane ruffling, which corresponds to an up-regulated Rac1 activity by oncogenic Ras.118 Furthermore, transient expression of oncogenic Ras was shown to increase Rac1-GTP levels in COS-1 cells.119 In contrast, Collard and colleagues determined that sustained activation of Ras down-regulated Rac activity in MDCK epithelial cells.117 Interestingly, activated Rac inhibited cell migration in these cells, suggesting an invasion-suppressing, rather than promoting, function for Rac.120 It was argued that while transient Ras activation can cause Rac activation, sustained Ras stimulation leads instead to down-regulation of Rac activity. Finally, Nur-E-Kamal and colleagues showed that Cdc42-GTP levels were indeed up-regulated in Ras-transformed NIH 3T3 cells and that stimulated receptor tyrosine kinases that activate Ras also activate Cdc42.121 In accordance, inhibition of Cdc42 by the Cdc42-specific GTPase binding domain of ACK-1 blocked Ras transforming activity. Thus, similar to Ras, Rho GTPases may have positive or negative roles in growth regulation that will be influenced by cell type differences and/or genetic context.
The signalling pathways that link Ras with Rho family GTPases are currently incompletely understood.122-124 Several possible links between Ras and Rac have been suggested (fig. 6). First, both Ras and Rac1 associate with the two subunits of phosphatidylinositol 3-kinase (PI3K) p110 and p85, respectively. Ras binding to p110 catalytic subunit may regulate the association of p85 with Rac1, which may determine Rac1 function.125,126 Additionally, Ras activation of PI3K leads to the production of lipid products such as phosphatidylinositol 3,4,5-trisphosphate (PIP3), which in turn may interact with the PH domain of Dbl family proteins to promote GEF activity. For example, PI3K activation has been described to cause activation of Vav and Tiam1.127,128 Second, the fact that the Ras exchange factor Son of sevenless (Sos), which in addition to the Cdc25 Ras GEF domain also contains a functional DH/PH unit that exhibits some activity towards Rac1 suggests that Sos-dependent Ras activation may potentially lead to Sos-dependent Rac1 activation.129 Another mechanism has been proposed where the activation of Grb2/Sos1 leads to the formation of a complex (Eps8-E3b1-Sos1) able to bind and possibly modulate Rac1 activity.130 Third, Ras activation of its effector RalGDS (for Ral guanine nucleotide dissociation stimulator) leads to the activation of the Ral small GTPase, which in turn binds a Rac/Cdc42 GAP (RalBP1/RIP1/RLIP76), and consequently, may modulate its activity towards Rac1.131,134 Several reports have emphasized the importance of the RalGDS pathway in Ras transformation of mouse and human cells.135,136 Depending on the cell type and the mechanisms used, these observations place Rho GTPases at the point of convergence of a multitude of membrane-triggered events ultimately determining cellular fate.
Rho GTPases are Involved in Tyrosine Kinase Oncoprotein Transformation
The implication of Ras in transformation caused by tyrosine kinase oncoproteins has also associated Rho GTPases with this process. For example, the BCR/Abl oncoprotein renders 32D mouse myeloid cells resistant to IL-3 deprivation-induced growth cessation and apoptosis, in part, by activation of Ras. Coexpression of dominant negative Rac1 rendered Ras-transformed 32D cells sensitive to IL-3 withdrawal growth inhibition and impaired invasion in vitro and leukemogenesis in vivo.94 Along the same line, tyrosine oncoproteins rely on multiple Rho GTPases to cause transformation via Ras. For example, Fps/Fes transformation of Rat-2 fibroblasts was impaired not only by dominant negative Ras, but also by Rac1(N17) and Cdc42(N17).93
A Ras-independent requirement of Rho GTPases in tyrosine kinase-mediated transformation has also been documented. For example, although Ras was not found to be required for the transforming activity of an avian viral mutant of EGFR, dominant negative RhoA, but not Rac1 nor Cdc42, was found to block its transforming activity.137 Thus, while tyrosine kinases can cause activation of Ras, the signalling pathways that link tyrosine kinase oncoproteins with Rho GTPases may not always involve Ras activation. This possibility is supported by earlier observations that oncogenic but not normal Ras triggers the activation of Rho GTPase cascades.
Dbl Family Oncoproteins Are Activators of Rho GTPases
Further evidence that up-regulated Rho GTPase activity promotes oncogenesis comes from the observations that many Dbl family proteins were identified initially as oncoproteins before they were characterized as Rho GEFs.15,16,138 Many Dbl family proteins were identified initially in biological screens for novel transforming (e.g., Dbl, Vav, Ect2, Tim, Net1, Lfc, Lsc) or invasion-inducing (Tiam1) oncogenes106,114,139,140 and in most cases, structural alterations were identified as the mechanism of activation of their transforming activities. For example, amino terminal truncation of negative regulatory sequences accounts for the formation of constitutively activated and transforming mutants of Dbl, Vav, Ect2, and Tiam1. However, despite their identification in screens of tumor cell-derived DNA or RNA, these genetic alterations occurred during the transfection procedure and did not originate from the patient tumor sample.
Despite the tantalizing links between Dbl family proteins and oncogenesis, evidence that Dbl family proteins are aberrantly activated in human cancers has been limited to three examples (Table 1). Recently, mutations in Tiam1 have been reported in human renal carcinomas leading to constitutive activation of the protein.141 The two other Dbl family members were identified initially as proteins whose genes are rearranged in human leukemias. First, the product of the breakpoint cluster region (BCR) gene is truncated and forms a fusion protein with the Abl tyrosine kinase caused by the Philadelphia chromosome translocation, an aberration associated with about 95% of chronic myelogenous leukemias.142 Although some versions of the BCR-Abl fusion protein do contain the Rho GEF catalytic domain, these versions are less transforming than those that lack this domain. While BCR-Abl has been found to activate Rac, this has been attributed to its association with and activation of the Vav Dbl family protein.143 Second, the leukemia associated RhoGEF (LARG) was also identified as a gene rearranged in mixed lineage leukemia (MLL) and encoded a chimeric fusion protein with MLL.144 LARG has been subsequently shown to cause growth transformation of rodent fibroblasts via activation of its target GTPase RhoA.145,146 While these rearrangements mimic those that render other Dbl family proteins constitutively activated, there is no current evidence that these BCR or LARG fusion proteins promote oncogenesis by causing persistent activation of Rho GTPases.
Table 1
Aberrant Rho GTPase activity in human cancers.
Finally, there are preliminary observations that altered expression of RhoGDI may be involved in oncogenesis (Table 1). Decreased mRNA expression of RhoGDI2 was associated with a more invasive variant of the T24 bladder cancer cell line147 suggesting a role for Rho activation in metastasis and invasion. In contrast, proteomic analyses identified RhoGDI protein overexpression in invasive ovarian cancers, suggesting that loss of Rho activity may promote tumor progression.148 However, whether Rho GTPase activity was altered by changes in RhoGDI activity were not determined in these studies.
Transforming G Protein-Coupled Receptors Are Linked to Rho GTPases
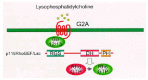
Investigations into the mechanisms of receptor signalling have uncovered a pivotal role for Rho GTPases in heterotrimeric G protein-coupled receptor (GPCR) oncogenesis. The orphan GPCR Mas, initially identified in a genetic screen for transforming proteins, provided the first evidence for the transforming potential of GPCRs.149 Mas was later shown to cause lamellipodia formation as well as Rac-dependent transformation of NIH 3T3 cells.99 However, the fact that Mas is normally expressed in nonmitotic neural cells raises the possibility that it may not play an important role in oncogenesis.
