

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

In this chapter, polyomaviruses will be presented in an immunological context. Principal observations will be discussed to elucidate humoral and cellular immune responses to different species of the polyomaviruses and to individual viral structural and regulatory proteins. The role of immune responses towards the viruses or their proteins in context of protection against polyomavirus induced tumors will be described. One central aspect of this presentation is the ability of polyomaviruses, and particularly large T-antigen, to terminate immunological tolerance to nucleosomes, DNA and histones. Thus, in the present chapter we will focus on clinical, experimental and theoretical aspects of the immunity to polyomaviruses.
Introduction
Polyomaviruses are small naked viruses with an icosahedral capsid and a circular dsDNA genome. The name polyomavirus derives from early observations that these viruses may cause tumors when inoculated into newborn mice. Polyomaviruses are widespread among vertebrates, but the different species have a narrow host and cell range. Two polyomaviruses solely infecting humans were discovered in 1971 by two independent groups. Gardner et al1 originally described BK virus (BKV) in the urine of a renal transplant patient, while JC virus (JCV) was isolated and partially characterized by Padgett et al2 from the brain tissue of a patient with progressive multifocal leukencephalopathy. A third species, Simian virus 40 (SV40) was discovered already in 1960, and was originally discovered as a contaminant of inactivated Salk polio vaccine3 that was distributed world-wide in the years between 1955 and 1963.4
The association between polyomavirus infection and immunity to the viral particles and their polypeptides has been determined in worldwide epidemiological surveys. This aspect has been studied extensively over the past 3-4 decades following the detection of BKV, JCV and SV40, the species dealt with in this chapter. These studies span from determination of humoral immunity against the virus in context of monitoring seroepidemiology, over humoral immunity of individual viral proteins, to T cell mediated immunity. A less known, but potentially important role of the polyomaviruses resides in the ability of polyomavirus encoded proteins to bind host autologous molecules and thereby induce antigen-selective autoimmunity to p53, histones and double-stranded (ds)DNA. This will be described in detail in the last part of this chapter.
Polyomaviruses — Structure and Genomic Organization
To understand how polyomaviruses induce immunity and autoimmunity, a short description of the genomic and structural organization of the virus particle is essential.
BKV is a nonenveloped virus with an icosahedral capsid, and the virions are roughly made up of 88% proteins and 12% DNA. The viral genome consists of a single copy of a circular dsDNA molecule of approximately 5,300 base-pairs. The BKV genome shares 75% overall homology with JCV and 70% homology with the SV40 genome (reviewed in ref. 5). During virus replication, newly replicated viral DNA associates with host cell histones to form minichromosomes. Hence, in mature virions, viral DNA is complexed with the cellular histones to form a nucleosomal structure,6,7 the core mononucleosome thus containing an octamer of histones H2A, H2B, H3 and H4, analogous with the structure of nucleosomes in mammals.8-10 On the average, each polyomavirus genome contains 21 nucleosomes.6
The viral circular genome can be divided into three functional regions (fig. 1): (i) the so-called early region encodes the regulatory proteins large tumor antigen (large T-antigen, in this chapter denoted T-ag) and small tumor antigen (small t-ag); (ii) the late region containing the genetic information for the structural (capsid) proteins VP1, VP2, VP3 and the agnoprotein; and (iii) the noncoding control region (NCCR) which harbor the origin of replication and the sequences involved in the transcriptional regulation of both the early and the late genes.
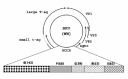
The early region is expressed at the initiation of the viral replication. T-ag is a multifunctional protein with distinct domains fulfilling different roles. Major crucial properties are its helicase activity and its ability to bind host cell regulatory proteins like the retinoblastoma protein family, p53 and others (for review, see refs. 5,11). These activities are important as they may control cell function and undermine the infected cell's destiny for apoptosis. Thus, T-ag controls both viral DNA replication, early and late gene transcription, and interferes with host cell transcription factors.12
The capsids are composed of 72 capsomers each consisting of the three major structural proteins, VP1, VP2 and VP3. For intact, infectious polyomaviruses, VP1 is the dominant solvent phase protein, and is the most redundant protein of the capsid and accounts for 70-80% of the total protein mass of the virus particle.5 In fact, VP1 can self-assemble into virus-like particles in vitro.13 Therefore, antibodies against this protein dominate immune responses to polyomaviruses, at least as detected in assays using intact virus particles as antigens, e.g., in hemagglutination inhibition assay (HI, an assay using antibodies to inhibit the potential for polyomaviruses BKV and JCV to agglutinate human blood group 0 erythrocytes), enzyme linked immunosorbent assay (ELISA), or immune electron microscopy. Important from an immunological point of view is the fact that the capsid proteins VP1-3 also bind DNA directly or indirectly. This is basically depending on two features of the polypeptides: i. due to nuclear localization signals present in the capsid proteins, and ii. to the fact that these proteins may associate directly with DNA structures or with other nucleosome/chromatin bound proteins like transcription factors.5,14-17 In an immunological context, this information is important, as it may provide an explanation as to why and how polyomavirus infection may transform host cell nucleosomes immunogenic from a natural state of nonimmunogenicity of these structures.18
Serological studies have revealed that there exist several subtypes of BKV. The heterogeneity is due to differences in the sequences of the amino acids 61-83 of VP1.19 The highest genetic diversity between different BKV isolates is, however, found in the NCCR. While the NCCR of the archetypal BKV strain WW has a linear arrangement of the transcription factor binding sequence blocks O-P-Q-R (fig.1), deletions, duplications and rearrangements have been reported in the NCCR of other BKV strains. It is generally believed that this polymorphism in the VP1 and NCCR may offer advantages to the virus in its host.5
Immunology of Polyomaviruses
Except for seroepidemiological studies and observations in polyomavirus large T-ag transgenic mice, there is little information about the role of innate and adaptive immunity related to these viruses. The main interest in this regard has been linked to humoral immune responses to trace the distribution of each of the polyomavirus species among human individuals, to try to establish the role of T cells in fighting the virus and to establish latent infection, and the role of virus-encoded proteins in the generation of autoimmunity to nucleosomes and DNA. Few studies describe how the innate immune system handle this virus, and which receptors on antigen-presenting cells (APC) bind this virus, and whether this interaction results in up-regulation of costimulatory signals necessary for full activation of the adaptive immune system. In the next sections, seroepidemiology of BKV, JCV and SV40, T cell recognition of virus-derived proteins and their role in initiating autoimmunity will be discussed.
Seroepidemiology of Polyomaviruses
Since these viruses were detected, their prevalence in the population has been thoroughly investigated. As all these viruses have been shown to inherit the potential to establish latent infections20 and to induce tumors, at least in heterologous hosts (reviewed in ref. 21), it became important to study their prevalence in the human populations globally and locally to establish a base of information potentially important for studies of their clinical impact. Also, to perceive information of their distribution in the human population, an origin could be created to detect new possible diseases caused by such viruses based on cell tropism, cytopathogenic and oncogenic potential, and, as recently has been demonstrated, their potential to terminate immunological tolerance to DNA and to nucleosomes.18,22-25
Already the first studies demonstrated a high incidence of BKV and JCV in different populations, 26-29 although the incidence in remote areas could differ from this generalized picture.30 There exist a large body of studies and literature covering this field (see e.g, refs. 13, 19, 31-33). In the context of this chapter, important aspects to understand the role of immunity to the polyomaviruses will be discussed.
Cross-Reactivity of Human Polyomavirus Antibodies
A high degree of amino acid sequence identity exists between the functional and structural proteins of the human polyomaviruses SV40, BKV, and JCV. Amino acid sequence identity varies between 58% up to 83% when comparing the different corresponding proteins of BKV (Dunlop), JCV (CY) and SV40 766 strains (see Table 1). Stretches of 10 consecutive or more identical amino acids were found in several regions of the distinct proteins (fig. 2). Because of the high degree of identity between SV40, BKV, and JCV proteins, antibodies raised against polyomaviruses proteins have the potential to cross-react (reviewed in refs. 19, 34). Results of seroepidemiological studies to determine the prevalence of antibodies against a certain species among human polyomaviruses should therefore be interpreted with care as serum antibodies can react with all three human polyomavirus species.
Table 1
Amino acid identity between the proteins of BKV (Dunlop strain, accession number V01108), JCV (CY strain, accession number AB038249) and SV40 (766 strain, accession number J02400).

Seroepidemiology of BKV
Serological data obtained over 30 years demonstrate that infection with BKV is established in early childhood, and occurs globally with similar frequency in industrial and developing countries. Several immunology-based assays have been developed to monitor the infectivity of BKV. Of these, HI,26,35 complement fixation,26 indirect immunofluorescence (IIF),36 RIA,37 ELISA,38,39 and western blot40 have been the most widely used assays. These tests may have quite different sensitivities, and may detect different spectra of virus antibodies, a fact that could affect the interpretation of the prevalence determined in the different studies. Surprisingly, in light of the different assays used for antibody determination, and the technical development of sensitive assays for antibody detection over the last three decades, the observed prevalence for BKV in different populations is consistently high (see Table 2 for examples). Comparing two studies, one early of Gardner et al in 1973,26 and one late of Stolt et al in 2003,41 determining the prevalence for BKV antibodies in different age groups, the results are surprisingly consistent and comparable (Table 3), although performed with different assays (HI versus ELISA using recombinant viral proteins assembled into virus-like particles) to detect the antibodies, and timely distant from each other (30 years).
Table 2
Examples of data on prevalence of BK virus in the human population. Data are selected that compare techniques and time for antibody detection.
The prevalence of BKV varies in different studies between 40 and 95%. The overall differences between the reports cannot be explained by the use of different assay procedures, as results determined by HI spans from 40% (data published in 197642) to 94% (197443). These prevalences, obtained with HI, covers the figures obtained with most of the assays used in later studies. One explanation for this diversity may be that the cut off levels of the assays are not standardized internationally, and that exchange of sera for inter-laboratory standardization has not been performed or thoroughly documented. However, due to the nature of the study, one should expect differences in the prevalence of anti-BKV antibodies in different more or less well defined human populations. Analyzing the results collected over time, the discrepancies cannot be explained by improvements of the assays from early to late studies (see examples of data presented in Tables 22 and 3 collected over 30 years).
Table 3
Comparison of age dependent prevalence of antibodies to BKV and JCV using hemagglutination inhibition (26) or ELISA using virus-like particle (VLP) containing recombinant BKV or JCV VP1 (41).
