

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

We provide in this chapter an overview of the basic steps to reconstruct evolutionary relationships through standard phylogeny estimation approaches as well as network approaches for sequences more closely related. We discuss the importance of sequence alignment, selecting models of evolution, and confidence assessment in phylogenetic inference. We also introduce the reader to a variety of software packages used for such studies. Finally, we demonstrate these approaches throughout using a data set of 33 whole genomes of polyomaviruses. A robust phylogeny of these genomes is estimated and phylogenetic relationships among the polyomaviruses determined using Bayesian and maximum likelihood approaches. Furthermore, population samples of SV40 are used to demonstrate the utility of network approaches for closely related sequences. The phylogenetic analysis suggested a close relationship among the BK viruses, JC viruses, and SV40 with a more distant association with mouse polyomavirus, monkey polymavirus (LPV) and then avian polyomavirus (BFDV).
Introduction
Polyomaviruses are small, nonenveloped, double-stranded DNA viruses that are widely distributed among vertebrates. Each polyomavirus is exquisitely adapted to a single species, or to a group of closely related species. The polyomaviruses are often described as having coevolved with their hosts. As a rule, primary infections occur early in life and are asymptomatic and harmless. The viruses remain latent in the kidney after primary infection, and are reactivated in conditions associated with T-cell deficiency. Almost all the diseases caused by these viruses occur in immunodeficient hosts. The viruses multiply in the nucleus and virus-induced pathology is characterized by nuclei that are enlarged and have basophilic inclusions.
The human polyomaviruses, BKV and JCV, and macaque polyomavirus, SV40, are very similar biologically, and each virus has over 70 % nucleotide sequence similarity with the other two. In its natural host, SV40 produces an illness similar to JCV-PML in humans, while the cynomolgus virus produces a nephropathy similar to BKV-nephropathy in humans. Other viruses that might be biologically similar, although little is actually known about them, are SA12 of the chacma baboon, the recently described cynomolgus macaque virus, the bovine and the rabbit polyomaviruses, and perhaps the mouse polyoma virus. The avian budgerigar virus, which produces liver and spleen necrosis, and a rat polyomavirus, which produces salivary gland pathology in athymic animals, both produce lesions that are characteristic of viruses of the polyomavirus family.
The simian lymphotropic polyomavirus of African green monkeys seems to have a different biology from these viruses. In the mouse, the mouse polyomavirus (referred to above) seems biologically very different from K virus, which grows in endothelial cells of the lung. The tropism of K virus-infected endothelial cells is unique among polyomaviruses. The oddest polyomavirus is the hamster polyomavirus, which produces skin tumors and was at one time thought to be a papillomavirus. Thus, lymphotropic virus of African green monkeys, K virus of mouse and the hamster polyomavirus seem not to fit the general pattern.
In order to explore the diversity of polyomaviruses, both genomic diversity at the molecular level and diversity in host specificity, a phylogenetic perspective is essential. A phylogeny represents the evolutionary history among organisms or their parts. In our case, we will attempt to reconstruct evolutionary histories using whole genome analyses. Our analysis will provide a robust estimate of phylogenetic relationships among the polyomaviruses. Additionally, we will examine the population dynamics of SV40 in particular using only partial sequence and a network genealogical approach. We discuss the basics of sequence alignment, model selection, and recombination detection and their importance in terms of phylogenetic estimation. We introduce the reader to a variety of useful software packages for performing these analyses. Throughout this chapter, we demonstrate these important components of evolutionary analyses using the polyomaviruses as our model system. Our study results in a robust estimate of the evolutionary relationships among the polyomaviruses based on both maximum likelihood and Bayesian analyses. Finally, we introduce the reader to network approaches to estimating gene genealogies using an SV40 data set and suggest approaches for testing hypotheses of this virus associated with cancer.