The link between GPCRs and Rho GTPases has been best delineated for the p115 RhoGEF/ Lsc, where association of activated Gα13 with the NH2-terminal RGS (regulator of G protein signalling) domain up-regulates the DH catalytic activity towards RhoA124,150 (fig. 7). In agreement, constitutively activated variants of Gα13 and Lsc have also been shown to cause RhoA-dependent transformation of NIH 3T3 cells.151 Other GPCRs coupled to the Gα13 heterotrimeric subunit are G2A and Par-1. Like Mas, G2A and Par-1 were also identified in NIH 3T3 expression library screens for novel transforming proteins. Both G2A and Par-1 activate RhoA and their transforming activities were shown to require Rho GTPase activation.100,152 Interestingly, G2A expression is up-regulated by BCR-Abl, and Par-1 overexpression has been associated with tumor cell invasion.153,154
Aberrant Expression of Rho GTPases in Oncogenesis
Evidence for the direct involvement of Rho GTPases in human cancer development, although less well documented, is rapidly accumulating (Table 1). For example, human breast, colon, and lung tumors exhibit high protein expression levels of Rac1, Cdc42, and in particular RhoA protein, when compared to normal tissue from the same patient.155 Increased levels of RhoA mRNA was detected in testicular germ cell tumor tissue and correlated with tumor progression.156 Similarly, a recent study also indicated that RhoA and Rac2 were largely overexpressed in head and neck cancer tissues and proposed their use as markers for squamous cell carcinoma of the head and neck.157 In addition, Rac3 was found to be hyperactive in human breast tumor cell lines and appears to be required for breast cancer cell proliferation.158
A role for another Rho family member, RhoC, in human cancer metastasis and invasion has been uncovered in melanoma cell lines.159 RhoC overexpression was found in 90% of inflammatory breast cancer and may promote tumor angiogenesis.160,161 RhoC was also found to be overexpressed in adenocarcinomas of the pancreas and its high levels of expression correlated with poor prognosis.162
A recent addition to the Rho family, a Cdc42-homolog termed Wrch-1, was found to be regulated by Wnt-1 oncogenic signalling in human mammary tumors.163 Overexpression of Wrch-1, like Wnt-1, promoted morphological transformation of mouse mammary epithelial cells. Taken together, Wrch-1 could mediate the transforming effects of Wnt-1 signalling in the regulation of cell morphology, cytoskeletal organization, and cell proliferation.
The identification of mutant Rho proteins in human cancer specimens has so far been limited to two examples. The first represents a fusion of the Rho family member RhoH/TTF to the lymphoma-associated LAZ3/BCL6 gene, a mutation identified in nonHodgkin's lymphoma (NHL) cell lines164,165 and was later found in patients with NHL and multiple myelomas.166 The second example is that of Rac1b, a novel splice variant of Rac1, found to be aberrantly expressed in human colorectal167 and breast168 carcinomas, establishing a direct link between Rho GTPase activation in general, and Rac in particular, and tumor development in humans. Rac1b contains an additional 19 amino acid insert just before the switch II region of Rac1, and in vitro analyses indicated that this variant exhibits a fast-cycling guanine nucleotide exchange activity, suggesting that it may be an activated variant of Rac1.
Rho GTPases Mediate Transformation through Multiple Effectors
The conformations adopted by switch 1 and switch 2 in the active GTP-bound state enable high affinity interactions with multiple downstream molecules termed effectors which transmit their signals downstream.1,6,7,169 For example, over 25 known or candidate effectors of Rac1 have been identified to date. In addition to the complexity provided by multiple effectors, the fact that effector utilization can be shared by different Rho GTPases adds another level of regulatory complication to Rho GTPase function. For instance, despite their distinct functions, Rac1 and Cdc42 share a wide spectrum of effectors.1 The majority of these effector-mediated signalling pathways remain to be fully delineated, but enough evidence points to the involvement of few key effectors in Rho GTPase transformation. In the following sections, we summarize what is known regarding the effectors and signalling activities important for the transforming actions of RhoA, Rac1, and Cdc42.
RhoA Transformation and ROCK
Effector domain mutants have provided critical clues regarding what effector signalling pathway is important for RhoA transformation. These analyses have dissociated transformation from a variety of effector-mediated signalling pathways such as stress fiber formation and SRE activation.170,171 Interestingly, these studies have also shown strong correlations between the ability of RhoA to bind ROCK, and its ability to cooperate with Raf to cause transformation of NIH 3T3 cells.170 Indeed, treatment with the Y-27632 pharmacologic inhibitor of ROCK impaired RhoA transforming activity, as well as the transforming activity of oncoproteins (Ras, Dbl, and Net) that activated RhoA.170 In addition, a role for ROCK in facilitating RhoA-mediated invasion has been documented. Itoh and colleagues showed that dominant active ROCK promoted invasion of MM1 hepatoma cells, whereas inhibition of ROCK activity by Y-27632 reduced the ability of these cells to invade in mice.172 On the other hand, the fact that activated ROCK does not reproduce the same transforming activity seen with activated RhoA suggests that additional effectors are likely to contribute to RhoA transformation. This may involve, for example, an effector(s) that interacts with the unique insert domain of RhoA, a region required for transformation but not for ROCK binding.173
Role of Reactive Oxygen Species (ROS) NF- κB and Par-6 in Rac1 Transformation
Effector domain mutants have also been used to address the importance of specific effector signalling functions in Rac1 transformation. PAK binding, as well as induction of lamellipodia, activation of the JNK and p38 MAPKs, SRF, the MEKK1 and p70 S6 kinases, were all shown to be dispensable for Rac1-induced transformation of NIH 3T3 cells.34,174,175 PAK and JNK activation were also dissociated from the ability of Rac1 to induce cell cycle progression.176 Since a role in growth induction as well as apoptosis has been established for several of these pathways, it is likely that multiple signalling pathways may also contribute to Rac1-induced cellular transformation.
Rac1 is distinguished from RhoA and Cdc42 in its ability to activate the phagocyte NADPH oxidase system177,178 by interacting with and modulating the activity of the p67 subunit of the complex.179 Rac also activates a related oxidase system in nonphagocytic cells by an as yet to be identified effector. The reactive oxygen species (ROS) by-products of this reaction induce NF-κB activation, a process inhibited by ROS inhibitors such as N-acetylcystine (NAC).58 ROS have been shown to be important second messengers for Ras-mediated transformation180 and Rac-mediated cell cycle progression.181 Deletion of the Rho insert region of Rac1 impaired its mitogenic activity and ROS production in certain cell types. This finding, together with the observation that inhibitors of ROS production inhibit Rac1 mitogenic activity, support an important contribution of ROS in Rac transformation, possibly by its ability to activate the NF-κB-mediated cell survival pathway. NF-κB was found to be required for the transforming activity of two Dbl oncoproteins, Dbl and Dbs (GEFs for RhoA and Cdc42), providing further evidence for the importance of this signalling pathway in Rho GTPase transformation.182
A novel effector for Rac1 and Cdc42 has been identified as the human homologue of the cell polarity determinant in Caenorhabditis elegans, hPar-6, and has also been linked to cellular transformation.183,184 Rac1 and Par-6 associate with an atypical protein kinase C isoform, PKCζ, an interaction that releases the kinase activity of PKCζ leading to transformation. Additionally, Par-6 has been suggested to play an important role in the polarization of mammalian cells by functioning as an adaptor protein that links activated Rac and Cdc42 to PKCζ signalling.185,186 While it was found that PKCζ potentiated Rac1 transformation, whether PKCζ is a critical effector of Rac and Cdc42 transformation remains to be clarified.183
Cross-Talk between Vesicular Transport and Cdc42 Transformation
Cerione and colleagues identified the γ-subunit (γ-COP) of COPI, a cytoplasmic ‘coatomer’ protein complex as an effector important for Cdc42 transformation.187 The coatomer coats membrane regions that bud and form vesicles, a process controlled by the Arf small GTPase. Interactions between Arf and COPI direct the formation of vesicles that are involved in the selective transport of proteins from the Golgi complex back to the endoplasmic reticulum (ER) and in protein and lipid recycling from the plasma membrane through endosomes.3 Interestingly, Cdc42 mutants that no longer bound to γ-COP or affect coatomer interaction with the Golgi (or perhaps endosomal compartments) were also unable to cause growth transformation. However, the nontransforming insert mutant of Cdc42188 retained the ability to bind γ-COP and promote vesicle trafficking, suggesting that γ-COP may not be sufficient to mediate transformation by Cdc42.