The antibodies recognizing the whole viral particle (presumable dominated by antibodies to the solution phased VP1, see above) seem to persist once the immune system is stimulated. This is strengthened by the fact that the detection of infectious particles has a much lower prevalence than that described above for anti-BKV antibodies (see Table 4). Antibodies to polyomavirus T-ag are relatively rare, although most individuals are infected with the virus, indicating that anti-T-ag antibodies are more closely linked to episodes of productive infections. In one situation we could directly trace this intimate and timely association between productive infection and transient humoral immune response to T-ag.23 In that study, urinary secretion of polyomavirus DNA sequences was monitored weekly in 20 Systemic Lupus Erythematosus (SLE) patients over one year, and development of anti-T-ag antibodies was determined. There was an overall strong correlation between presence of anti-T-ag antibodies and productive infection as judged by BKV DNA PCR-positive urine samples. In one patient, the production of anti-T-ag antibodies coincided with the appearance of urinary BKV DNA sequences. As the BKV DNA sequences disappeared from the urine, the anti-T-ag antibody fainted over the next 6-8 weeks. This is consistent with the very low incidence of anti-T-ag antibodies in healthy individuals (<1%) we44,45 and others46 have detected. Thus, as VP antibodies seem to reflect an accumulative incidence of latent BKV infection, antibodies to T-ag seem to indicate present or recent productive infection with significant expression of T-ag as is essential for virus replication to take place.5,11 Summarizing data on prevalence of antibodies to structural proteins and to T-ag, it is likely that antibodies to the structural proteins VP1-3 can be used to determine accumulated prevalence of the sum of active and latent polyomavirus infections, while antibodies to T-ag can potentially be used to trace prevalence of productive infection.
Table 4
Prevalence of human polyomavirus replication in healthy individuals as determined by the presence of viral proteins, DNA sequences or virions in urine samplesa.
Seroepidemiology of JCV
As for BKV, seroepidemiology of JCV has been determined over the 3 decades since their detection. The first published study came from Padgett and Walker already in 1973,27 two years after the original description of the virus. In this study, different age groups were examined (3, data organized according to Walker and Padgett47 from ref. 27) and the anti-JCV antibodies had roughly the same age-related profile as that seen for BKV antibodies (3).26,47 Stolt et al determined the seroepidemiology of JCV using recombinant virus-like particles consisting of JCV VP1 as antigen in IgG-specific ELISA.41 In their study, antibodies to JCV peaked around the age of 9-11 years, and the accumulative incidence in humans aged 1-13 was 32% (3), while in the early study of Padgett and Walker,27,47 the accumulative incidence, using HI, in the age group 1-14 years was 28%. These results are strikingly consistent taken into account that the studies are performed with very different antibody assays (HI, versus VP1-specific ELISA, Table 3). and approximately 30 years apart. However, no attempts to ensure that the antibodies detected are specific for either BKV or JCV (or SV40 for that case) were undertaken.
That the frequency of the different polyomavirus species as determined by serological assays differs among humans is not an argument for species-specific virus antibodies, but may simply reflect the different distribution of the viruses. To ascertain that the antibodies indeed recognize one or another of the virus species, inhibition experiments should have been performed. In the work by Stolt et al, the authors describe different prevalence of BKV and JCV with lower frequency in the age groups of JCV in the same sera, indicating some species-specificity of the antibodies. Furthermore, in a report by Hogan et al,48 where they investigated seroconversion in renal transplant patients, more of the patients seroconverted for JCV than for BKV. This directly demonstrated that at least in some individuals antibodies are produced that specifically recognize JCV.48 Such results indicate presence of antibody subpopulations in the sera recognizing one single polyomavirus species, but do not rule out that antibodies may also recognize determinants shared by the different viruses. In one study, Taguchi et al49 determined the prevalence and age of acquisition of antibodies against JCV and BKV. About 50% of the children in this study acquired antibodies against BKV by 3 years of age and against JCV by 6 years of age. These results indicate that dual latent infections with both viruses are common, and that antibodies can be used to determine seroconversion of either of the viruses.49 More work is, however, needed to determine whether polyomavirus antibodies cross-react over the species barriers, as recently discussed by Knowles.34 This information is important to exactly determine the prevalence of each individual polyomavirus in the human population.
Seroepidemiology of SV40
Although originally described as a natural habitant of Asiatic macaques, SV40 can infect humans.50-52 Recovery of infectious SV40 virions indicates an established infection and implies that humans may function as a natural host for this polyomavirus. For almost a decade (1955-1963), millions of children worldwide were administered SV40-contaminated poliovirus vaccines and therefore it is not surprising that antibodies against SV40 could be present in the human population. However, seropositive individuals are also found amongst those that never received contaminated poliovirus vaccines.
Recent seroepidemiological studies using different detection methods by different groups revealed that approximately 10% (range 0-12%) of sera obtained from individuals around the world have low titer antibodies to SV40. No significant differences were found in the seropositive prevalence of sera obtained from individuals that most probably had received contaminated vaccine compared to those that were vaccinated with SV40-free vaccines.13,53-61 These results are in good agreement with older studies on SV40 seropositivity (3-13%) in individuals that had never received contaminated vaccines (see ref. 60).
These antibody titers may be low due to either limited viral replication in the human host or failure of the human system to recognize and respond robustly to SV40 infections.62 Alternatively, the low titers and low prevalence of SV40 antibodies may be due to cross-reactivation with the other human polyomaviruses BKV and JCV. Indeed, the study by Carter and colleges detected SV40 antibodies in 46 out of 699 serum samples tested using a virus particle-like ELISA. However, all these samples were also positive for BKV and JCV and in fact these antibodies were cross-reacting with these other polyomaviruses.57 Of a total of 3669 serum samples tested by Minor and collaborators, 187 were SV40 seropositive, but just one serum had only antibodies against SV40, suggesting that most seropositive individuals have cross-reactive antibodies generated by infection with human polyomaviruses BKV and/or JCV. However, this only positive sample argues against that all SV40 seropositive individuals are the result of cross reactivity.61 Because of these problems, results of serological studies should be interpreted carefully and be supplemented with data on the presence of SV40 DNA as an evidence of the presence of this virus in the normal human population. The low prevalence of SV40 viruria in this population (1%, see Table 4). reflects the low SV40 seropositive rates and argues against frequent presence of current SV40 infection in healthy individuals.
The origin of antibodies to SV40 remains to be explained but data from several groups suggest that these antibodies do not solely arise from exposure to SV40-contaminated poliovirus vaccines.54,60,61 A highly specific serological assay for SV40 is required for unambiguous assessment of SV40 prevalence in the human population.
Epidemiology of Productive Polyomavirus Infection in Healthy Individuals
After primary infection in early child-hood, BKV establishes a life-long latent infection in immunocompetent individuals. This latent infection in healthy individuals is supported by the low prevalence (3.6%, n=300) of BKV IgM antibodies in healthy adult blood donors63 and the low prevalence of BKV viral proteins, virions, or viral nucleic acid sequences in urine samples of immunocompetent individuals. BKV replication as evidence for reactivation has been monitored by the presence of virions as determined by electron microscopy or by Decoy cells (i.e., polyomavirus-laden uroepithelial cells), which are hallmarks of BK virus replication, or by propagation of urine samples on permissive cell cultures. In recent years, PCR has been applied to detect BKV nucleic acid sequences (reviewed in ref. 64). Using these methods, about 1% of healthy controls (n=19,845) showed signs of BKV productive infection (Table 4). JCV IgM antibodies were detected in 15% of healthy blood donors in England65 and there was a positive association between JCV seropositivity and age.34 Thus JCV immunity may be boosted throughout life by persistent infection, reactivation or reinfection. The higher prevalence of JCV IgM antibodies in immunocompetent individuals corresponds well with higher prevalence of JCV viruria in this human population. Almost 35% (n=870) of normal individuals showed PCR-based signs of JCV viruria, and there was a higher incidence of urinary JCV excretion in older individuals.66 The prevalence of SV40 seropositive individuals in healthy blood donors varied between 1.3-5% throughout all age groups examined.34 In accordance, urinary SV40 was detected in 1% (n=100) in immunocompetent persons (Table 4). All the epidemiological data taken together (seroepidemiology and epidemiology of productive infection) indicate that most human individuals are latently infected with polyomaviruses, while productive infection is rare among healthy individuals, and that antibodies to T-ag have an incidence similar to that of productive infection. This points at two phenomenona: anti-polyomavirus antibodies (presumably against VP1-3) are stable and long-lasting, while anti-T-ag antibodies seem to faint, as the productive infection is terminated, consistent with observations described above.23,44,67
Polyomaviruses and T Cell Responses
Antigen Processing and Viral Infection
To establish relevant and protective immune responses to a viral infection, peptides derived from viral proteins need to be presented to two main effector T cell populations, CD8+ and CD4+ T cells. The former are meant to kill virus-infected cells, while the latter provide help for B cells to produce anti-viral antibodies. Both systems are important in defense against viral infections.
The critical event in antigen recognition by T cells is the way antigenic peptides are presented and thus recognized by the T cell receptor (TCR). The central molecules in this presentation are the foreign antigen peptide and MHC molecules with two distinct classes: MHC class I and MHC class II. These two MHC classes deliver peptides to the cell surface from two different intracellular compartments reflecting the origins of the antigens that are processed; either intracellular, or taken up from the outside of the cell. MHC molecules are glycoproteins that localize to the cell surface. These two classes of MHC molecules differ both in function, structure and cell distribution. While MHC class I is present on all nucleated cells, MHC class II are present mainly on specialized antigen-presenting cells.
The peptides that bind to MHC class I molecules classically derive from virus-encoded proteins. Such proteins are generated by translation in the cytosol and transported to the endoplasmic reticulum (ER) by transporter proteins called Transporters associated with antigen Processing-1 and 2 (TAP1 and TAP2),68,69 where they associate with MHC class I complex. At this stage, the fully folded MHC class I-peptide complex is released from the TAP complex in the ER and is transported to the plasma membrane, where the peptides are presented to appropriate CD8+ T cells.
The antigen peptides that bind to MHC class II enter the cell by endocytosis and are arrested in the endosomes. This occurs in specialized APC, although not exclusively as e.g., nonprofessional APC may also present peptides in context of MHC class II molecules (see below). Endosomal and lysosomal proteases are activated by low pH, and thereby degrade the proteins into peptides, which now are ready to be bound by MHC class II molecules.70 Due to presence of peptides in this compartment that are not generated from (antigenic) proteins taken up by the cell, the peptide-binding cleft of the folded MHC class II molecules is protected from binding unwanted peptides through transient binding of the invariant chain (Ii). The Ii binds noncovalently to MHC class II71 and prevents binding of irrelevant (autologous) peptides. A second function of the invariant chain is to target the MHC class II-invariant chain complex to the endosomal compartment where the invariant chain is degraded by acidic proteases in successive steps and replaced by the peptides that will be presented at the cell surface (see refs. 70,72 for more details).