Sequence Alignment
Perhaps the most difficult and underappreciated aspects of phylogeny estimation is the sequence alignment phase. For population genetic studies using single gene regions, alignments are often trivial, especially for conserved gene regions. However, for whole genome analysis across the phylogenetic diversity shown in the polyomaviruses, alignment is far from trivial. A standard approach is to use the popular alignment tool Clustal X1 with the default parameters and then start using the output alignment for phylogeny estimation. One of the difficulties with this approach is that Clustal X does not take into account an amino acid reading frame. Thus gaps can be inserted within a genetic codon triplet breaking up an otherwise reasonable reading frame. Therefore, for coding sequences, it is important to review an alignment using a sequence editor that allows one to toggle between amino acids and nucleotides, for example Se-Al2 (fig. 1). Needless to say, the hand proofing of sequence alignments for large data sets becomes quickly unwieldy. An alternative is to use an alignment software that takes into account coding frame for nucleotide data, aligns by the amino acid sequence, and then converts back to the nucleotides. To our knowledge, such software does not exist that can handle a reasonable number of whole genomes. However, our group is currently developing software called AlignmentHelper that can perform this task. Even with the aid correcting for codons, there can still be significant ambiguity in the resulting alignment. The alignment provides the basis of positional homology (the assumption that each nucleotide in the same column of data shares a common ancestor) for phylogeny estimation. If the alignment is questionable in any region, the inferred phylogeny may be in error as a result. For example, in Figure 1, we have some assurance of the positioning of the gaps as a triplet due to the translation to amino acids. However, it is impossible to reasonably choose where that gap should be in sequence 3. As shown, it tends to link sequence 3 with sequences 1 and 2 (this is indeed the Clustal X output). An alternative would be to slide the gap to the right (3') two or even three amino acids to link sequence 3 with sequences 4 and 5. This ambiguity (and often even greater ambiguity exists especially for nonprotein coding regions) can lead to spurious inferred relationships. Therefore it is important to have some assessment of positional homology before attempting phylogeny estimation. A quick assessment can be accomplished using the software Gblocks.3 This software uses the number of contiguous conserved positions, lack of gaps, and high conservation of flanking positions to assess the “goodness” of different blocks of nucleotides within an alignment. Using this approach, one can quickly define regions of suspect positional homology and eliminate them from subsequent analyses.

Figure 1
Sample alignment with conversion to amino acids demonstrating the importance of this conversion for accurate alignment and the importance of positional homology for phylogeny estimation.
For our analysis of polyomavirus phylogeny, we selected 33 sequences from GenBank (Table 1). These sequences were aligned (each gene being aligned independently) using T-Coffee,4 which performs more accurate alignments than Clustal W although at a much slower speed for these large data sets. The resulting alignment was then adjusted using Se-Al and AlignmentHelper. Then GBlocks was used to identify regions of confident alignment throughout the genome of the polyomaviruses. Our final alignment included 8916 positions (larger than the average genome size!). After excluding blocks of ambiguous alignment as defined by GBlocks, our data set consisted of a total of 5298 nucleotides. Figure 2 shows the idealized polyomavirus genome with shading across the reading frames to indicate those sections eliminated by GBlocks analysis. The remainder of the coding sequence across the genome was then used for phylogenetic analysis. With a robust alignment, we moved to testing for recombination, since most phylogenetic approaches assume that recombination has not occurred throughout the history of the sequences under study. Thus, we must first statistically test for the possibility of recombination.
Table 1
Summary of the polyomavirus sequence data used in this analysis.