Conclusions and Future Directions
While establishing the importance of Rho GTPases in human carcinogenesis will require further experimental support and validation, the vital role these GTPases play in diverse cellular functions establish them as key components of malignant tumor progression. As mediators of a variety of oncoproteins, Rho proteins represent attractive targets for intervention and inhibition of oncogene function. GGTIs represent one possible approach for blocking the action of Rho GTPases. However, in light of the many other cellular proteins that are also substrates for GGTaseI, it remains to be determined whether more precise approaches will be needed to block Rho GTPase function. Information regarding the effectors and downstream signalling pathways that promote transformation by Rho GTPases remains limited. This task is certain to be complicated by the expanding ensemble of effectors and the plethora of cellular functions attributed to Rho GTPases. Regulation of gene expression is an important consequence of Rho GTPase function and the critical gene targets that mediate Rho GTPase activities will need to be identified. Further delineation of these pathways may characterize more precise points of therapeutic intervention. Finally, while most of the attention has been focused on Rac1, RhoA, and Cdc42, the emergence of other GTPases such as RhoC and Rac3 as important GTPases in cancer suggest that the involvement of other “nonclassical” members of the family deserves more attention.
References
- 1.
- Bishop AL, Hall A. Rho GTPases and their effector proteins. Biochem J. 2000;348(Pt 2):241–55. [PMC free article: PMC1221060] [PubMed: 10816416]
- 2.
- Bar-Sagi D, Hall A. Ras and Rho GTPases: A family reunion. Cell. 2000;103:227–38. [PubMed: 11057896]
- 3.
- Ridley AJ. Rho family proteins: Coordinating cell responses. Trends in Cell Biology. 2001;11:471–7. [PubMed: 11719051]
- 4.
- Aznar S, Lacal JC. Rho signals to cell growth and apoptosis. Cancer Letters. 2001;165:1–10. [PubMed: 11248412]
- 5.
- Pruitt K, Der CJ. Ras and Rho regulation of the cell cycle and oncogenesis. Cancer Letters. 2001;171:1–10. [PubMed: 11485822]
- 6.
- Van AelstL, D'Souza-Schorey C. Rho GTPases and signalling networks. Genes and Development. 1997;11:2295–322. [PubMed: 9308960]
- 7.
- Zohn IM, Campbell SL, Khosravi-Far R. et al. Rho family proteins and Ras transformation: The RHOad less traveled gets congested. Oncogene. 1998;17:1415–38. [PubMed: 9779988]
- 8.
- Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–14. [PubMed: 9438836]
- 9.
- Boguski MS, McCormick F. Proteins regulating Ras and its relatives. Nature. 1993;366:643–54. [PubMed: 8259209]
- 10.
- Lamarche N, Hall A. GAPs for rho-related GTPases. Trends Genet. 1994;10:436–40. [PubMed: 7871593]
- 11.
- Heisterkamp N, Kaartinen V, van Soest S. et al. Human ABR encodes a protein with GAPrac activity and homology to the DBL nucleotide exchange factor domain. J Biol Chem. 1993;268:16903–6. [PubMed: 8349582]
- 12.
- Wittinghofer F. Ras signalling. Caught in the act of the switch-on. Nature. 1998;394:317 319–20. [PubMed: 9690464]
- 13.
- Eva A, Aaronson SA. Isolation of a new human oncogene from a diffuse B-cell lymphoma. Nature. 1985;316:273–5. [PubMed: 3875039]
- 14.
- Eva A, Vecchio G, Rao CD. et al. The predicted DBL oncogene product defines a distinct class of transforming proteins. Proceedings of the National Academy of Sciences. 1988;85:2061–5. [PMC free article: PMC279928] [PubMed: 3281159]
- 15.
- Whitehead IP, Campbell S, Rossman KL. et al. Dbl family proteins. Biochem and Biophys Acta. 1997;1332:F1–F23. [PubMed: 9061011]
- 16.
- Cerione RA, Zheng Y. The Dbl family of oncogenes. Curr Opin Cell Biol. 1996;8:216–22. [PubMed: 8791419]
- 17.
- Olofsson B. Rho guanine dissociation inhibitors: Pivotal molecules in cellular signalling. Cell Signal. 1999;11:545–54. [PubMed: 10433515]
- 18.
- Fukumoto Y, Kaibuchi K, Hori Y. et al. Molecular cloning and characterization of a novel type of regulatory protein (GDI) for the rho proteins, ras p21-like small GTP-binding proteins. Oncogene. 1990;5:1321–8. [PubMed: 2120668]
- 19.
- Leonard D, Hart MJ, Platko JV. et al. The identification and characterization of a GDP-dissociation inhibitor (GDI) for the CDC42Hs protein. J Biol Chem. 1992;267:22860–8. [PubMed: 1429634]
- 20.
- Nomanbhoy TK, Cerione RA. Characterization of the interaction between RhoGDI and Cdc42Hs using fluorescence spectroscopy. J Biol Chem. 1996;271:10004–9. [PubMed: 8626553]
- 21.
- Hart MJ, Maru Y, Leonard D. et al. A GDP dissociation inhibitor that serves as a GTPase inhibitor for the ras-like protein CDC42Hs. Science. 1992;258:812–5. [PubMed: 1439791]
- 22.
- Cox AD, Der CJ. Protein prenylation: More than just glue? Curr Opin Cell Biol. 1992;4:1008–16. [PubMed: 1485954]
- 23.
- Prendergast GC, Rane N. Farnesyltransferase inhibitors: Mechanism and applications. Expert Opin Investig Drugs. 2001;10:2105–16. [PubMed: 11772308]
- 24.
- Cox AD. Farnesyltransferase inhibitors: Potential role in the treatment of cancer. Drugs. 2001;61:723–32. [PubMed: 11398905]
- 25.
- Sebti SM, Hamilton AD. Farnesyltransferase and geranylgeranyltransferase I inhibitors in cancer therapy: Important mechanistic and bench to bedside issues. Expert Opin Investig Drugs. 2000;9:2767–82. [PubMed: 11093352]
- 26.
- Capon DJ, Seeburg PH, McGrath JP. et al. Activation of Ki-ras 2 gene in human colon and lung carcinomas by two different point mutations. Nature. 1983;304:507–13. [PubMed: 6308467]
- 27.
- Rittinger K, Walker PA, Eccleston JF. et al. Crystal structure of a small G protein in complex with the GTPase-activating protein rhoGAP. Nature. 1997;388:693–7. [PubMed: 9262406]
- 28.
- Ihara K, Muraguchi S, Kato M. et al. Crystal structure of human RhoA in a dominantly active form complexed with a GTP analogue. J Biol Chem. 1998;273:9656–66. [PubMed: 9545299]
- 29.
- Khosravi-Far R, Solski PA, Kinch MS. et al. Activation of Rac and Rho, and mitogen activated protein kinases, are required for Ras transformation. Mol Cell Biol. 1995;15:6443–53. [PMC free article: PMC230895] [PubMed: 7565796]
- 30.
- Reinstein J, Schlichting I, Frech M. et al. p21 with a phenylalanine 28-leucine mutation reacts normally with the GTPase activating protein GAP but nevertheless has transforming properties. J Biol Chem. 1991;266:17700–6. [PubMed: 1894650]
- 31.
- Lin R, Bagrodia S, Cerione R. et al. A novel Cdc42Hs mutant induces cellular transformation. Cur Biol. 1997;7:794–7. [PubMed: 9368762]
- 32.