Thus, MHC class I-peptide complexes activate CD8+ T cells committed to kill e.g., virus-infected cells, while MHC class II-peptide complexes activate CD4+ T cells aimed at activating macrophages (Th1 cells) or B cells (Th2 cells). Both these arms of antigen presentation are operational in context of polyomavirus infections.
Role of T Cells in Polyomavirus Infection
From the nature of the immune system and its activation, both the innate and the adaptive immune systems are engaged in defense against virus infection. The innate system is important for proper presentation of antigenic peptides, and to provide appropriate costimulatory molecules to ensure activation of T cells recognizing the infectious-derived peptides. Costimulation is necessary for both CD4+ and CD8+ T cells. According to Janeway and Medzhitov,73-77 polyomaviruses belong to the group of infectious nonself agens delivering pathogen-associated molecular patterns (PAMP) that are recognized by cell surface pattern recognition receptors (PRR) on APC.78 Interaction between PAMP and PRR may be required for up-regulation of costimulatory molecules like CD80 and CD86. It is, however, not known by which pattern polyomaviruses may increase expression of these molecules on APC. However, one study may be relevant. Velupillay et al79 investigated the role of the innate immune system using the PERA/Ei mouse strain (PE mice), which is highly susceptible to tumor induction by polyomavirus. This susceptibility can be transmitted in a dominant manner in crosses with resistant C57BR/cdJ mice (BR mice). The authors demonstrated that PE and F1 mice infected by polyomavirus responded by increased costimulatory molecule B7.2 (CD86) expression on APC, whereas BR mice responded with increased expression of B7.1 (CD80) molecules.79 A system like this may, if pursued, provide information about the underlying processes determining if CD80 or CD86 are increasingly expressed.
T cell immunity to polyomaviruses has been studied in quite different natural and experimental conditions. Most of these studies focus on T-ag, and investigations of T cell responses to other virus-encoded proteins are rare. T-ag is, however, suitable to examine, as it is highly immunogenic and induces T cell as well as B cell responses.22,23,33,80-83 We have characterized T cell responses to T-ag in randomly selected healthy humans and in SLE patients. By stimulating peripheral blood mononuclear cells (PBMC) from these with purified SV40 T-ag, virtually all responded by T cell proliferation, and T cell lines could be established by T-ag and nucleosome-T-ag complexes.45,82,84 Since T-ag from BKV, JCV and SV40 demonstrate 73%-83% amino acid homology (see above), the data obtained using SV40 T-ag probably do not reflect a high incidence of SV40 infected individuals, but more likely that most individuals have cross-reactive T-ag-specific memory T cells. The T cell responses in this system were dominated by CD4+ T cells, as demonstrated by a substantial decrease in proliferation when adding anti-human CD4 antibodies to the cultures prior to antigenic stimulation. Using anti-CD8 antibodies in this system resulted in a weak reduction of T cell proliferation. Similar results were observed in mice immunized with SV40 T-ag.83 The CD4+ T cell lines established had the potential to induce weak anti-T-ag antibody production in vitro when cocultured with highly purified autologous B cells.82 Drummond et al85 found that all of healthy seropositive individuals had T cells proliferating in vitro in response to antigens prepared from BKV-infected fibroblasts. Seronegative individuals did not harbor such reactive T cells. Although not definitively established, some of these T cells may be CD4+, since they correlated with presence of antibodies to BKV.85
In one artificial study by Bates et al, the role of CD8+ T cells in abrogating SV40 infectious cycle in vitro has been the focus.86 In that study, they showed that SV40-encoded T-ag translocated into the cell membrane in addition to the nucleus of SV40-infected permissive monkey cells. The surface T-ag in SV40-transformed mouse cells provided a target for the cytotoxic T lymphocytes (CTL) which recognized SV40 T-ag in association with experimentally transfected murine K/D, (MHC) class I H-2 antigens. Treatment of SV40-infected TC-7/H-2Db and TC-7/H-2Kb with T-ag specific CD8+ T cell clones abrogated the virus lytic cycle. This opened for the possibility that this could take place also in vivo, and that CD8+ T cells could remove polyomavirus-infected cells.86
Additional studies have been performed in determining T cell mediated immunity against T-antigen induced tumors. This field has recently been extensively reviewed by Tevethia and Schell,33 and principal observations only will be summarized here. It has been known for a long time that polyomaviruses and T-ag of these viruses can induce tumors. Early evidence that SV40 encoded antigens could serve as a tumor specific antigen was provided by Tevethia et al in 1980, as they demonstrated that prior immunization of hamsters with SV40 particles, or SV40 transformed cells induced T cell-mediated resistance to a subsequent tumor challenge (reviewed in ref. 33). Interestingly, in constitutive T-ag transgenic mice, this protection against tumorogenesis was not obtained. While T-ag has the potential to induce tumors in diverse organs relative to the organ-specificity of the promoters introduced, T cell immunity to T-ag does not develop in these mice. This is most probably due to tolerance development, either central or peripheral.
An interesting demonstration of this was provided in our laboratory when developing T-ag transgenic mice under the control of a tetracycline-responsive transcriptional activator (tTA). We tested two opposite tetracycline-dependent transgenic systems, i. gene activation in the absence of tetracycline (tet-off ), and ii. activation in the presence of tetracycline (tet-on). In control experiments, we examined the tendency for leakage of the T-ag gene by direct visualization of T-ag under nonexpressing conditions, and tested T cell responses after turning the gene on. In the tet-on system, spontaneous expression was observed in absence of tetracycline, as determined by RT-PCR and immune electron microscopy, and these mice did not respond to immunization with T-ag. The tet-off system was tight, and in the nonexpressing situation, the mice responded to T-ag immunization by producing antibodies to T-ag, and T cells responded in vitro to T-ag by vivid proliferation (manuscript in preparation).
Thus, tumorogenesis in constitutively expressing T-ag transgenic mice cannot be inhibited by the immunity to T-ag in such mice, simply due to tolerance development. In an interesting study by Ye et al87 a correlate to this observation was done in unconditioned T-ag transgenic mice. If the promoter used to control T-ag expression (the RIPI T-ag2 mouse) allowed expression in embryonic life, T cell tolerance developed. If another promoter was selected (the RIPI T-ag4 mouse), this situation changed as expression started several weeks after birth. These mice were immunologically responsive to T-ag. Furthermore, immunization of these mice with T-ag by SV40 infection delayed the T-ag induced tumor growth by up to one year. When the tumors in such mice appeared, the T cell responses to T-ag had fainted.87 In RT3 mice, expressing T-ag as a transgene controlled by the insulin promoter, crossed with H2-Kk-restricted TCR transgenic mice, CD8+ T cells developed normally, and did not possess tolerance, even after several months of observations.88,89 As above, this may be due to the prolonged delay after birth for the transgene to be expressed. Extrapolating from these data, T cells may become autoantigenic to determinants expressed a substantial (not exactly defined) time interval after birth.89 These few examples demonstrate that tumors induced by polyomavirus T-ag may be suppressed if CD8+ T cells specific for T-ag are not rendered tolerant. This may occur through expression of T-ag before immune competency is reached, i.e., during fetal life. From this, one may speculate whether acquired latent infection with potentially oncogenic polyomaviruses may perpetuate T cell mediated immunity to e.g., T-ag that may protect against tumor development in the infected organism. This may actually explain why it is so difficult to equivocally prove the oncogenic potential of polyomaviruses in (at least healthy) humans.90
The role of CD4+ T lymphocytes in immunity to SV40 induced tumors has been less studied, but is thought to provide help for MHC class I-restricted CD8+ cytotoxic (anti-tumor) T lymphocytes. These latter T cells, according to e.g., observations referred to above, seem to serve as the predominant effector cell in killing tumor cells. In a recent study, Kennedy et al91 evaluated the role of T lymphocyte subsets in tumor immunity induced by recombinant SV40 T-ag within an experimental murine pulmonary metastasis model of SV40 T-ag-expressing tumors. By depleting BALB/c mice of either CD4+ or CD8+ T cells in the induction phase of the immune response to SV40 T-ag, indications were found that CD4+ T cells but not CD8+ T cells were critical in the production of antibodies to SV40 T-ag and in tumor immunity after SV40 T-ag immunization. Among the anti-T-ag antibodies, IgG1 was the dominating IgG subclass, indicating that Th2 type T helper cells were involved. Those results suggested that CD4+ T cells, along with antibody responses, indeed may play a role in the induction of tumor immunity to e.g., an SV40 encoded tumor antigen.
In another polyomavirus-mediated disease, JCV can reactivate and cause progressive multifocal leukencephalopathy (PML), a fatal demyelinating disease of the central nervous system. For this to develop, deficit of cell-mediated immunity must occur, which seems to be the case in acquired immunodeficiency syndrome (AIDS), malignancies or in organ transplant recipients. The humoral immune response, measured by the presence of JCV-specific IgG antibodies in the blood or intrathecally, as detected in the cerebrospinal fluid, is inversely related to progression of PML. Consistent with the underlying immunosuppression, the proliferative response of CD4+ T lymphocytes to mitogens or JCV antigens is reduced in PML patients.92 CD8+ cytotoxic T lymphocytes recognize intracellularly synthesized viral proteins in context of MHC class I molecules (see above). One JCV peptide, the VP1(p100) ILMWEAVTL, has been characterized as a cytotoxic T cell epitope in HLA-A *0201 positive PML survivors. Studies demonstrated that VP1(p100)-stimulated peripheral blood mononuclear cells from 5 out of 7 PML survivors had JCV-specific cytotoxic T cells, versus none of 6 PML progressors. The cellular immune response against the VP1(p100) peptide may therefore be crucial in the prevention of PML disease progression (reviewed in92).
In another recent study, Gasnault et al confirmed these results93 by demonstrating that all of nine healthy donors and seven of thirteen nonPML HIV-infected patients with urinary JCV excretion had positive JCV-specific CD4+ T cell responses. No significant response was found in 14 patients with active PML, while nine of 10 PML survivors had positive responses. A restoration of JCV-specific CD4+ T cell responses was associated with JCV clearance from the cerebrospinal fluid. Thus, JCV-specific CD4+ T cell responses appeared also in this study to play a critical role in the control of cerebral JCV infection, preventing PML development. Such responses can be restored in PML survivors possibly following effective and prolonged antiretroviral therapy.