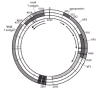
Recombination
Recombination in polyomaviruses, at least in JC viruses, has been controversial but seemingly does occur.5,6 Recombination can have a large impact on our ability to accurately estimate evolutionary relationships7 and population genetic parameters such as genetic diversity and substitution rates.8 Furthermore, recombination can be an important evolutionary force that should be taken into account when considering drug and vaccine design.9 Thus it is essential to test for recombination in a given data set. The question then becomes, what test should be used? There are a wide variety of methods for detecting recombination. Many of them are easily fooled by other phenomena such as population structure or rate heterogeneity. Posada and Crandall10 evaluated a number of different methods that claim to detect recombination using a computer simulation approach. They found that phylogenetic methods (those most commonly used to test for recombination in viral sequences) typically performed poorer than methods that use substitution patterns or incompatibility among sites as a criterion for the inference of recombination. The “best” method for detecting recombination also greatly depended on the overall amount of genetic diversity in the sample. Posada11 found similar results using empirical data sets. Therefore, there is still no easy guideline for choosing a method to apply to any particular data set to detect recombination. We therefore ran our 33-sequence data set through a variety of recombination detection algorithms and found no evidence for recombination (at least among the major clades). Given a lack of evidence for recombination coupled with a robust model of evolution, we can now proceed to estimation the evolutionary relationships among these viruses via phylogeny reconstruction.
Phylogeny Estimation
Our first decision in reconstructing evolutionary histories is what optimality criterion we should use. There are both algorithmic methods like neighbor-joining12 and methods that optimize solutions based on some criterion like parsimony (minimizing the branch length) or likelihood (maximizing the likelihood). Optimality methods are generally better than algorithmic methods because they find not only the optimal solution but a variety of solutions close to the optimum whereas algorithmic methods provide simply a point estimate of the solution. There could possibly be a number of solutions that look quite different that are just as good as the point estimate provided by the algorithmic approach. Unfortunately, because the algorithmic approaches like neighbor-joining are computationally very fast, many researchers choose this approach despite its limitations. We highly recommend to the reader to use the more thorough optimality methods discussed below.
Optimality Criteria
There are two fundamentally different optimality criteria that are typically used in phylogeny estimation, minimize the branch lengths (parsimony) or maximize the likelihood scores. There are a variety of ways to implement these different criteria. Maximum parsimony can be performed with a “weighting matrix” that effectively incorporates a more realistic model of evolution within the parsimony framework. Such weighting matrices can be justified by using empirical estimates from the data for observed patterns of nucleotide substitutions. This approach has the advantage of being able to take into account gaps as characters in phylogeny estimation. However, the parsimony approach cannot accommodate rate heterogeneity (different substitution rates at different sites along the sequence) and it therefore performs poorly when there is great rate heterogeneity.13 The alternative then becomes maximum likelihood.14 This approach does incorporate a model of evolution (see below) allowing for rate heterogeneity, invariable sites, differences in base frequencies (none of which are accommodated in a parsimony framework), as well as differences in substitution rates. This approach does not, however, accommodate gap characters. These are typically treated as missing data. The other weakness of the maximum likelihood approach is in computational time. It is a very slow approach in general, especially with reasonably large data sets. However, alternative methods have been developed to speed the likelihood searches including genetic algorithms,15,16 parallel algorithms,17 and Bayesian approaches to assess relative likelihoods.18 We have used both standard maximum likelihood as implemented by PAUP*,19 as well as a Bayesian approach implemented in Mr. Bayes.20
Bayesian analysis differs from maximum likelihood in that the standard likelihood is defined as the probability of the data given the tree and the model, or L = Prob(Data | Tree).21 The Bayesian inference of phylogeny, on the other hand, is based on the posterior probability of a tree defined as. pr(Tree.Data) = Pr(Data.Tree)|r(Tree)/Pr(Data)Both methods incorporate models of evolution as discussed above. One great advantage of the Bayesian approach is that the posterior probability is also used as a confidence assessment (see below), thus eliminating the need to repeat an analysis 100s-1000s of times to obtain a bootstrap value as an assessment of nodal confidence. Computationally, Bayesian approaches tend to be much faster and find very similar trees (both in terms of the topology as well as branch lengths). Bayesian approaches have also been implemented in a variety of other contexts including detecting selection, estimating divergence times, testing for a molecular clock, and evaluating models of evolution (see Huelsenbeck et al18 for a review). Our preferred optimality criteria require some way of modeling the evolutionary changes in the sequence data along a tree. We need to not only model the changes, but determine if our selected model is a reasonable estimate of the true underlying changes. The next section offers insights into models of evolution and model selection.