- Cepko CL, Roberts B, Mulligan RC. Construction and applications of a highly transmissible murine retrovirus shuttle vector. Cell. 1984;37:1053–62. [PubMed: 6331674]
- 33.
- Feig LA. Tools of the trade: Use of dominant-inhibitory mutants of Ras-family GTPase. Nature Cell Biol. 1999;1:E25–E27. [PubMed: 10559887]
- 34.
- Westwick JK, Lambert QT, Clark GJ. et al. Rac regulation of transformation, gene expression, and actin organization by multiple, PAK-independent pathways. Mol Cell Biol. 1997;17:1324–35. [PMC free article: PMC231857] [PubMed: 9032259]
- 35.
- Sahai E, Alberts AS, Treisman R. RhoA effector mutants reveal distinct effector pathways for cytoskeletal reorganization, SRF activation and transformation. EMBO Journal. 1998;17:1350–61. [PMC free article: PMC1170483] [PubMed: 9482732]
- 36.
- Valencia A, Chardin P, Wittinghofer A. et al. The ras protein family: Evolutionary tree and role of conserved amino acids. Biochemistry. 1991;30:4637–48. [PubMed: 2029511]
- 37.
- Karnoub AE, Der CJ, Campbell SL. The insert region of rac1 is essential for membrane ruffling but not cellular transformation. Mol Cell Biol. 2001;21:2847–57. [PMC free article: PMC86914] [PubMed: 11283263]
- 38.
- Wu W-J, Leonard DA, Cerione RA. et al. Interaction between Cdc42Hs and Rho GDI is mediated through the Rho insert region. J Biol Chem. 1997;272:26153–8. [PubMed: 9334181]
- 39.
- Zong H, Raman N, Mickelson-Young LA. et al. Loop 6 of RhoA confers specificity for effector binding, stress fiber formation, and cellular transformation. J Biol Chem. 1999;274:4551–60. [PubMed: 9988689]
- 40.
- Symons M, Settleman J. Rho family GTPases: More than simple switches. Trends Cell Biol. 2000;10:415–9. [PubMed: 10998597]
- 41.
- Ridley AJ, Paterson HF, Johnston CL. et al. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–10. [PubMed: 1643658]
- 42.
- Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389–99. [PubMed: 1643657]
- 43.
- Guasch RM, Scambler P, Jones GE. et al. RhoE regulates actin cytoskeleton organization and cell migration. Mol Cell Biol. 1998;18:4761–71. [PMC free article: PMC109062] [PubMed: 9671486]
- 44.
- Nobes CD, Lauritzen I, Mattei MG. et al. A new member of the Rho family, Rnd1, promotes disassembly of actin filament structures and loss of cell adhesion. J Cell Biol. 1998;141:187–97. [PMC free article: PMC2132722] [PubMed: 9531558]
- 45.
- Kozma R, Ahmed S, Best A. et al. The Ras-related proteins Cdc42Hs and bradykinin promote formation of peripheral actin microspikes and filopodia in Swiss 3T3 fibroblasts. Mol Cell Biol. 1995;15:1942–52. [PMC free article: PMC230420] [PubMed: 7891688]
- 46.
- Lamaze C, Chuang T-H, Terlecky LJ. et al. Regulation of receptor-mediated endocytosis by Rho and Rac. Nature. 1996;382:177–9. [PubMed: 8700210]
- 47.
- Massol P, Montcourrier P, Guillemot JC. et al. Fc receptor-mediated phagocytosis requires CDC42 and Rac1. EMBO J. 1998;17:6219–29. [PMC free article: PMC1170948] [PubMed: 9799231]
- 48.
- Caron E, Hall A. Identification of two distinct mechanisms of phagocytosis controlled by different Rho GTPases. Science. 1998;282:1717–21. [PubMed: 9831565]
- 49.
- Ridley AJ, Comoglio PM, Hall A. Regulation of scatter factor/hepatocyte growth factor responses by Ras, Rac and Rho in MDCK cells. Mol Cell Biol. 1995;15:1110–22. [PMC free article: PMC232019] [PubMed: 7823927]
- 50.
- Lanzetti L, Di FiorePP, Scita G. Pathways linking endocytosis and actin cytoskeleton in mammalian cells. Experimental Cell Research. 2001;271:45–56. [PubMed: 11697881]
- 51.
- Jou TS, Schneeberger EE, Nelson WJ. Structural and functional regulation of tight junctions by RhoA and Rac1 small GTPases. J Cell Biol. 1998;142:101–15. [PMC free article: PMC2133025] [PubMed: 9660866]
- 52.
- Allen WE, Zicha D, Ridley AJ. et al. A role for Cdc42 in macrophage chemotaxis. J Cell Biol. 1998;141:1147–57. [PMC free article: PMC2137177] [PubMed: 9606207]
- 53.
- Mabuchi I, Hamaguchi Y, Fujimoto H. et al. A rho-like protein is involved in the organisation of the contractile ring in dividing sand dollar eggs. Zygote. 1993;1:325–31. [PubMed: 8081830]
- 54.
- Johnson DI. Cdc42: An essential Rho-type GTPase controlling eukaryotic cell polarity. Microbiol Mol Biol Rev. 1999;63:54–105. [PMC free article: PMC98957] [PubMed: 10066831]
- 55.
- Van AelstL, D'Souza-Schorey C. Rho GTPases and signalling networks. Genes and Development. 1997;11:2295–322. [PubMed: 9308960]
- 56.
- Hill CS, Wynne J, Treisman R. The Rho family GTPases RhoA, Rac1 and Cdc42Hs regulate transcriptional activation by SRF. Cell. 1995;81:1159–70. [PubMed: 7600583]
- 57.
- Treisman R. The SRE: A growth factor responsive transcriptional regulator. Semin Cancer Biol. 1996;1:47–58. [PubMed: 2133110]
- 58.
- Sulciner DJ, Irani K, Yu Z-X. et al. Rac1 regulates a cytokine-stimulated, redox-dependent pathway necessary for NF-KB activation. Mol Cell Biol. 1996;16:7115–21. [PMC free article: PMC231715] [PubMed: 8943367]
- 59.
- Perona R, Montaner S, Saniger L. et al. Activation of the nuclear factor-KB by Rho, CDC42, and Rac-1 proteins. Genes and Dev. 1997;11:463–75. [PubMed: 9042860]
- 60.
- Montaner S, Perona R, Saniger L. et al. Multiple signalling pathways lead to the activation of the nuclear factor kappaB by the Rho family of GTPases. J Biol Chem. 1998;273:12779–85. [PubMed: 9582304]
- 61.
- Gjoerup O, Lukas J, Bartek J. et al. Rac and Cdc42 are potent stimulators of E2F-dependent transcription capable of promoting retinoblastoma susceptibility gene product hyperphosphorylation. J Biol Chem. 1998;273:18812–8. [PubMed: 9668055]
- 62.
- Coso OA, Chiariello M, Yu J-C. et al. The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signalling pathway. Cell. 1995;81:1137–46. [PubMed: 7600581]
- 63.
- Minden A, Lin A, Claret F-X. et al. Selective activation of the JNK signalling cascade and c-Jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell. 1995;81:1147–57. [PubMed: 7600582]
- 64.
- Clarke N, Arenzana N, Hai T. et al. Epidermal growth factor induction of the c-jun promoter by a Rac pathway. Mol Cell Biol. 1998;18:1065–73. [PMC free article: PMC108819] [PubMed: 9448004]
- 65.
- Chang JH, Pratt JC, Sawasdikosol S. et al. The small GTP-binding protein Rho potentiates AP-1 transcription in T cells. Mol Cell Biol. 1998;18:4986–93. [PMC free article: PMC109083] [PubMed: 9710582]
- 66.
- Simon AR, Vikis HG, Stewart S. et al. Regulation of STAT3 by direct binding to the Rac1 GTPase. Science. 2000;290:144–7. [PubMed: 11021801]
- 67.