From these few examples, it may be clear that both CD4+ and CD8+ T cells are important in establishing control of polyomavirus induced tumor progression and metastasis, and in controlling other consequences of productive polyomavirus infection like PML. A possible link between latent polyomavirus infection and protective immunity controlling tumorogenesis is important to establish. To eventually settle this, well planned prospective studies must be performed particularly comparing seropositive healthy individuals that acquire immune deficiencies with normal seropositive healthy controls. T cells operational in context of polyomavirus-related induction of autoimmunity to DNA and nucleosomes will be discussed in relevant sections below.
Polyomaviruses, SLE and Autoimmunity to Nucleosomes and dsDNA
Systemic Lupus Erythematosus (SLE) and Anti-dsDNA Antibodies
SLE, the prototype of a systemic rheumatic syndrome, is characterized by production of a wide array of autoantibodies.94 Dominant proportions among these are antibodies directed against nuclear constituents95 (antinuclear antibodies, ANA). Aside from their diagnostic importance, subpopulations of ANA, especially those binding dsDNA may have the additional effect as initiators of glomerulonephritis typical for this autoimmune syndrome.96-103 Not all anti-dsDNA antibodies are involved in SLE nephritis, but those that are nephritogenic seem to recognize mammalian dsDNA, although some reports indicate that antibodies specific for nucleosomes also may have nephritogenic potential.99,104-106
The ethiology of SLE is, although the disease has been described for at least 15 centuries ago,107 an unresolved matter. One may, according to the pleotropic picture of the disease, question whether SLE represents one disease entity, or represents a continuous overlap of individual, etiologically unrelated, organ manifestations (discussed in ref. 25). Hence, the term SLE may theoretically represent a heading for a wide variety of intrinsically unrelated disease manifestations, explaining the highly diverse picture of the disorder,108 and may therefore principally be meaningless to use to define a single disease entity. On this background, it is important to try to understand at least the molecular and cellular origin of one of the major pathogenic and disease modifying factors characterizing this disease, namely antibodies to dsDNA. In this section, the cellular and molecular impact of polyomaviruses and polyomavirus-encoded DNA binding proteins on production of anti-DNA antibodies will be discussed in terms of their direct and measurable effect on initiation and sustained production of this antibody population. We have during recent years continuously developed in vivo and in vitro experimental systems employing intact BKV or SV40 T-ag to describe processes that have shed light on this enigmatic autoimmune response (reviewed in refs. 22,24,25,109,110).
B Cells Specific for dsDNA Can Be Activated by Polyomaviruses—the Phenomenon
This research program was initiated in 1986 based on an unintended observation done by Christie and colleagues in their attempts to induce antibodies to BKV in rabbits.40 Their main focus was to develop an ELISA to trace polyomavirus antibodies in humans. After deliberate immunization of rabbits with purified, infectious BKV particles, development of serum antibodies were assayed by several detection methods, including western blots. As expected, they observed antibodies to capsid proteins, in addition to several low molecular weight polypeptides.40 These were assumed to be host cell histones, which are used by viral DNA to form minichromosomes.6,7 Extrapolating from these data, we reconsidered an old idea to explain how autoimmunity to DNA and nucleosomes could be initiated. This idea originated from early studies on immunogenicity of haptens, defined as molecules that could bind antibodies, but not by themselves stimulate to antibody production. In other words, they were nonimmunogenic. For haptens to gain immunogenic potential there is a need (i) for B cells to recognize that particular hapten, and (ii) for the hapten to be coupled to a carrier protein in order for cognate interacting T cells to be stimulated and thereby to provide help for hapten-specific B cells.
The Hapten-Carrier System—Formal Requirements
The first prerequisite relevant to use a hapten-carrier system to explain how polyomaviruses may initiate autoimmunity to DNA is the presence of B cells specific for dsDNA. The B cell receptors for antigens are generated by several stochastic events during somatic maturation of the B cells to ensure generation of all for the organism necessary antibody specificities. These events include (i) random recombination of one each of a large repertoire of V, D, and J genes for the immunoglobulin heavy chains and V and J genes for the light ones, (ii) insertion of nontemplate nucleotides between the variable region genes, and (iii) the use of full length, truncated or inverted D genes in all three reading frames.111 Combination of heavy and light chains to constitute intact immunoglobulin molecules adds to this manifold of specificities.72 Such random processes generate large arrays of immunoglobulin specificities, including specificity for autologous constituents. Provided naïve DNA-specific B cells have high affinity receptors for the autoantigen, they may be deleted, preferentially in the bone marrow,112 or their receptors may be revised, a process called receptor editing, implying that the B cell substitute one light chain by another, thus changing its antigenic specificity.113 However, due to the adaptive nature of the B cell receptor, antigen stimulation of low-affinity B cells may through somatic mutations linked to progression of the immune response, result in higher affinity for that given antigen.114 Deletion of high-affinity B cells specific for autoantigens is therefore not a guarantee against development of high-affinity autoantibodies.
Secondly, for B cells to be stimulated, they need cognate interacting T helper cells.115 T cells specific for autologous ligands may be physically or functionally inactivated in the thymus,72 or rendered nonresponsive in the periphery (see e.g., refs. 25, 73). Thus, although B cells may recognize autologous ligands, they still will not be activated by a given autoantigen due to the lack of sufficient T cell help. This may be circumvented if an autoantigen form complexes in vivo with a nonself antigen. In this situation, a scenario may be created that fulfill all demands to activate autoimmune B cells, provided the auto-specific B cell process and present peptides derived from the complexed nonself ligand. In other words, this model is in harmony with the hapten-carrier system to induce anti-hapten antibodies by otherwise nonimmunogenic haptens.116,117 In this context, the autoantigen is analogous to the (nonimmunogenic) hapten, while the nonself complexed ligand represents the carrier protein. The conceptual framework for the research program described here for polyomavirus dependent termination of tolerance to DNA, histones and nucleosomes relied on this hapten-carrier model.
The Hapten—Carrier System for Induction of Anti-dsDNA Antibodies —Experimental Systems and Clinical Observations
In a prospective set of experiments, the first series of evidences that polyomaviruses had the potential to induce antibodies to the major components of nucleosomes, DNA and histones, were provided.118 That this immunization regime resulted in antibodies recognizing mammalian dsDNA in both ELISA and the Crithidia luciliae assay, regarded as specific for SLE,119 was important as the current view at that time was that mammalian dsDNA was nonimmunogenic.120-123 In subsequent experiments we verified this observation,124,125 and unequivocally proved that the antibodies bound different forms of DNA; mammalian ssDNA and dsDNA, and different synthetic single-stranded and double-stranded analogous of DNA126,127 In the latter experiments, the induced antibodies possessed DNA specificities reflecting the DNA used for immunization. If polyomavirus DNA complexed with methylated bovine serum albumin (mBSA) was used as immunogen, the emerging antibodies were mainly specific for that DNA, and did not crossreact with mammalian DNA, similar to what had been observed in other experimental systems.121-123 However, if experimental animals were primed with viral DNA-carrier protein complexes, they responded to subsequent immunization using calf thymus (CT) dsDNA-mBSA by producing antibodies cross-reacting with mammalian dsDNA including human and CT dsDNA.127
In a next set of experiments, we tested whether natural infection with BKV resulted in similar sets of anti-nucleosomal antibodies. In that study we demonstrated that the earlier described anti-dsDNA responses to BKV in experimental animals also appeared during natural BKV infection in man.128 Fifty-nine children were examined over time for serological signs (development of IgG and IgM anti-VP antibodies) of primary BKV infection. Of eight children found to undergo primary infection with BKV, anti- BKV-dsDNA specific antibodies appeared in all. In 4 of the 8 patients the antibodies cross-reacted significantly with mammalian dsDNA, and weak antibody binding to mammalian dsDNA was also noted in at least three other patients.128 The antibodies resembled those induced in the experimental model with regard to their high relative affinity for BK dsDNA, and somewhat lower, but definitive, affinity for mammalian dsDNA. In contrast, most, but not all, anti-dsDNA antibodies from 10 SLE patients cross-reacted extensively with dsDNA from viral and mammalian origin. Thus, a dsDNA virus like BKV may provoke immunological intolerance to mammalian dsDNA, with features similar to those encountered in SLE. Furthermore, these observations demonstrated that induction of anti-dsDNA antibodies was not restricted to experimental immunization of animals, but did also take place in humans during naturally acquired BKV infection. A logic question following these observations was how autoimmune (e.g., (NZB/NZW)F1 mice) mice responded to polyomaviruses compared to responses in normal mice. After inoculation with polyomavirus or polyomavirus-dsDNA complexed with mBSA, the normal Balb/C mice responded by producing anti-DNA antibodies mostly recognizing polyomavirus ssDNA and dsDNA, while the autoimmune mice readily produced nephritogenic anti-mammalian dsDNA antibodies.129 Highly relevant, and similar to our results, Gilkeson et al130 compared anti-DNA antibody responses in normal and (NZBxNZW)F1 mice after immunization with bacterial DNA-mBSA complexes. Whereas the immunologically normal mice produced antibodies that were specific for the immunizing bacterial DNA, (NZBxNZW)F1 mice produced antibodies that also bound CT dsDNA. Furthermore, the induced antibodies resembled lupus anti-DNA antibodies in their fine specificity for synthetic analogous of ss/dsDNA or Crithidia luciliae kinetoplast DNA.130
All together, these data demonstrated that ubiquitous human viruses like polyomaviruses, when activated in vivo, had the potential to induce the production of pathogenic anti-DNA antibodies in disposed individuals. In a subsequent study, we generated anti-dsDNA producing B cell hybridomas from mice hyper-immunized with polyomavirus BK. The structure and gene usage of the variable regions of heavy and light chains of these induced anti-dsDNA antibodies were determined in order to compare the structural features of the induced antibodies with those characterized in murine SLE131-141 This study revealed that the polyomavirus induced anti-dsDNA antibodies were highly similar to potentially pathogenic anti-DNA antibodies produced in context of murine SLE, particularly with respect to the presence of the basic amino acid arginine at amino acid positions 99-101 in the heavy chain variable regions.142 Thus, both polyomavirus and bacterial DNA, when complexed with an immunogenic carrier protein, had the potential to induce pathogenic anti-dsDNA antibodies with variable regions structures similar to those described in SLE.118,129,130
These results were all in agreement with the hapten-carrier model, and opened for aimed studies to determine the origin and nature of the carrier protein involved in the immune response to DNA. It became important to search for the origin of in vivo-produced proteins that rendered DNA immunogenic, instead of searching for immunogenic DNA.