Models of Evolution
Models of evolution represent a probability statement for the change from one nucleotide to another (e.g., G ⇒ A). The model is often represented as a relative rate of change from one nucleotide to another, leaving five free rates with one fixed at a relative rate of 1.0 in a symmetrical model (e.g., G ⇒ A) (fig. 3). The first model of evolution developed was that of Jukes and Cantor,22 which accounted for multiple changes at a single site with equal rate parameters for all rates of change. Later, Kimura noticed that in many data sets transitions (change from purine to purine or pyrimidine to pyrimidine) occurred much more frequently than transversions (changes from purine to pyrimidine or vise versa). He then developed the Kimura 2-parameter model to allow for differences in the transition and transversion rates.23 Subsequent models have been developed that incorporate differences for all relative rates (the general time reversible or GTR model)24 as well as differences in nucleotide frequencies among base pairs,14 rate heterogeneity among sites,25 invariable sites,26 and even codon position.27,28 While the Kimura 2-parameter model is the default model for many studies for both historical reasons and simply because it is the default model in the population software package PHYLIP,29 it has often been shown to be too simplistic to reasonably model the molecular evolution of viral systems.30
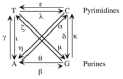
Given this plethora of models to choose from, one is left with a decision on how to make a reasonable choice of models. One could simply choose the most complex model of evolution available knowing that models by definition are simplifications of biological reality thereby hoping that the most complex model might come closest to a true underlying model. One problem with this approach is that highly complex models require many parameters. All these parameters need to be estimated from the data. One then needs to worry about having enough data to accurately estimate all these parameters. The errors in these estimates are typically not incorporated into a model of evolution that is subsequently used for phylogeny estimation. Therefore, an alternative is to fit the model to the data using some criterion like maximum likelihood through likelihood ratio testing,31 an Akaike information criterion, or a Bayesian information criterion.32 This approach allows one to determine statistically the relative gain in likelihood for adding more parameters to the model. There is now software available, for example ModelTest,33 to assist in the evaluation of different models for a given data set.
One might ask if the model of evolution can really make much of a difference in the resulting estimated phylogeny and conclusions based on that tree. In a study on the origins of primate T-cell leukemia/lymphoma viruses (PTLVs), Kelsey et al34 found that previous researchers had used the Kimura 2-parameter model without justification but that a model selected using ModelTest resulted in not only a different model of evolution, it changed the conclusion of the origin of PTLVs from Asia to Africa. Thus the main conclusions of studies can be severely affected by the wrong choice of model of evolution. It is therefore essential to justify one's choice in model and demonstrate that that model reasonably fits the data.
We used ModelTest to select a model of evolution for our 33-sequence data set of polyomaviruses. The resulting model was the general-time reversible model (GTR) with invariable sites and rate heterogeneity. The model parameters for this analysis were as follows: Base frequencies (A, C, G, T) = (0.3166 0.1973 0.2273), Nst = 6, Rmat = (1.8508 2.8908 1.5896 2.0755 3.9402), Rates = gamma, Shape = 1.5685, Pinvar = 0.1011. Now that we have a model of evolution in hand, it is time to estimate a phylogeny.