- Faruqi TR, Gomez D, Bustelo XR. et al. Rac1 mediates STAT3 activation by autocrine IL-6. Proc Natl Acad Sci USA. 2001;98:9014–9. [PMC free article: PMC55365] [PubMed: 11470914]
- 68.
- Yamamoto M, Marui N, Sakai T. et al. DP-ribosylation of the rhoA gene product by botulinum C3 exoenzyme causes Swiss 3T3 cells to accumulate in the G1 phase of the cell cycle. J Biol Chem. 1993;268:1449–55. [PubMed: 8502473]
- 69.
- Olson MF, Ashworth A, Hall A. An essential role for Rho, Rac and Cdc42 GTPases in cell cycle progression through G1. Science. 1995;269:1270–2. [PubMed: 7652575]
- 70.
- Westwick JK, Lee RJ, Lambert QT. et al. Transforming potential of Dbl family proteins correlates with transcription from the cyclin D1 promoter but not with activation of Jun NH2-terminal kinase, p38/Mpk2, serum response factor or c-Jun. J Biol Chem. 1998;273:16739–47. [PubMed: 9642229]
- 71.
- Adnane J, Bizouarn FA, Qian Y. et al. p21(WAF1/CIP1) is upregulated by the geranylgeranyltransferase I inhibitor GGTI-298 through a transforming growth factor beta- and Sp1- responsive element: Involvement of the small GTPase rhoA. Mol Cell Biol. 1998;18:6962–70. [PMC free article: PMC109279] [PubMed: 9819384]
- 72.
- Olson MF, Paterson HF, Marshall CJ. Signals from Ras and Rho GTPases interact to regulate expression of p21Waf1/Cip1. Nature. 1998;394:295–9. [PubMed: 9685162]
- 73.
- Embade N, Valeron PF, Aznar S. et al. Apoptosis induced by Rac GTPase correlates with induction of FasL and ceramides production. Mol Biol Cell. 2000;11:4347–58. [PMC free article: PMC15077] [PubMed: 11102528]
- 74.
- Jiménez B, Arends M, Esteve P. et al. Induction of apoptosis in NIH3T3 cells after serum deprivation by overexpressin of rho-p21, a GTPase protein of the ras superfamily. Oncogene. 1995;10:811–6. [PubMed: 7898922]
- 75.
- Esteve P, Embade N, Perona R. et al. Rho-regulated signals induce apoptosis in vitro and in vivo by a p53- independent, but Bcl2 dependent pathway. Oncogene. 1998;17:1855–69. [PubMed: 9778052]
- 76.
- Liu AX, Du W, Liu J-P. et al. RhoB alteration is necessary for apoptoic and antineoplastic responses to farnesyltransferase inhibiotrs. Mol Cell Biol. 2000;20:6105–13. [PMC free article: PMC86086] [PubMed: 10913192]
- 77.
- Hutchinson J, Jin J, Cardiff RD. et al. Activation of Akt (protein kinase B) in mammary epithelium provides a critical cell survival signal required for tumor progression. Mol Cell Biol. 2001;21:2203–12. [PMC free article: PMC86854] [PubMed: 11238953]
- 78.
- Ruggieri R, Chuang YY, Symons M. The small GTPase Rac suppresses apoptosis caused by serum deprivation in fibroblasts. Mol Med. 2001;7:293–300. [PMC free article: PMC1950038] [PubMed: 11474575]
- 79.
- Coniglio SJ, Jou TS, Symons M. Rac1 protects epithelial cells against anoikis. J Biol Chem. 2001;276:28113–20. [PubMed: 11369774]
- 80.
- Barone MV, Sepe L, Melillo RM. et al. RET/PTC1 oncogene signalling in PC Cl 3 thyroid cells requires the small GTP-binding protein Rho. Oncogene. 2001;20:6973–82. [PubMed: 11704822]
- 81.
- Tu S, Cerione RA. Cdc42 is a substrate for caspases and influences Fas-induced apoptosis. J Biol Chem. 2001;276:19656–63. [PubMed: 11278572]
- 82.
- Qiu R-G, Chen J, McCormick F. et al. A role for Rho in Ras transformation. Proc Natl Acad Sc. 1995;92:11781–5. [PMC free article: PMC40486] [PubMed: 8524848]
- 83.
- Qiu R-G, Chen J, Kirn D. et al. An essential role for Rac in Ras transformation. Nature. 1995;374:457–9. [PubMed: 7700355]
- 84.
- Qiu R-G, Abo A, McCormick F. et al. Cdc42 regulates anchorage-independent growth and is necessary for Ras transformation. Mol Cell Biol. 1997;17:3449–58. [PMC free article: PMC232198] [PubMed: 9154844]
- 85.
- Prendergast GC, Khosravi-Far R, Solski PA. et al. Critical role of RhoB in cell transformation by oncogenic Ras. Oncogene. 1995;10:2289–96. [PubMed: 7784077]
- 86.
- Roux P, Gauthier-Rouvière C, Doucet-Brutin S. et al. The small GTPases Cdc42Hs, Rac1 and RhoG delineate Raf-independent pathways that cooperate to transform NIH3T3 cells. Curr Biol. 1997;7:629–37. [PubMed: 9285711]
- 87.
- Lebowitz PF, Du W, Prendergast GC. Prenylation of RhoB is required for its cell transforming function but not its ability to activate serum response element-dependent transcription. J Biol Chem. 1997;272:16093–5. [PubMed: 9195903]
- 88.
- Murphy GA, Solski PA, Jillian SA. et al. Cellular functions of TC10, a Rho family GTPase: Regulation of morphology, signal transduction, and cell growth. Oncogene. 1999;18:3831–45. [PubMed: 10445846]
- 89.
- Urich M, Senften M, Shaw PE. et al. A role for the small GTPase Rac in polyomavirus middle-T antigen-mediated activation of the serum response element and in cell transformation. Oncogene. 1997;14:1235–41. [PubMed: 9121774]
- 90.
- Connolly JO, Soga N, Guo XL. et al. Rac is essential in the transformation of endothelial cells by polyoma middle T. Cell Adhes Commun. 2000;7:409–22. [PubMed: 10830619]
- 91.
- Renshaw MW, Lea-Chou E, Wang JY. Rac is required for v-Abl tyrosine kinase to activate mitogenesis. Curr Biol. 1996;6:76–83. [PubMed: 8805224]
- 92.
- Rodrigues GA, Park M, Schlessinger J. Activation of the JNK pathway is essential for transformation by the Met oncogene. EMBO Journal. 1997;16:2634–45. [PMC free article: PMC1169874] [PubMed: 9184210]
- 93.
- Li J, Smithgall TE. Fibroblast transformation by Fps/Fes tyrosine kinases requires Ras, Rac, and Cdc42 and induces extracellular signal-regulated and c-Jun N-terminal kinase activation. J Biol Chem. 1998;273:13828–34. [PubMed: 9593727]
- 94.
- Skorski T, Wlodarski P, Daheron L. et al. BCR/ABL-mediated leukemogenesis requires the activity of the small GTP-binding protein Rac. Proc Natl Acad Sci USA. 1998;95:11858–62. [PMC free article: PMC21730] [PubMed: 9751755]
- 95.
- Boerner JL, Danielsen A, McManus MJ. et al. Activation of Rho is required for ligand-independent oncogenic signalling by a mutant epidermal growth factor receptor. J Biol Chem. 2001;276:3691–5. [PubMed: 11110781]
- 96.
- Sachdev P, Jiang YX, Li W. et al. Differential requirement for Rho family GTPases in an oncogenic insulin-like growth factor-I receptor-induced cell transformation. J Biol Chem. 2001;276:26461–71. [PubMed: 11346642]
- 97.
- Nguyen KT, Zong CS, Uttamsingh S. et al. The role of phosphatidylinositol 3-kinase, Rho family GTPases, and Stat3 in Ros-induced cell transformation. J Biol Chem. 2002 in press [PubMed: 11799110]
- 98.