Polyomavirus T-ag: A Natural Carrier Protein for dsDNA
Since polyomaviruses obviously had the potential to induce the production of anti-dsDNA antibodies in experimental animals,118,129,142 this pointed at virus-encoded proteins as potential carrier molecules rendering DNA immunogenic. As described above, e.g., VP1 has both nuclear localization signals (NLS) and potential to bind DNA directly and indirectly through interaction with DNA bound proteins.5,14-17 On the other hand, in the permissive host another virus encoded protein could be more relevant, as it is required for viral transcription and replication, and binds both viral and host DNA — polyomavirus large T-ag.5 Thus, T-ag could represent a nonself DNA-bound protein that served as the T cell determinant. This assumed process would require that DNA-specific B cells indeed had the potential to present T-ag-derived peptides in context of sufficient costimulatory signals, and that T cells were primed by T-ag presented by conventional antigen-presenting cells.
In two different experimental systems these presumptions were verified. The first was based on immunizations with plasmids encoding T-ag under control of eukaryotic promoters22 Immunologically normal mice inoculated with plasmids encoding wild-type, DNA-binding T-ag produced antibodies to this protein. These antibodies were kinetically linked to significant production of antibodies to dsDNA, histones, and to certain transcription factors, deduced to be produced according to the basic idea of the model: all autologous ligands linked to T-ag could theoretically be rendered immunogenic provided the presence of a (functional) repertoire of B cells.22 Injection of plasmids expressing irrelevant nonDNA-binding proteins like luciferase, or plasmids containing T-ag sequences but lacking a promoter, did not result in such antibodies,22 indicating that plasmid DNA itself was nonimmunogenic. In the second experimental approach,23 we could directly demonstrate i. that SLE patients were highly susceptible to persistent productive polyomavirus infections, or recurrent virus reactivation, as opposed to normal individuals; and ii. that linked to virus expression, antibodies to T-ag, DNA and to transcription factors like TBP and CREB, but not to other nonnucleosomal autoantigens, were produced. Interestingly, antibodies to mammalian dsDNA correlated with persistent infection in the SLE patients.23,109 These studies22,23 have resulted in novel knowledge about a possible source of these antibodies and about a process that may explain how antibodies to dsDNA can be generated.
The SLE-Related Polyomaviruses Belong to Wild-Type Strains
As polyomaviruses demonstrated a strong tendency to productive infection in SLE,23,67 but not in normal individuals, nor in rheumatoid arthritis patients, it was important to assess whether these viruses differed from those latently infecting normal individuals. A detailed characterization of the NCCR of the virus genome containing the promoter/enhancer region,67 and the gene encoding the main capsid protein VP1,143 potentially important for binding to, and thereby infecting, cells was therefore undertaken. However, for both genomic regions mostly sequences were detected identical to strains circulating in the healthy human population, demonstrating that such regions probably were not responsible for the strong tendency for productive infection in SLE patients.67,143 A provisional explanation is therefore that SLE patients have lost their ability to control this virus, as described for e.g., immune deficient individuals.5,11
Virus-Induced Anti-dsDNA Antibodies—A Model Not Restricted to Polyomaviruses
Collectively, the data generated so far demonstrate that in vivo expression of the polyomavirus DNA-binding T-ag resulted in generation of IgG antibodies to T-ag and nucleosomal ligands like DNA, histones and transcription factors, but not to other autoantigens not linked to nucleosomes, indicating an antigen-selective T cell dependent B cell response. Complexes formed in vivo and in vitro of T-ag and nucleosomes created a molecular basis for antigen-selective interaction of T-ag specific T cells and nucleosome (DNA) specific B cells.82 In harmony with this, B cells cocultured with T-ag specific T cells and stimulated with T-ag or nucleosome-T-ag complex, could present T-ag-derived peptides to T cells and produce antibodies with specificity reflecting the nature of the stimulating antigens, e.g., against T-ag or DNA.82 Thus, T-ag may both initiate and maintain an autoantibody response to e.g., DNA in situations where T-ag is actively expressed.
This hapten-carrier model has been proven valid in other, but not all, studies of termination of immunological tolerance to DNA by viruses. Whether human cytomegalovirus (HCMV) was expressed in SLE and correlated with development of autoimmunity was examined in our laboratory using the same biological material as in the foregoing studies.23,143 The result of this study was that HCMV did not correlate with autoimmunity, as active infection was not detected in these patients as judged from lack of both viruria and lack of increased IgM and IgG anti-HCMV antibody titers.144 These data, however, do by no means rule out that other viruses, and even HCMV, may participate in generating autoimmunity to nucleosomal antigens.
Immunization of immunologically normal mice with complexes of the DNA-binding domain of the human papillomavirus E2 protein and a DNA fragment encompassing the E2-binding site resulted in antibodies against the E2 protein and dsDNA. The latter antibodies reacted with free and E2-bound dsDNA but not with the protein.145 Similar to data discussed above,22 mice inoculated with a vector expressing the single DNA-binding protein EBNA-1 of the Epstein—Barr virus, generated autoantibodies to both dsDNA and the Sm antigen,146 probably in context of a carrier function of the EBNA-1 antigen. Similarly, a recent case report presented evidence that a 22-year old woman developed SLE following infection with Epstein-Barr virus. Antibodies to dsDNA and EBNA-1 characterized the antibody profile of this patient.147
In a study by Dong et al148 high titer antibodies specific for the p53 tumor suppressor protein were induced in mice immunized with purified complexes of murine p53 and the SV40 T-ag, but not in mice immunized with either protein separately. The autoantibodies to p53 in these mice were primarily of the IgG1 isotype and were not cross-reactive with T-ag. The high levels of autoantibodies to p53 in mice immunized with p53-T-ag complexes were transient, similar to the induced anti-dsDNA antibodies described above, but low levels of the anti-p53 antibodies persisted. The latter may have been maintained by the self antigen, since the anti-p53, but not the anti-T-ag response could be restimulated with murine p53. One explanation for their results may be that antigen processing of the complex of T-ag and p53 could activate both T-ag-specific and autoreactive p53-specific T helper cells, thus driving anti-p53 autoantibody production. The induction of autoantibodies during the course of an immune response directed against this naturally occurring complex of self and nonself antigens may therefore be relevant to the generation of specific autoantibodies in context of viral infections, irrespective whether the individual suffers from SLE or not.
In the interpretation of their data, Reeves and Dong et al148,149 indicated that humoral autoimmunity can be initiated by a “hit and run” mechanism in which the binding of a viral antigen to a self protein triggers an immune response that subsequently can be perpetuated by self antigen. For this to occur, however, autoimmune T cells must also be engaged in context of the viral infection, and not be rendered tolerant once the infection (and production of autoantigen-binding viral proteins) is terminated. This has been an important focus in advancing our studies on polyomavirus-induced tolerance to autoantigens, and experiments described below explain how T cell tolerance to nucleosomes may be terminated in context of stimulation with nucleosome-T-ag complexes. The results so far have provided us with a carrier protein, large T-ag, that has the potential to render DNA/nucleosomes immunogenic, and which may be constitutively expressed in vivo in SLE patients, but transiently and rarely in normal individuals. This insight opened for the study of determinant spreading, or better: the consequence of linked presentation of self nucleosomal peptides and nonself T-ag derived peptides to activate dormant autoimmune T cells.
Indirect Activation of Nucleosome-Specific T Cells Through Determinant Spreading Requires Complex Formation of T-ag and Nucleosomes
Based on results described above, regulation of T cell tolerance to nucleosomes became important, as T cells in SLE, but not in healthy humans, are intolerant to this complex.45,82,84,150-152 Central in this perspective was to test whether autoimmune, nucleosome-specific T cells are physically eliminated in the thymus through deletion, or whether such cells are entering the periphery where they may receive stimuli that result either in activation, anergy or in deletion. We approached these problems by testing whether T-ag expression also could have the potential to terminate histone-specific T cell anergy as a consequence of linked presentation by APC of histones and T-ag. This was likely to occur in vivo, as we succeeded in determining and characterizing this process in vitro.45 Thus, by stimulating PBMC with nucleosome-T-ag complexes, but not with nucleosomes or T-ag, such T cell cultures could functionally be restimulated in the next round by nucleosomes or histones.45 This would be in accordance with a model that implies APC-mediated linked presentation of nonself T-ag and (self ) histone peptides. In this situation, responder T-ag-specific T cells may, through secretion of IL-2, activate nonselectively autoimmune, histone-specific T cells present in the microenvironment.153,154 Subsequently, these T cells may clonally expand provided that histone-derived peptides are presented by APC in context of HLA class II, and that sufficient costimulatory signals are available.155-159 The full picture of this model, including activation of autoimmune, histone-specific T cells and initiation of autoimmune DNA-specific B cells is outlined in Figure 3.
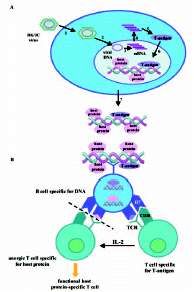
The results obtained using T cells from healthy human individuals and human SLE were reproduced experimentally in mice immunized with nucleosomes and nucleosome-T-ag complexes.83 Only mice immunized with nucleosome-T-ag complexes harbored T cells that subsequently responded to pure nucleosomes or histones, presumably due to a linked presentation of nucleosomal peptides and T-antigen derived peptides by the same APC in vivo.83 Thus, in this latter study, we obtained experimental results mimicking those described for the human system.44,45,82 These results therefore strongly indicate that all individuals may harbor autoimmune nucleosome-specific T cells similar to those detected in active SLE.