Here we used both maximum likelihood and Bayesian approaches to estimate phylogenetic relationships among the polyomaviruses. Both methods used the same model of evolution (see above) and both methods estimated the same tree (with an identical likelihood score) (fig. 4). The comparative speeds, however, were quite different. The Bayesian analysis took 24.5 hours to run on a Lunix 3.0 GHz Xeon PC computer, whereas the maximum likelihood analysis took ∼79.5 hours. The resulting tree shows a major grouping of each of the well-characterized polyomaviruses, e.g., JCVs form a clade sister to the BK viruses. The (BK, JCV) clade is sister to the SV40 clade. There is a robust clade of mouse polyomavirus related to the hamster papovavirus (HaPV). That clade is then sister to a clade of virus infecting monkeys. More distantly related are the goose hemorrhagic polyomavirus (GHPV) and the avian polyomaviruses (BFPV).
Confidence Assessment
Many studies mistakenly stop at this point (having a tree) and start telling stories about their tree and its wonderful significance, as we have just done. However, it is important to recognize that phylogeny estimation is a difficult problem and a single point estimate is not to be trusted, even when using a robust optimality criterion. Therefore, some measure of confidence is desired to judge the statistical validity of the inferred relationships. Confidence assessment is typically performed by using either posterior probabilities (for Bayesian approaches), or through a bootstrap35 or jackknife procedure. Bootstrapping is the most common form of confidence assessment and consists of resampling the data with replacement, reevaluating that new (resampled) data set using the same optimality criterion and same model of evolution and then repeating this many times. The resulting trees are then evaluated by a majority-rule consensus procedure with the bootstrap proportions being associated with the number of times that node is represented in the family of bootstrapped trees. Bootstrap values have been shown to be highly biased (seemingly in a conservative way).36 The obvious difficulty with bootstrap values (as mentioned above) is that one needs to reestimate a phylogeny for each resampled data set. For reasonable bootstrap values, typically 100 — 1000 bootstrap replications are required. Thus if your original search takes any time at all (in our case 79.5 hours), your bootstrap evaluation of confidence will simply take too long (∼9 years!).
The alternative approach then is to use a method like Bayesian analysis that performs an assessment of confidence at the same time as estimating the tree. In the Bayesian analysis, the search continues until the likelihood score plateaus, including the tree and substitution parameters. At this point there is typically a large set of trees with very similar likelihood scores. These trees are then used to create consensus trees with the percentage of times a particular node shows up in that group of trees related as the posterior probably. In our Figure 4, these posterior probabilities are shown on the major branches and range from 0.29 to 1.0. All of the major clades (those nodes leading to monophyletic groups of distinct viruses, e.g., BK) are supported by posterior probabilities of 1.0. There is a growing literature on the relationship between posterior probabilities and bootstrap values37,38 and no consensus seems to have been reached at the moment.39,40
Population Variation of SV40
Notice that in Figure 4, there is very little resolution within the SV40 clade and parts of the JCV clade. Most of the nodes have no posterior probabilities associated with them (because we did not label nodes with less than 0.5 posterior probability). When one is working with closely related sequences with little divergence (and a greater potential for recombination), network approaches for visualizing genealogical relationships become preferred representations.41 These network approaches allow for the simultaneous visualization of multiple solutions, the biological reality of nonbifurcating genetic exchange (e.g., through recombination, hybridization, etc.). They also allow for greater resolution when sequence divergences are low.42,43
We, therefore, used the statistical parsimony approach44 as implemented in the software TCS45 to estimate evolutionary relationships among the SV40 viruses. Using this approach, minimum connections are made using a 95% confidence assessment based on a statistical assessment of the conditional probability of the change of more than one nucleotide at a particular site. With a high probability (>95%) that a multiple change has not occurred, minimum connections are made to infer genealogical relationships. This approach has been tested using empirical data from a known bacteriophage phylogeny and shown to be robust and outperform other approaches such as parsimony,42 likelihood and distance46 approaches. Indeed, the resulting relationships (fig. 5) show a great more resolution that the SV40 clade in Figure 4. One can also use the network structure to help interpret the data. For example, population genetic theory argues that sequences with high frequency in the sample and those in the interior of the network are older in evolutionary age.47,48 Thus we can infer that isolates OPC/MEN, Rh911, and some of 777* are older in age (perhaps the oldest in the sample) relative to the other haplotypes that appear on the tips of the network. Using such reasoning it becomes possible to test hypotheses, for example, about the association with SV40 with cancer.49 Clearly the methodology for testing such hypotheses is available. We only await an appropriate data set for such an analysis.