- Gutkind JS, Novotny EA, Brann MR. et al. Muscarinic acetylcholine receptor subtypes as agonist-dependent oncogenes. Proc Natl Acad Sci. 1991;88:4703–7. [PMC free article: PMC51734] [PubMed: 1905013]
- 99.
- Zohn IE, Symons M, Chrzanowska-Wodnicka M. et al. Mas oncogene signalling and transformation require the small GTP- binding protein Rac. Mol Cell Biol. 1998;18:1225–35. [PMC free article: PMC108835] [PubMed: 9488437]
- 100.
- Zohn IE, Klinger M, Karp X. et al. G2A is an oncogenic G protein-coupled receptor. Oncogene. 2000;19:3866–77. [PubMed: 10951580]
- 101.
- Duncia JV, Santella JB3, Higley CA. et al. MEK inhibitors: The chemistry and biological activity of U0126, its analogs, and cyclization products. Bioorg Med Chem Lett. 1998;8:2839–44. [PubMed: 9873633]
- 102.
- Shaw RJ, Paez JG, Curto M. et al. The Nf2 tumor suppressor, merlin, functions in Rac-dependent signalling. Dev Cell. 2001;1:63–72. [PubMed: 11703924]
- 103.
- Ingram DA, Hiatt K, King AJ. et al. Hyperactivation of p21(ras) and the hematopoietic-specific Rho GTPase, Rac2, cooperate to alter the proliferation of neurofibromin-deficient mast cells in vivo and in vitro. J Exp Med. 2001;194:57–69. [PMC free article: PMC2193446] [PubMed: 11435472]
- 104.
- Avraham H, Weinberg RA. Characterization and expression of the human rhoH12 gene product. Mol Cell Biol. 1989;9:2058–66. [PMC free article: PMC362999] [PubMed: 2501657]
- 105.
- Perona R, Esteve P, Jiméz B. et al. Tumorigenic activity of rho genes from Aplysia californica. Oncogene. 1993;8:1285–92. [PubMed: 8479750]
- 106.
- Habets GGM, Scholtes EHM, Zuydgeest D. et al. Identification of an invasion-inducing gene, Tiam-1, that encodes a protein with homology to GDP-GTP exchangers for Rho-like proteins. Cell. 1994;77:537–49. [PubMed: 7999144]
- 107.
- Keely PJ, Westwick JK, Whitehead IP. et al. Cdc42 and Rac1 induce integrin-mediated cell motility and invasiveness through PI(3)K. Nature. 1997;390:632–6. [PubMed: 9403696]
- 108.
- Imamura F, Mukai M, Ayaki M. et al. Involvement of small GTPases Rho and Rac in the invasion of rat ascites hepatoma cells. Clin Exp Metastasis. 1999;17:141–8. [PubMed: 10411106]
- 109.
- Zhuge Y, Xu J. Rac1 mediates type I collagen-dependent MMP-2 activation. role in cell invasion across collagen barrier. J Biol Chem. 2001;276:16248–56. [PubMed: 11340084]
- 110.
- Murphy GA, Solski PA, Jillian SA. et al. Cellular functions of TC10, a Rho family GTPase: Regulation of morphology, signal transduction and cell growth. Oncogene. 1999;18:3831–45. [PubMed: 10445846]
- 111.
- Cobellis G, Missero C, Di LauroR. Concomitant activation of MEK-1 and Rac-1 increases the proliferative potential of thyroid epithelial cells, without affecting their differentiation. Oncogene. 1998;17:2047–57. [PubMed: 9798676]
- 112.
- Fischer RS, Zheng Y, Quinlan MP. Rac1 and extracellularly regulated kinase activation are sufficient for E1A-dependent cooperative transformation of primary epithelial cells, but progression can only be modulated by E1A or Rac1. Cell Growth Differ. 1998;9:209–21. [PubMed: 9543387]
- 113.
- Joneson T, Bar-Sagi D. Suppression of Ras-induced apoptosis by the Rac GTPase. Mol Cell Biol. 1999;19:5892–901. [PMC free article: PMC84438] [PubMed: 10454536]
- 114.
- Michiels F, Habets GG, Stam JC. et al. A role for Rac in Tiam1-induced membrane ruffling and invasion. Nature. 1995;375:338–40. [PubMed: 7753201]
- 115.
- Izawa I, Amano M, Chihara K. et al. Possible involvement of the inactivation of the Rho-Rho-kinase pathway in oncogenic Ras-induced transformation. Oncogene. 1998;17:2863–71. [PubMed: 9879992]
- 116.
- Cleverley SC, Costello PS, Henning SW. et al. Loss of Rho function in the thymus is accompanied by the development of thymic lymphoma. Oncogene. 2000;19:13–20. [PubMed: 10644975]
- 117.
- Zondag GC, Evers EE, ten Klooster JP. et al. Oncogenic Ras downregulates Rac activity, which leads to increased Rho activity and epithelial-mesenchymal transition. J Cell Biol. 2000;149:775–82. [PMC free article: PMC2174558] [PubMed: 10811819]
- 118.
- Bar-Sagi D, Feramisco JR. Induction of membrane ruffling and fluid-phase pinocytosis in quiescent fibroblasts by ras proteins. Science. 1986;233:1061–8. [PubMed: 3090687]
- 119.
- Walsh AB, Bar-Sagi D. Differential activation of the Rac pathway by Ha-Ras and K-Ras. J Biol Chem. 2001;276:15609–15. [PubMed: 11278702]
- 120.
- Hordijk PL, ten KloosterJP, van der Kammen RA. et al. Inhibition of invasion of epithelial cells by Tiam1-Rac signalling. Science. 1997;278:1464–6. [PubMed: 9367959]
- 121.
- Kamal MS, Qureshi MM, Kamal JM. et al. Construction of a cell-permeable CDC42 binding fragment of ACK that inhibits v-Ha-Ras transformation. Ann N Y Acad Sci. 1999;886:285–8. [PubMed: 10667241]
- 122.
- Scita G, Tenca P, Areces LB. et al. An effector region in Eps8 is responsible for the activation of the Rac-specific GEF activity of Sos-1 and for the proper localization of the Rac-based actin-polymerizing machine. J Cell Biol. 2001;154:1031–44. [PMC free article: PMC2196181] [PubMed: 11524436]
- 123.
- Scita G, Nordstrom J, Carbone R. et al. EPS8 and E3B1 transduce signals from Ras to Rac. Nature. 1999;401:290–3. [PubMed: 10499589]
- 124.
- Scita G, Tenca P, Frittoli E. et al. Signalling from Ras to Rac and beyond: Not just a matter of GEFs. EMBO J. 2000;19:2393–8. [PMC free article: PMC212757] [PubMed: 10835338]
- 125.
- Hu Q, Klippel A, Muslin AJ. et al. Ras-dependent induction of cellular responses by constitutively active phosphatidylinositol-3 kinase. Science. 1995;268:100–2. [PubMed: 7701328]
- 126.
- Rodriguez-Viciana P, Warne PH, Khwaja A. et al. Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell. 1997;89:457–67. [PubMed: 9150145]
- 127.
- Han J, Luby-Phelps K, Das B. et al. Role of substrates and products of PI 3-kinase in regulating activation of Rac-related guanosine triphosphatases by Vav. Science. 1998;279:558–60. [PubMed: 9438848]
- 128.
- Fleming IN, Gray A, Downes CP. Regulation of the Rac1-specific exchange factor Tiam1 involves both phosphoinositide 3-kinase-dependent and -independent components. Biochem J. 2000;351:173–82. [PMC free article: PMC1221348] [PubMed: 10998360]
- 129.
- Nimnual AS, Yatsula BA, Bar-Sagi D. Coupling of Ras and Rac guanosine triphosphatases through the Ras exchanger Sos. Science. 1998;279:560–3. [PubMed: 9438849]
- 130.