In a recent study,84 the complementary determining region 3 (CDR3) structures of the TCR V(α) and/or V(β) chains were determined. Histone- and T-ag- specific T cells were generated by stimulation of PBMCs with nucleosome-T-ag complexes and subsequently maintained by pure histones. T-ag-specific T cell clones were initiated and maintained by T-ag. The frequencies of circulating histone- or T-ag-specific T cells were determined in healthy individuals and in SLE patients by limiting dilution of PBMCs, and TCR gene usage and variable-region structures were determined by complementary DNA sequencing. These sequences were compared between T-ag- and histone-specific T cells and between normal individuals and SLE patients for each specificity. Individual in vitro-expanded histone-specific T cells from normal individuals displayed identical TCR V(α) and/or V(β) CDR3 regions sequences, indicating that they were clonally expanded in vivo. Essentially the same was observed for T-ag-specific T cells. The frequencies of in vitro antigen-responsive T-ag- or histone-specific T cells from normal individuals were similar to those from SLE patients. Although heterogeneous for variable-region structure and gene usage, histone- specific T cells from healthy individuals and SLE patients selected the acidic amino acids aspartic and/or glutamic acids at positions 99 and/or 100 in the V(β) CDR3 regions which may be important in recognizing basic amino acids within histone polypeptides. Thus, autoimmune T cells from healthy individuals can be activated by nucleosome- T-ag complexes and maintained by histones in vitro and in vivo. Such T cells possessed TCR structures similar to those from SLE patients, demonstrating that T cell autoimmunity to nucleosomes may be an inherent property of the normal immune system. This is further strengthened by the fact that such autoimmune T cells were clonally expanded in vivo, although nonresponsive at the time they were isolated.84
Thus, complexes formed by virus-encoded DNA-binding T-ag and host cell nucleosomes are directly immunogenic to DNA-specific B cells,22,23 and indirectly to autoimmune, histone or nucleosome-specific T cells through linked presentation of nonself and self peptides in context of HLA II molecules.45,83,84
Precedence for this model relates to diversification of T cell responses to include responses to determinants between different polypeptides contained within molecular complexes or within mixtures of solitaire molecules, provided that T cells are present which are able to respond to one of the components.160,161 For example, Lin et al161 observed that autologous cytochrome C was nonimmunogenic with regard to T cell proliferation. However, T cells purified from mice that were immunized with self and nonself cytochrome C subsequently proliferated to isolated self cytochrome C. Similar results have been obtained after immunization of normal mice with self/nonself snRNP.162
Why Is a NonSelf Viral Protein Necessary for Induction of B Cell and T Cell Autoimmunity to Nucleosomes?
There may be several reasons why pure autologous nucleosomes may not be immunogenic although B cells specific for e.g., dsDNA, histones or transcription factors are present in the body and amenable to antigenic stimulation.22,163 First, T cells specific for nucleosomes or nucleosome-derived peptides may be sorted out in the thymus, one of the places in the body where nucleosomes must be generated in large amounts due to clonal deletion of autoreactive T cells.164-166 Whether the T cells specific for nucleosomes or histones, as those described above, have escaped clonal deletion due to low affinity for their autologous ligands remains to be established. Alternatively, autoimmune T cells may have escaped deletion merely based on stochastic events — for them a fortunate event, for the body, however, an unlucky one as they now can participate in unwanted potentially pathogenic autoimmune responses. In the periphery, on the other hand, several control mechanisms may be involved to control autoimmune T cells that have escaped clonal deletion in the thymus. These mechanisms are linked to both the innate and the adaptive immune system.
In this sense, we must differentiate between infectious pathogens and autologous molecules or complexes of these. Invading antigens in the form of e.g., infectious agens are mostly drained into the lymph nodes, where antigens can be processed and presented properly by professional APCs delivering appropriate costimulatory signals and activation signals for antigen-selective T cells. This may initiate meaningful immune responses with the elimination of the infectious agens as the intended goal. In certain situations, a potentially pathogenic side effect of this process is, however, that (in our context) nucleosomes may be rendered immunogenic both for B cells and T cells along pathways described above (see fig. 3 for details). Thus, viral infections may induce potentially pathogenic autoimmunity. Subsequently, pure nucleosomes devoid of nonself polypeptides could theoretically stimulate and expand this autoimmune response further, provided that such T cells are not functionally down-regulated. This may, however, actually be achieved by APCs not providing costimulatory signals or by MHC class II expressing cells of nonlymphoid organs. For example, stimulation of Toll-like receptors by infectious ligands seems to trigger dendritic cells to maturate, subsequently resulting in up-regulation of costimulatory molecules and increased antigen-presenting capacity for T cells.75-77,167 Lack of stimulation of these receptors may result in lack of expression of costimulatory molecules. This may be the case for e.g., antigens like human autologous nucleosomes/chromatin that may lack PAMPs recognized by cell surface PRR (see above, and ref. 76). In situations where autologous nucleosomes are available to e.g., dendritic cells, nucleosomes may therefore tolerate T cells by presenting e.g., nucleosome-derived peptides rather than activating them.
Furthermore, nonlymphoid cells like endothelial or epithelial cells may have the capacity to present peptides in context of HLA class II,168- 170 but without providing costimulatory signals mediated by CD80/86.171-173 This is, however, controversial, as others have detected also costimulatory signal molecules on e.g., endothelial cells.174-176 In situations where APCs present autologous peptides in context of HLA class II but lacking costimulation, T cells will not respond properly,156,177 and may instead be antigen-selectively anergized.155,178
If this holds to be true, evolution has responded immensely meaningfully to an unexpected threat exerted by e.g., viruses aside from their cytopathogenic effects. These may directly be able to initiate autoimmunity, as complexes formed by autoantigens (here: nucleosomes) and viral proteins (like polyomavirus T-ag) may be drained from the focus of the infection into lymph nodes. Following this, germinal centers may be established, with autoimmune responses as a result (fig. 3). However, once such autoimmune T cells receive their stimuli, they proliferate and recirculate. During this latter process, they may encounter the same nucleosomal peptides in the periphery, now presented by inappropriate APCs like immature dendritic cells or HLA II expressing nonlymphoid cells lacking costimulatory molecules, thus bringing the active (functional) state of the T cells back to a state of tolerance.
Is a Protein like the DNA-Binding Polyomavirus Encoded T-ag Essential for Terminating Immune Nonresponsiveness to Nucleosomes?
Apoptosis is an event that characterizes the living organism, and must represent a heavy load of work for the phagocytic system. As all the apoptotic material will be accessible to cells capable of ingesting it, evolution must have developed control mechanisms protecting the body against autoimmunity to such material and against development of autoimmune disorders. Since it is quite obvious that autoimmune B cells and antigen-related T cells exist in the periphery, and since they can be activated antigen-selectively, the determination whether the immune system shall respond or not cannot rely solely on these cells. The intriguing ideas of Janeway and Medzhitov, describing PAMPs and PRRs as systems to differentiate between infectious nonself and noninfectious self,73,77 directly point at the innate immune system as a main control station to prevent autoimmunity. Although not yet clearly established, one may assume, also from the logic of evolution, that the receptors of cells of the innate system (e.g., dendritic cells), such as Toll like receptors (the membrane bound version of the PRR) do not recognize for example autologous nucleosomes, dsDNA or histones, as they simply do not have PAMPs. This is in contrast to e.g., bacterial DNA that differs from autologous DNA by having substantially more of PAMPs like CpG. From this, one may speculate whether the innate immune system may protect against immune responses to autoantigens like those present on chromatin.
As tolerance to nucleosomes is maintained in a healthy body it must be effectively controlled by the innate and/or the adaptive immune system. There are, however, several problems that need to be solved. For example, in DNAse 1179 or Nrf2180 knock-out mice, autoimmunity to dsDNA spontaneously develops, followed by antibody deposition in kidneys and development of nephritis. In the normal counterpart of these mice, this does not take place. One potential explanation for this is that e.g., DNAse 1 may be important for degradation and elimination of nucleosomes. DNAse1 deficiency may correlate to decreased degradation, decreased elimination and increased extra-cellular storage of nucleosomes. This, according to others,100,181 may result in altered degradation of chromatin, resulting in presentation of cryptic or altered self-determinants not present in the normal organism.181,182 If such neo-antigens can stimulate e.g., dendritic cells to up-regulate costimulatory molecules is not, however, established. Nevertheless, if true, such altered determinants may, due to increased extra-cellular amounts, also be processed and presented globally in the organism, and thereby potentially also by nonprofessional APCs. These may, provided they do not up-regulate costimulatory molecules, reverse the activated state of the T cells to a state of anergy. Thus, although results from the DNAse 1 knock out mouse indicate that autologous nucleosomes may stimulate the immune system to produce anti-DNA antibodies, a concise and direct experimental system confirming this model is still missing. This problem, whether pure self (in context of apoptosis, altered self may also be self ) can induce anti-self, needs, in light of the problems discussed here, further studies.
Irrespective Whether Self-Derived or Infectious-Derived Proteins Serve As Carrier Proteins for (Nucleosomal) DNA, Stimulation of DNA-Specific May Be the Net Result
The development of the induced anti-DNA antibodies, and their qualities, is described in Figure 4. If stimulated properly, the immune system produces antibodies to dsDNA that may develop from two principally different sources. One may be somatic mutation of variable regions of antibodies to ssDNA, with introduction of highly basic amino acids (particularly arginines) at distinct positions, yielding specificity for dsDNA.139,144,146,148 The other may be represented by stimulation of B cells with an inherent (initial) specificity for dsDNA normally deleted in the bone marrow112 If such B cells escape deletion, they may be adequately stimulated by dsDNA, and clonally expanded, provided it is complexed with an immunogenic carrier protein.
Concluding Remarks
Polyomaviruses are strong stimulators of the immune system. The immune responses that evolve have all the characteristics of a secondary, adaptive, T cell dependent response. This is also true for the autoimmune responses against chromatin constituents induced through the involvement of polyomavirus-encoded ligands. These principally dual responses, anti-self and anti-nonself, reflect the Janus face of polyomaviruses, in the healthy body they are dormant and try to hide without irritating the immune system, but in situations where they are activated, they may initiate autoimmunity, and even induce tumors after prolonged productive infection.
References
- 1.
- Gardner SD, Field AM, Coleman DV. et al. New human papovavirus (B.K.) isolated from urine after renal transplantation. Lancet. 1971;1(712):1253–1257. [PubMed: 4104714]
- 2.
- Padgett BL, Walker DL, ZuRhein GM. et al. Cultivation of papova-like virus from human brain with progressive multifocal leucoencephalopathy. Lancet. 1971;1(7712):1257–1260. [PubMed: 4104715]
- 3.
- Sweet BH, Hilleman MR. The vacuolating virus, S.V. 40. Proc Soc Exp Biol Med. 1960;105:420–427. [PubMed: 13774265]
- 4.
- Shah K, Nathanson N. Human exposure to SV40: Review and comment. Am J Epidemiol. 1976;103(1):1–12. [PubMed: 174424]
- 5.
- Moens U, Rekvig OP. Molecular biology of BK virus and clinical and basic aspects of BK virus renal infectionIn: Khalili K, Stoner GL, eds.Human polyomaviruses. Molecular and clinical perspectivesNew York: Wiley-Liss Inc.,2001359–408.
- 6.
- Meneguzzi G, Pignatti PF, Barbanti-Brodano G. et al. Minichromosome from BK virus as a template for transcription in vitro. Proc Natl Acad Sci USA. 1978;75(3):1126–1130. [PMC free article: PMC411421] [PubMed: 26051]
- 7.