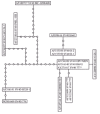
Figure 5
A network of genealogical relationships among the SV40 viruses where zeros are missing intermediates and each line represents a single mutational change.
Summary
Phylogenetic methods are of great utility for a wide variety of hypotheses in infectious disease studies.50 Here we have hopefully provided a useful introduction to a variety of phylogenetic methods and the complexity of phylogenetic analyses in general. We have done so using the polyomavirus as a model system and generated a novel phylogeny for the relationships among the viruses associated with this group. We hope that this work will stimulate further interest in phylogenetic inference with infectious diseases and in the proper use of phylogenetic methodology. We refer the reader to the new (and excellent) text by Felsenstein21 for further details on phylogenetic inference.
Acknowledgements
We thank Nasimul Ahsan for the invitation to present this chapter and his patience in editing. We would also like to thank the NIH for supporting our work through grants R01 AI50217 (RPV, KAC) and GM66276 (KAC). This work was also supported by the Brigham Young University Cancer Research Center (RGC).
References
- 1.
- Thompson JD, Gibson TJ, Plewniak F. et al. The clustalX windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nuc Acid Res. 1997;24:4876–4882. [PMC free article: PMC147148] [PubMed: 9396791]
- 2.
- Se-A.Sequence Alignment Editor [computer program]. Version 2.0Oxford: http://evolve.zoo.ox.ac.uk.2002 .
- 3.
- Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis MBE 200017|(4):540–552. [PubMed: 10742046]
- 4.
- Notredame C, Higgins D, Heringa J. T-Coffee: A novel method for multiple sequence alignments. J Mol Biol. 2000;302:205–217. [PubMed: 10964570]
- 5.
- Hatwell JN, Sharp PM. Evolution of human polyomavirus JC. J Gen Virol. 2000;81:1191–1200. [PubMed: 10769060]
- 6.
- Jobes DV, Chima SC, Ryschkewitsch CF. et al. Phylogenetic analysis of 22 complete genomes of the human polyomavirus JC virus. J Gen Virol. 1998;79:2491–2498. [PubMed: 9780056]
- 7.
- Posada D, Crandall KA. The effect of recombination on the accuracy of phylogeny estimation. JME. 2002;54:396–402. [PubMed: 11847565]
- 8.
- Schierup MH, Hein J. Consequences of recombination on traditional phylogenetic analysis. Genetics. 2000;156:879–891. [PMC free article: PMC1461297] [PubMed: 11014833]
- 9.
- Rambaut A, Posada D, Crandall KA. et al. The causes and consequences of HIV evolution. Nat Rev Genet. 2004;5(1):52–61. [PubMed: 14708016]
- 10.
- Posada D, Crandall KA. Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. PNAS. 2001;98(24):13757–13762. [PMC free article: PMC61114] [PubMed: 11717435]
- 11.
- Posada D. On the performance of methods for detecting recombination from DNA sequences: Real data. MBE. 2002;19(5):708–717. [PubMed: 11961104]
- 12.
- Saitou N, Nei M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. MBE. 1987;4(4):406–425. [PubMed: 3447015]
- 13.
- Huelsenbeck JP, Hillis DM. Success of phylogenetic methods in the four-taxon case. Syst Biol. 1993;42(3):247–264.
- 14.
- Felsenstein J. Evolutionary trees from DNA sequences: A maximum likelihood approach. JME. 1981;17:368–376. [PubMed: 7288891]
- 15.