- Scita G, Nordstrom J, Carbone R. et al. EPS8 and E3B1 transduce signals from Ras to Rac. Nature. 1999;401:290–3. [PubMed: 10499589]
- 131.
- Jullien-Flores V, Dorseuil O, Romero F. et al. Bridging Ral GTPase to Rho pathways. J Biol Chem. 1995;270:22473–7. [PubMed: 7673236]
- 132.
- Cantor SB, Urano T, Feig LA. Identification and characterization of Ral-binding protein 1, a potential downstream target of Ral GTPases. Mol Cell Biol. 1995;15:4578–84. [PMC free article: PMC230698] [PubMed: 7623849]
- 133.
- Park SH, Weinberg RA. A putative effector of Ral has homology to Rho/Rac GTPase activating proteins. Oncogene. 1995;11:2349–55. [PubMed: 8570186]
- 134.
- Feig LA, Urano T, Cantor S. Evidence for a Ras/Ral signalling cascade. Trends Bio Sci. 1996;21:438–41. [PubMed: 8987400]
- 135.
- Ward Y, Wang W, Woodhouse E. et al. Signal pathways which promote invasion and metastasis: Critical and distinct contributions of extracellular signal-regulated kinase and Ral-specific guanine exchange factor pathways. Mol Cell Biol. 2001;21:5958–69. [PMC free article: PMC87314] [PubMed: 11486034]
- 136.
- Gupta S, Stuffrein S, Plattner R. et al. Role of phosphoinositide 3-kinase in the aggressive tumor growth of HT1080 human fibrosarcoma cells. Mol Cell Biol. 2001;21:5846–56. [PMC free article: PMC87304] [PubMed: 11486024]
- 137.
- Boerner JL, Danielsen A, McManus MJ. et al. Activation of Rho is required for ligand-independent oncogenic signalling by a mutant epidermal growth factor receptor. J Biol Chem. 2001;276:3691–5. [PubMed: 11110781]
- 138.
- Zheng Y. Dbl family guanine nucleotide exchange factors. Trends Bio Sci. 2001;26:724–32. [PubMed: 11738596]
- 139.
- Zheng Y, Olson MF, Hall A. et al. Direct involvement of the small GTP-binding protein Rho in lbc oncogene function. J Biol Chem. 1995;270:9031–4. [PubMed: 7721814]
- 140.
- Glaven JA, Whitehead IP, Nomanbhoy T. et al. Lfc and Lsc oncoproteins represent two new guanine nucleotide exchange factors for the Rho GTP-binding protein. J Biol Chem. 1996;271:27374–81. [PubMed: 8910315]
- 141.
- Engers R, Zwaka TP, Gohr L. et al. Tiam1 mutations in human renal-cell carcinomas. Inter J Cancer. 2000;88:369–76. [PubMed: 11054665]
- 142.
- Kalidas M, Kantarjian H, Talpaz M. Chronic myelogenous leukemia. JAMA. 2001;286:895–8. [PubMed: 11509034]
- 143.
- Bassermann F, Jahn T, Miething C. et al. Association of BCR-ABL with the protooncogene VAV is implicated in activation of the RAC-1 pathway. J Biol Chem. 2002 in press [PubMed: 11790798]
- 144.
- Kourlas PJ, Strout MP, Becknell B. et al. Identification of a gene at 11q23 encoding a guanine nucleotide exchange factor: Evidence for its fusion with MLL in acute myeloid leukemia. Proc Natl Acad Sci USA. 2000;97:2145–50. [PMC free article: PMC15768] [PubMed: 10681437]
- 145.
- Fukuhara S, Chikumi H, Gutkind JS. Leukemia-associated Rho guanine nucleotide exchange factor (LARG) links heterotrimeric G proteins of the G(12) family to Rho. FEBS Letters. 2000;485:183–8. [PubMed: 11094164]
- 146.
- Reuther GW, Lambert QT, Booden MA. et al. Leukemia-associated Rho guanine nucleotide exchange factor, a Dbl family protein found mutated in leukemia, causes transformation by activation of RhoA. J Biol Chem. 2001;276:27145–51. [PubMed: 11373293]
- 147.
- Seraj MJ, Harding MA, Gildea JJ. et al. The relationship of BRMS1 and RhoGDI2 gene expression to metastatic potential in lineage related human bladder cancer cell lines. Clin Exp Metastasis. 2000;18:519–25. [PubMed: 11592309]
- 148.
- Jones MB, Krutzsch H, Shu H. et al. Proteomic analysis and identification of new biomarkers and therapeutic targets for invasive ovarian cancer. Proteomics. 2002;2:76–84. [PubMed: 11788994]
- 149.
- Young D, Waitches G, Birchmeier C. et al. Isolation and characterization of a new cellular oncogene encoding a protein with multiple potential transmembrane domains. Cell. 1986;45:711–9. [PubMed: 3708691]
- 150.
- Kozasa T, Jiang X, Hart MJ. et al. p115 RhoGEF, a GTPase activating protein for Galpha12 and Galpha13. Science. 1998;280:2109–11. [PubMed: 9641915]
- 151.
- Fukuhara S, Murga C, Zohar M. et al. A novel PDZ domain containing guanine nucleotide exchange factor links heterotrimeric G proteins to Rho. J Biol Chem. 1999;274:5868–79. [PubMed: 10026210]
- 152.
- Martin CB, Mahon GM, Klinger MB. et al. The thrombin receptor, PAR-1, causes transformation by activation of Rho-mediated signalling pathways. Oncogene. 2001;20:1953–63. [PubMed: 11360179]
- 153.
- Kabarowski J, Feramisco J, Le LQ. et al. Direct genetic demonstration of Gα13 coupling to the orphan G protein-coupled receptor G2A leading to RhoA-dependent actin rearrangement. PNAS. 2000;97:12109–14. [PMC free article: PMC17302] [PubMed: 11050239]
- 154.
- D'Andrea MR, Derian CK, Santulli RJ. et al. Differential expression of protease-activated receptors- 1 and -2 in stromal fibroblasts of normal, benign, and malignant human tissues. Am J Path. 2001;158:2031–41. [PMC free article: PMC1891970] [PubMed: 11395381]
- 155.
- Fritz G, Just I, Kaina B. Rho GTPases are over-expressed in human tumors. International Journal on Cancer. 1999;81:682–7. [PubMed: 10328216]
- 156.
- Kamai T, Arai K, Tsujii T. et al. Overexpression of RhoA mRNA is associated with advanced stage in testicular germ cell tumour. BJU Int. 2001;87:227–31. [PubMed: 11167647]
- 157.
- Abraham MT, Kuriakose MA, Sacks PG. et al. Motility-related proteins as markers for head and neck squamous cell cancer. Laryngoscope. 2001;111:1285–9. [PMC free article: PMC3616334] [PubMed: 11568556]
- 158.
- Mira JP, Benard V, Groffen J. et al. Endogenous, hyperactive Rac3 controls proliferation of breast cancer cells by a p21-activated kinase-dependent pathway. Proc Natl Acad Sci USA. 2000;97:185–9. [PMC free article: PMC26637] [PubMed: 10618392]
- 159.
- Clark EA, Golub TR, Lander ES. et al. Genomic analysis of metastasis reveals an essential role for RhoC. Nature. 2000;406:532–5. [PubMed: 10952316]
- 160.
- van GolenKL, Wu ZF, Qiao XT. et al. RhoC GTPase overexpression modulates induction of angiogenic factors in breast cells. Neoplasia. 2000;2:418–25. [PMC free article: PMC1507979] [PubMed: 11191108]
- 161.
- van GolenKL, Wu ZF, Qiao XT. et al. RhoC GTPase, a novel transforming oncogene for human mammary epithelial cells that partially recapitulates the inflammatory breast cancer phenotype. Cancer Research. 2000;60:5832–8. [PubMed: 11059780]
- 162.