- Varshavsky A, Levinger L, Sundin O. et al. Cellular and SV40 chromatin: Replication, segregation, ubiquitination, nuclease-hypersensitive sites, HMG-containing nucleosomes, and heterochromatin-specific protein. Cold Spring Harb Symp Quant Biol. 1983;47(Pt 1):511–528. [PubMed: 6305564]
- 8.
- Kornberg RD. Structure of chromatin. Annu Rev Biochem. 1977;46:931–954. [PubMed: 332067]
- 9.
- McGhee JD, Felsenfeld G. Nucleosome structure. Annu Rev Biochem. 1980;49:1115–1156. [PubMed: 6996562]
- 10.
- Kornberg RD, Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell. 1999;98(3):285–294. [PubMed: 10458604]
- 11.
- Moens U, Seternes OM, Johansen B. et al. Mechanisms of transcriptional regulation of cellular genes by SV40 large T- and small t-antigens. Virus Genes. 1997;15(2):135–154. [PubMed: 9421878]
- 12.
- Cole CN. Polyomaviruses: The viruses and their replicationIn: Fields BN, Knipe DM, Howley PM, eds.Fields virologyPhiladelphia: Lippincot-Raven Publishers,19961997–2025.
- 13.
- Viscidi RP, Rollison DE, Viscidi E. et al. Serological cross-reactivities between antibodies to simian virus 40, BK virus, and JC virus assessed by virus-like-particle-based enzyme immunoassays. Clin Diagn Lab Immunol. 2003;10(2):278–285. [PMC free article: PMC150538] [PubMed: 12626455]
- 14.
- Wychowski C, Benichou D, Girard M. The intranuclear location of simian virus 40 polypeptides VP2 and VP3 depends on a specific amino acid sequence. J Virol. 1987;61(12):3862–3869. [PMC free article: PMC256004] [PubMed: 2824820]
- 15.
- Wychowski C, Benichou D, Girard M. A domain of SV40 capsid polypeptide VP1 that specifies migration into the cell nucleus. EMBO J. 1986;5(10):2569–2576. [PMC free article: PMC1167154] [PubMed: 3023047]
- 16.
- Gharakhanian E, Kasamats H. Two independent signals, a nuclear localization signal and a Vp1-interactive signal, reside within the carboxy-35 amino acids of SV40 Vp3. Virology. 1990;178(1):62–71. [PubMed: 2167562]
- 17.
- Gharakhanian E, Takahashi J, Clever J. et al. In vitro assay for protein-protein interaction: Carboxyl-terminal 40 residues of simian virus 40 structural protein VP3 contain a determinant for interaction with VP1. Proc Natl Acad Sci USA. 1988;85(18):6607–6611. [PMC free article: PMC282026] [PubMed: 2842781]
- 18.
- Rekvig OP, Moens U, Fredriksen K. et al. Human polyomavirus BK and immunogenicity of mammalian DNA: A conceptual framework. Methods. 1997;11(1):44–54. [PubMed: 8990088]
- 19.
- Knowles WA. The epidemiology of BK virus and the occurence of antigenic and genomic subtypesIn: Khalili K, Stoner GL, eds.Human polyomaviruses. Molecular and clinical perspectivesNew York: Wiley-Liss,2001527–559.
- 20.
- Dorries K. Latent and persistant polyomavirus infectionIn: Khalili K, Stoner GL, eds.Human polyomaviruses. Molecular and clinical perspectives New York: Wiley-Liss Inc.,2001197–235.
- 21.
- Tooze J. DNA tumor viruses. Molecular biology of tumor viruses. Cold Spring Harbor: Cold Spring Harbor Laboratory. 1980
- 22.
- Moens U, Seternes OM, Hey AW. et al. In vivo expression of a single viral DNA-binding protein generates systemic lupus erythematosus-related autoimmunity to double-stranded DNA and histones. Proc Natl Acad Sci USA. 1995;92(26):12393–12397. [PMC free article: PMC40364] [PubMed: 8618908]
- 23.
- Rekvig OP, Moens U, Sundsfjord A. et al. Experimental expression in mice and spontaneous expression in human SLE of polyomavirus T-antigen. A molecular basis for induction of antibodies to DNA and eukaryotic transcription factors. J Clin Invest. 1997;99(8):2045–2054. [PMC free article: PMC508030] [PubMed: 9109450]
- 24.
- Van GhelueM, Moens U, Bendiksen S. et al. Autoimmunity to nucleosomes related to viral infection: A focus on hapten-carrier complex formation. J Autoimmun. 2003;20(2):171–182. [PubMed: 12657530]
- 25.
- Rekvig OP, Nossent JC. Anti-double-stranded DNA antibodies, nucleosomes, and systemic lupus erythematosus: A time for new paradigms? Arthritis Rheum. 2003;48(2):300–312. [PubMed: 12571837]
- 26.
- Gardner SD. Prevalence in England of antibody to human polyomavirus (B.K.). BMJ. 1973;i:77–78. [PMC free article: PMC1588750] [PubMed: 20791873]
- 27.
- Padgett BL, Walker DL. Prevalence of antibodies in human sera against JC virus, an isolate from a case of progressive multifocal leukoencephalopathy. J Infect Dis. 1973;127(4):467–470. [PubMed: 4571704]
- 28.
- Shah KV, Daniel RW, Wazawski RM. High prevalence of antibodies to BK virus, an SV40-related papovavirus, in residents of Maryland. J Infect Dis. 1973;128(6):784–787. [PubMed: 4587749]
- 29.
- Mantyjarvi RA, Meurman OH, Vihma L. et al. A human papovavirus (B.K.), biological properties and seroepidemiology. Ann Clin Res. 1973;5(5):283–287. [PubMed: 4365866]
- 30.
- Brown P, Tsai T, Gajdusek DC. Seroepidemiology of human papovaviruses. Discovery of virgin populations and some unusual patterns of antibody prevalence among remote peoples of the world. Am J Epidemiol. 1975;102(4):331–340. [PubMed: 233851]
- 31.
- Rollison DEM, Shah KV. The epidemiology of SV40 infection due to contaminated polio vaccines: Relation of the virus to human cancerIn: Khalili K, Stoner GL, eds.Human polyomaviruses. Molecular and clinical perspectivesNew York: Wiley-Liss,2001561–584.
- 32.
- Agostini HT, Jobes DV, Stoner GL. Molecular evolution and epidemiology of JC virusIn: Khalili K, Stoner GL, eds.Human polyomaviruses. Molecular and clinical perspectivesNew York: Wiley-Liss,2001491–526.
- 33.
- Tevethia SS, Schell TD. The immune response to SV40, JCV, and BKVIn: Khalili K, Stoner GL, eds.Human polyomaviruses. Molecular and clinical perspectivesNew York: Wiley-Liss,2001585–610.
- 34.
- Knowles WA, Pipkin P, Andrews N. et al. Population-based study of antibody to the human polyomaviruses BKV and JCV and the simian polyomavirus SV40. J Med Virol. 2003;71(1):115–123. [PubMed: 12858417]
- 35.
- Flower AJ, Banatvala JE, Chrystie IL. BK antibody and virus-specific IgM responses in renal transplant recipients, patients with malignant disease, and healthy people. Br Med J. 1977;2(6081):220–223. [PMC free article: PMC1631345] [PubMed: 195667]
- 36.
- Portolani M, Marzocchi A, Barbanti Brodano G. et al. Prevalence in Italy of antibodies to a new human papovavirus (BK virus). J Med Microbiol. 1974;7(4):543–546. [PubMed: 4375196]
- 37.
- Zapata M, Mahony JB, Chernesky MA. Measurement of BK papovavirus IgG and IgM by radioimmunoassay (RIA). J Med Virol. 1984;14(2):101–114. [PubMed: 6092527]
- 38.
- Flaegstad T, Traavik T, Kristiansen BE. Age-dependent prevalence of BK virus IgG and IgM antibodies measured by enzyme-linked immunosorbent assays (ELISA). J Hyg Lond. 1986;96(3):523–528. [PMC free article: PMC2129697] [PubMed: 3016078]
- 39.
- Flaegstad T, Ronne K, Filipe AR. et al. Prevalence of anti BK virus antibody in Portugal and Norway. Scand J Infect Dis. 1989;21(2):145–147. [PubMed: 2543061]
- 40.
- Christie KE, Flaegstad T, Traavik T. Characterization of BK virus-specific antibodies in human sera by Western immunoblotting: Use of a zwitterionic detergent for restoring the antibody-binding capacity of electroblotted proteins. J Med Virol. 1988;24(2):183–190. [PubMed: 2832537]
- 41.
- Stolt A, Sasnauskas K, Koskela P. et al. Seroepidemiology of the human polyomaviruses. J Gen Virol. 2003;84(Pt 6):1499–1504. [PubMed: 12771419]
- 42.
- Corallini A, Barbanti-Brodano G, Portolani M. et al. Antibodies to BK virus structural and tumor antigens in human sera from normal persons and from patients with various diseases, including neoplasia. Infect Immun. 1976;13(6):1684–1691. [PMC free article: PMC420820] [PubMed: 184044]
- 43.
- Dougherty RM, Distefano HS. Isolation and characterization of a papovavirus from human urine. Proc Soc Exp Biol Med. 1974;146(2):481–487. [PubMed: 4600925]
- 44.
- Bredholt G, Olaussen E, Moens U. et al. Linked production of antibodies to mammalian DNA and to human polyomavirus large T antigen: Footprints of a common molecular and cellular process? Arthritis Rheum 1999; 42(12):2583-2592. [published erratum appears in Arthritis Rheum. 2000;43(4):929]. [PubMed: 10616004]
- 45.
- Andreassen K, Moens U, Nossent H. et al. Termination of human T cell tolerance to histones by presentation of histones and polyomavirus T antigen provided that T antigen is complexed with nucleosomes. Arthritis Rheum. 1999;42(11):2449–2460. [PubMed: 10555041]
- 46.
- Takemoto KK. Human polyomaviruses: Evaluation of their possible involvement in human cancerIn: Essex M, Todaro G, zur Hausen M, eds.Viruses in Naturally Occuring CancersCold Spring Harbor, New York: Cold Spring Harbor Conference on cell proliferation,19807311–318.
- 47.
- Walker DL, Padgett BL. The epidemiology of human polyomaviruses. Prog Clin Biol Res. 1983;105:99–106. [PubMed: 6304775]
- 48.