- Lewis PO. A genetic algorithm for maximum-likelihood phylogeny inference using nucleotide sequence data. MBE. 1998;15(3):277–283. [PubMed: 9501494]
- 16.
- Lemmon AR, Milinkovitch MC. The metapopulation genetic algorithm: An efficient solution for the problem of large phylogeny estimation. PNAS. 2002;99(16):10516–10521. [PMC free article: PMC124960] [PubMed: 12142465]
- 17.
- Brauer MJ, Holder MT, Dries LA. et al. Genetic algorithms and parallel processing in maximum-likelihood phylogeny inference. MBE. 2002;19(10):1717–1726. [PubMed: 12270898]
- 18.
- Huelsenbeck JP, Ronquist F, Nielsen R. et al. Bayesian inference of phylogeny and its impact on evolutionary biology. Science. 2001;294:2310–2314. [PubMed: 11743192]
- 19.
- PAUP*.Phylogenetic analysis using parsimony (*and other methods) [computer program]. Version 4 Sunderland: Sinauer Associates,2000 .
- 20.
- Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17(8):754–755. [PubMed: 11524383]
- 21.
- Felsenstein J. Inferring phylogenies. Sunderland: Sinauer Associates. 2003
- 22.
- Jukes TH, Cantor CR. Evolution of protein moleculesIn: Munro HM, ed.Mammalian Protein MetabolismNew York: Academic Press,196921–132.
- 23.
- Kimura M. A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. JME. 1980;16:111–120. [PubMed: 7463489]
- 24.
- Rodríguez F, Oliver JL, Marin A. et al. The general stochastic model of nucleotide substitution. J Theor Biol. 1990;142:485–501. [PubMed: 2338834]
- 25.
- Yang Z. Among-site rate variation and its impact on phylogenetic analyses. Trends Eco Evol. 1996;11(9):367–372. [PubMed: 21237881]
- 26.
- Steel M, Huson D, Lockhart P. Invariable sites models and their use in phylogeny reconstruction. Syst Biol. 2000;49(2):225–232. [PubMed: 12118406]
- 27.
- Muse SV, Gaut BS. A likelihood approach for comparing synonymous and nonsynonymous nucleotide substitution rates, with application to the chloroplast genome. MBE. 1994;11(5):715–724. [PubMed: 7968485]
- 28.
- Yang Z, Goldman N, Friday A. Comparison of models for nucleotide substitution used in Maximum-likelihood phylogenetic estimation. MBE. 1994;11(2):316–324. [PubMed: 8170371]
- 29.
- PHYLIP [computer program]. Version 3.6 Seattle: Department of Genome Sciences, University of Washington,2002 .
- 30.
- Posada D, Crandall KA. Selecting models of nucleotide substitution: An application to human immunodeficiency virus 1 (HIV-1). MBE. 2001;18(6):897–906. [PubMed: 11371577]
- 31.
- Huelsenbeck JP, Crandall KA. Phylogeny estimation and hypothesis testing using maximum likelihood. Ann Rev Ecol Syst. 1997;28:437–466.
- 32.
- Posada D, Crandall KA. A comparison of different strategies for selecting models of DNA substitution. Syst Biol. 2001;50(4):580–601. [PubMed: 12116655]
- 33.
- Posada D, Crandall KA. Modeltest: Testing the model of DNA substitution. Bioinformatics. 1998;14(9):817–818. [PubMed: 9918953]
- 34.
- Kelsey CR, Crandall KA, Voevodin AF. Different models, different trees: The geographic origin of PTLV-I. Mol Phylogenet Evol. 1999;13(2):336–347. [PubMed: 10603262]
- 35.
- Felsenstein J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution. 1985;39:783–791. [PubMed: 28561359]
- 36.
- Hillis DM, Bull JJ. An empirical test of bootstrapping as a method for assessing confidence in phylogenetic analysis. Syst Biol. 1993;42:182–192.
- 37.