- Suwa H, Ohshio G, Imamura T. et al. Overexpression of the rhoC gene correlates with progression of ductal adenocarcinoma of the pancreas. Br J Cancer. 1998;77:147–52. [PMC free article: PMC2151257] [PubMed: 9459160]
- 163.
- Tao W, Pennica D, Xu L. et al. Wrch-1, a novel member of the Rho gene family that is regulated by Wnt-1. Genes and Dev. 2001;15:1796–807. [PMC free article: PMC312736] [PubMed: 11459829]
- 164.
- Dallery E, Galiegue-Zouitina S, Collyn-d'Hooghe M. et al. TTF, a gene encoding a novel small G protein, fuses to the lymphoma-associated LAZ3 gene by t(3;4) chromosomal translocation. Oncogene. 1995;10:2171–8. [PubMed: 7784061]
- 165.
- Dallery-Prudhomme E, Roumier C, Denis C. et al. Genomic structure and assignment of the RhoH/ TTF small GTPase gene (ARHH) to 4p13 by in situ hybridization. Genomics. 1997;43:89–94. [PubMed: 9226377]
- 166.
- Preudhomme C, Roumier C, Hildebrand MP. et al. Nonrandom 4p13 rearrangements of the RhoH/ TTF gene, encoding a GTP-binding protein, in nonHodgkin's lymphoma and multiple myeloma. Oncogene. 2000;19:2023–32. [PubMed: 10803463]
- 167.
- Jordan P, Brazao R, Boavida MG. et al. Cloning of a novel human Rac1b splice variant with increased expression in colorectal tumors. Oncogene. 1999;18:6835–9. [PubMed: 10597294]
- 168.
- Schnelzer A, Prechtel D, Knaus U. et al. Rac1 in human breast cancer: Overexpression, mutation analysis, and characterization of a new isoform, rac1b. Oncogene. 2000;19:3013–20. [PubMed: 10871853]
- 169.
- Wittinghofer A, Nassar N. How Ras-related proteins talk to their effectors. Trends Bio Sci. 1996;21:488–91. [PubMed: 9009833]
- 170.
- Sahai E, Ishizaki T, Narumiya S. et al. Transformation mediated by RhoA requires activity of ROCK kinases. Curr Biol. 1999;9:136–45. [PubMed: 10021386]
- 171.
- Zohar M, Teramoto H, Katz BZ. et al. Effector domain mutants of Rho dissociate cytoskeletal changes from nuclear signalling and cellular transformation. Oncogene. 1998;17:991–8. [PubMed: 9747878]
- 172.
- Itoh K, Yoshioka K, Akedo H. et al. An essential part for Rho-associated kinase in the transcellular invasion of tumor cells. Nat Med. 1999;5:221–5. [PubMed: 9930872]
- 173.
- Zong H, Kaibuchi K, Quilliam LA. The insert region of RhoA is essential for Rho kinase activation and cellular transformation. Mol Cell Biol. 2001;21:5287–98. [PMC free article: PMC87252] [PubMed: 11463812]
- 174.
- Joneson T, Bar-Sagi D. Ras effectors and their role in mitogenesis and oncogenesis. J Mol Med. 1997;75:587–93. [PubMed: 9297626]
- 175.
- Lambert JM, Karnoub AE, Graves LM. et al. Role of MLK3-mediated activation of p70 S6 kinase in Rac1 transformation. J Biol Chem. 2001 in press [PubMed: 11713255]
- 176.
- Lamarche N, Tapon N, Stowers L. et al. Rac and Cdc42 induce actin polymerization and G1 cell cycle progression independently of p65PAK and the JNK/SAPK MAP kinase cascade. Cell. 1996;87:519–29. [PubMed: 8898204]
- 177.
- Abo A, Pick E, Hall A. et al. Activation of the NADPH oxidase involves the small GTP-binding protein p21rac1. Nature. 1991;353:668–70. [PubMed: 1922386]
- 178.
- Knaus UG, Heyworth PG, Evans T. et al. Regulation of phagocyte oxygen radical production by the GTP-binding protein Rac2. Science. 1991;254:1512–5. [PubMed: 1660188]
- 179.
- Nisimoto Y, Freeman JLR, Motalebi SA. et al. Rac binding to p67phox. J Biol Chem. 1997;272:18834–41. [PubMed: 9228059]
- 180.
- Irani K, Xia Y, Zweier JL. et al. Mitogenic signalling mediated by oxidants in Ras-transformed fibroblasts. Science. 1997;275:1649–52. [PubMed: 9054359]
- 181.
- Joneson T, Bar-Sagi D. A Rac1 effector site controlling mitogenesis through superoxide production. J Biol Chem. 1998;273:17991–4. [PubMed: 9660749]
- 182.
- Whitehead IP, Lambert QT, Glaven JA. et al. Dependence of Dbl and Dbs transformation on MEK and NF-KB activation. Mol Cell Biol. 1999;19:7759–70. [PMC free article: PMC84831] [PubMed: 10523665]
- 183.
- Qiu RG, Abo A, Steven MG. A human homolog of the C. elegans polarity determinant Par-6 links Rac and Cdc42 to PKCzeta signalling and cell transformation. Curr Biol. 2000;10:697–707. [PubMed: 10873802]
- 184.
- Johansson A, Driessens M, Aspenstrom P. The mammalian homologue of the Caenorhabditis elegans polarity protein PAR-6 is a binding partner for the Rho GTPases Cdc42 and Rac1. J Cell Sci. 2000;113:3267–75. [PubMed: 10954424]
- 185.
- Noda Y, Takeya R, Ohno S. et al. Human homologues of the Caenorhabditis elegans cell polarity protein PAR6 as an adaptor that links the small GTPases Rac and Cdc42 to atypical protein kinase C. Genes Cells. 2001;6:107–19. [PubMed: 11260256]
- 186.
- Kay AJ, Hunter CP. CDC-42 regulates PAR protein localization and function to control cellular and embryonic polarity in C. elegans. Curr Biol. 2001;11:474–81. [PubMed: 11412996]
- 187.
- Wu WJ, Erickson JW, Lin R. et al. The gamma-subunit of the coatomer complex binds Cdc42 to mediate transformation. Nature. 2000;405:800–4. [PubMed: 10866202]
- 188.
- Wu WJ, Lin R, Cerione RA. et al. Transformation activity of Cdc42 requires a region unique to Rho- related proteins. J Biol Chem. 1998;273:16655–8. [PubMed: 9642217]
- Introduction
- The Rho Family of GTPases Is a Major Branch of the Ras Superfamily
- Rho GTPases Function As Membrane-Associated GDP/GTP-Regulated Molecular Switches
- Rho Family GTPases Are Critical Components of Diverse Signalling Pathways
- Rho GTPases Promote Oncogenic Transformation
- Rho GTPases Mediate Transformation through Multiple Effectors
- Conclusions and Future Directions
- References
- Rho Family GTPases and Cellular Transformation - Madame Curie Bioscience Databas...Rho Family GTPases and Cellular Transformation - Madame Curie Bioscience Database
- Deciphering the Complex Enzymatic Pathway for Biosynthesis of Wyosine Derivative...Deciphering the Complex Enzymatic Pathway for Biosynthesis of Wyosine Derivatives in Anticodon of tRNAPhe - Madame Curie Bioscience Database
- Tests of a Stereochemical Genetic Code - Madame Curie Bioscience DatabaseTests of a Stereochemical Genetic Code - Madame Curie Bioscience Database
- Origins and Evolution of Cotranslational Transport to the ER - Madame Curie Bios...Origins and Evolution of Cotranslational Transport to the ER - Madame Curie Bioscience Database
- Thermo-Chemo-Radiotherapy Association: Biological Rationale, Preliminary Observa...Thermo-Chemo-Radiotherapy Association: Biological Rationale, Preliminary Observations on Its Use on Malignant Brain Tumors - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...