- Erixon P, Svennnblad B, Britton T. et al. Reliability of bayesian posterior probabilities and bootstrap frequencies in phylogenetics. Syst Biol. 2003;52(5):665–673. [PubMed: 14530133]
- 38.
- Cummings M, Handley S, Myers D. et al. Comparing bootstrap and posterior probability values in the four-taxon case. Syst Biol. 2003;52(4):477–487. [PubMed: 12857639]
- 39.
- Alfardo M, Zoller S, Lutzoni F. Bayes or bootstrap? A simulation study comparing the performance of bayesian markov chain monte carlo sampling and boostrapping in assessing phylogenetic confidence. MBE. 2003;20(2):255–266. [PubMed: 12598693]
- 40.
- Douady C, Delsue F, Boucher Y. et al. Comparison of bayesian and maximum likelihood bootstrap measures of phylogenetic reliability. MBE. 2003;20(2):248–254. [PubMed: 12598692]
- 41.
- Posada D, Crandall KA. Intraspecific gene genealogies: Trees grafting into networks. Trends Ecol Evol. 2001;16(1):37–45. [PubMed: 11146143]
- 42.
- Crandall KA. Intraspecific cladogram estimation: Accuracy at higher levels of divergence. Syst Biol. 1994;43(2):222–235.
- 43.
- Crandall KA. Intraspecific phylogenetics: Support for dental transmission of human immunodeficiency virus. J Virol. 1995;69(4):2351–2356. [PMC free article: PMC188907] [PubMed: 7884881]
- 44.
- Templeton AR, Crandall KA, Sing CF. A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data. III. Cladogram estimation. Genetics. 1992;132:619–633. [PMC free article: PMC1205162] [PubMed: 1385266]
- 45.
- Clement M, Posada D, Crandall KA. TCS: A computer program to estimate gene genealogies. Molecular Ecology. 2000;9:1657–1659. [PubMed: 11050560]
- 46.
- Crandall KA. Multiple interspecies transmissions of human and simian T-cell leukemia/lymphoma virus type I sequences. MBE. 1996;13(1):115–131. [PubMed: 8583886]
- 47.
- Crandall KA, Templeton AR. Empirical tests of some predictions from coalescent theory with applications to intraspecific phylogeny reconstruction. Genetics. 1993;134:959–969. [PMC free article: PMC1205530] [PubMed: 8349118]
- 48.
- Castelloe J, Templeton AR. Root probabilities for intraspecific gene trees under neutral coalescent theory. Mol Phylogenet Evol. 1994;3(2):102–113. [PubMed: 8075830]
- 49.
- Ferber D. Monkey virus link to cancer grows stronger. Science. 2002;296:1012–1015. [PubMed: 12004103]
- 50.
- Crandall KA, Posada D. Phylogenetic approaches to molecular epidemiologyIn: Leitner T, ed.The Molecular Epidemiology of Human Viruses: Kluwer Academic Publishers 200225–39.
- Phylogenomics and Molecular Evolution of Polyomaviruses - Madame Curie Bioscienc...Phylogenomics and Molecular Evolution of Polyomaviruses - Madame Curie Bioscience Database
- Rho Family GTPases and Cellular Transformation - Madame Curie Bioscience Databas...Rho Family GTPases and Cellular Transformation - Madame Curie Bioscience Database
- Deciphering the Complex Enzymatic Pathway for Biosynthesis of Wyosine Derivative...Deciphering the Complex Enzymatic Pathway for Biosynthesis of Wyosine Derivatives in Anticodon of tRNAPhe - Madame Curie Bioscience Database
- Tests of a Stereochemical Genetic Code - Madame Curie Bioscience DatabaseTests of a Stereochemical Genetic Code - Madame Curie Bioscience Database
- Origins and Evolution of Cotranslational Transport to the ER - Madame Curie Bios...Origins and Evolution of Cotranslational Transport to the ER - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...