- Hogan TF, Borden EC, McBain JA. et al. Human polyomavirus infections with JC virus and BK virus in renal transplant patients. Ann Intern Med. 1980;92(3):373–378. [PubMed: 6243896]
- 49.
- Taguchi F, Kajioka J, Miyamura T. Prevalence rate and age of acquisition of antibodies against JC virus and BK virus in human sera. Microbiol Immunol. 1982;26(11):1057–1064. [PubMed: 6300615]
- 50.
- Kravetz HM, Knight V, Chanock RM. et al. Respiratory syncytial virus. III. Production of illness and clinical observations in adult volunteers. JAMA. 1961;176:657–663. [PubMed: 13754148]
- 51.
- Morris JA, Johnson KM, Aulisio CG. et al. Clinical and serologic responses in volunteers given vacuolating virus (SV-40) by respiratory route. Proc Soc Exp Biol Med. 1961;108:56–59. [PubMed: 14476249]
- 52.
- Melnick JL, Stinebaugh S. Excretion of vacuolating SV-40 virus (papova virus group) after ingestion as a contaminant of oral poliovaccine. Proc Soc Exp Biol Med. 1962;109:965–968. [PubMed: 14472428]
- 53.
- Basetse HR, Lecatsas G, Gerber LJ. An investigation of the occurrence of SV40 antibodies in South Africa. S Afr Med J. 2002;92(10):825–828. [PubMed: 12432810]
- 54.
- Butel JS, Wong C, Vilchez RA. et al. Detection of antibodies to polyomavirus SV40 in two central European countries. Cent Eur J Public Health. 2003;11(1):3–8. [PubMed: 12690795]
- 55.
- Butel JS, Arrington AS, Wong C. et al. Molecular evidence of simian virus 40 infections in children. J Infect Dis. 1999;180(3):884–887. [PubMed: 10438386]
- 56.
- Rollison DE, Helzlsouer KJ, Alberg AJ. et al. Serum antibodies to JC virus, BK virus, simian virus 40, and the risk of incident adult astrocytic brain tumors. Cancer Epidemiol Biomarkers Prev. 2003;12(5):460–463. [PubMed: 12750243]
- 57.
- Carter JJ, Madeleine MM, Wipf GC. et al. Lack of serologic evidence for prevalent simian virus 40 infection in humans. J Natl Cancer Inst. 2003;95(20):1522–1530. [PubMed: 14559874]
- 58.
- De SanjoseS, Shah K, Engels EA. et al. Lack of serological evidence for an association between simian virus 40 and lymphoma. Int J Cancer. 2003;107(3):507–508. [PubMed: 12584752]
- 59.
- Hamilton RS, Gravell M, Major EO. Comparison of antibody titers determined by hemagglutination inhibition and enzyme immunoassay for JC virus and BK virus. J Clin Microbiol. 2000;38(1):105–109. [PMC free article: PMC86031] [PubMed: 10618072]
- 60.
- Jafar S, Rodriguez-Barradas M, Graham DY. et al. Serological evidence of SV40 infections in HIV-infected and HIV-negative adults. J Med Virol. 1998;54(4):276–284. [PubMed: 9557293]
- 61.
- Minor P, Pipkin P, Jarzebek Z. et al. Studies of neutralising antibodies to SV40 in human sera. J Med Virol. 2003;70(3):490–495. [PubMed: 12767016]
- 62.
- Vilchez RA, Butel JS. Polyomavirus SV40 infection and lymphomas in Spain. Int J Cancer. 2003;107(3):505–506. [PubMed: 14506754]
- 63.
- Brown DW, Gardner SD, Gibson PE. et al. BK virus specific IgM responses in cord sera, young children and healthy adults detected by RIA. Arch Virol. 1984;82(3-4):149–160. [PubMed: 6095788]
- 64.
- Hirsch HH, Steiger J. Polyomavirus BK. Lancet Infect Dis. 2003;3(10):611–623. [PubMed: 14522260]
- 65.
- Knowles WA, Gibson PE, Hand JF. et al. An M-antibody capture radioimmunoassay (MACRIA) for detection of JC virus-specific IgM. J Virol Methods. 1992;40(1):95–105. [PubMed: 1331163]
- 66.
- Kitamura T, Aso Y, Kuniyoshi N. et al. High incidence of urinary JC virus excretion in nonimmunosuppressed older patients. J Infect Dis. 1990;161(6):1128–1133. [PubMed: 2161040]
- 67.
- Sundsfjord A, Osei A, Rosenqvist H. et al. BK and JC viruses in patients with systemic lupus erythematosus: Prevalent and persistent BK viruria, sequence stability of the viral regulatory regions, and nondetectable viremia. J Infect Dis. 1999;180(1):1–9. [PubMed: 10353854]
- 68.
- Song R, Harding CV. Roles of proteasomes, transporter for antigen presentation (TAP), and beta 2-microglobulin in the processing of bacterial or particulate antigens via an alternate class I MHC processing pathway. J Immunol. 1996;156(11):4182–4190. [PubMed: 8666786]
- 69.
- Uebel S, Tampe R. Specificity of the proteasome and the TAP transporter. Curr Opin Immunol. 1999;11(2):203–208. [PubMed: 10322157]
- 70.
- Pieters J. MHC class II-restricted antigen processing and presentation. Adv Immunol. 2000;75:159–208. [PubMed: 10879284]
- 71.
- Brachet V, Raposo G, Amigorena S. et al. Ii chain controls the transport of major histocompatibility complex class II molecules to and from lysosomes. J Cell Biol. 1997;137(1):51–65. [PMC free article: PMC2139866] [PubMed: 9105036]
- 72.
- Janeway CA, Travers P, Walport M. et al. Immunobiology. The immune system in health and disease. 5th ed. New York: Churchill Livingstone. 2003
- 73.
- Janeway JrCA. The immune system evolved to discriminate infectious nonself from noninfectious self. Immunol Today. 1992;13(1):11–16. [PubMed: 1739426]
- 74.
- Janeway JrCA, Bottomly K. Signals and signs for lymphocyte responses. Cell. 1994;76(2):275–285. [PubMed: 7904901]
- 75.
- Janeway JrCA, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. [PubMed: 11861602]
- 76.
- Medzhitov R, Janeway JrCA. Decoding the patterns of self and nonself by the innate immune system. Science. 2002;296(5566):298–300. [PubMed: 11951031]
- 77.
- Medzhitov R, Janeway JrCA. How does the immune system distinguish self from nonself? Semin Immunol. 2000;12(3):185–188. [PubMed: 10910738]
- 78.
- Perniok A, Wedekind F, Herrmann M. et al. High levels of circulating early apoptic peripheral blood mononuclear cells in systemic lupus erythematosus. Lupus. 1998;7(2):113–118. [PubMed: 9541096]
- 79.
- Velupillai P, Carroll JP, Benjamin TL. Susceptibility to polyomavirus-induced tumors in inbred mice: Role of innate immune responses. J Virol. 2002;76(19):9657–9663. [PMC free article: PMC136524] [PubMed: 12208944]
- 80.
- Tevethia SS, Flyer DC, Tjian R. Biology of simian virus 40 (SV40) transplantation antigen (TrAg). VI. Mechanism of induction of SV40 transplantation immunity in mice by purified SV40 T antigen (D2 protein). Virology. 1980;107(1):13–23. [PubMed: 6255675]
- 81.
- Tevethia SS, Lewis M, Tanaka Y. et al. Dissection of H-2Db-restricted cytotoxic T-lymphocyte epitopes on simian virus 40 T antigen by the use of synthetic peptides and H-2Dbm mutants. J Virol. 1990;64(3):1192–1200. [PMC free article: PMC249233] [PubMed: 1689391]
- 82.
- Andreassen K, Bredholt G, Moens U. et al. T cell lines specific for polyomavirus T-antigen recognize T- antigen complexed with nucleosomes: A molecular basis for anti- DNA antibody production. Eur J Immunol. 1999;29(9):2715–2728. [PubMed: 10508246]
- 83.
- Bredholt G, Rekvig OP, Andreassen K. et al. Differences in the reactivity of CD4+ T-cell lines generated against free versus nucleosome-bound SV40 large T antigen. Scand J Immunol. 2001;53(4):372–380. [PubMed: 11285117]
- 84.
- Andreassen K, Bendiksen S, Kjeldsen E. et al. T cell autoimmunity to histones and nucleosomes is a latent property of the normal immune system. Arthritis Rheum. 2002;46(5):1270–1281. [PubMed: 12115233]
- 85.
- Drummond JE, Shah KV, Donnenberg AD. Cell-mediated immune responses to BK virus in normal individuals. J Med Virol. 1985;17(3):237–247. [PubMed: 2999323]
- 86.
- Bates MP, Jennings SR, Tanaka Y. et al. Recognition of simian virus 40 T antigen synthesized during viral lytic cycle in monkey kidney cells expressing mouse H-2Kb- and H-2Db-transfected genes by SV40-specific cytotoxic T lymphocytes leads to the abrogation of virus lytic cycle. Virology. 1988;162(1):197–205. [PubMed: 2827378]
- 87.
- Ye X, McCarrick J, Jewett L. et al. Timely immunization subverts the development of peripheral nonresponsiveness and suppresses tumor development in simian virus 40 tumor antigen-transgenic mice. Proc Natl Acad Sci USA. 1994;91(9):3916–3920. [PMC free article: PMC43693] [PubMed: 8171012]
- 88.
- Geiger T, Soldevila G, Flavell RA. T cells are responsive to the simian virus 40 large tumor antigen transgenically expressed in pancreatic islets. J Immunol. 1993;151(12):7030–7037. [PubMed: 8258707]
- 89.
- Geiger T, Gooding LR, Flavell RA. T-cell responsiveness to an oncogenic peripheral protein and spontaneous autoimmunity in transgenic mice. Proc Natl Acad Sci USA. 1992;89(7):2985–2989. [PMC free article: PMC48788] [PubMed: 1532662]
- 90.
- Arrington AS, Butel JS. SV40 and human tumorsIn: Khalili K, Stoner GL, eds.Human polyomaviruses. Molecular and clinical perspectivesNew York: Wiley-Liss,2001461–489.
- 91.
- Kennedy RC, Shearer MH, Watts AM. et al. CD4+ T lymphocytes play a critical role in antibody production and tumor immunity against simian virus 40 large tumor antigen. Cancer Res. 2003;63(5):1040–1045. [PubMed: 12615720]
- 92.
- Koralnik IJ. Overview of the cellular immunity against JC virus in progressive multifocal leukoencephalopathy. J Neurovirol. 2002;8(Suppl 2):59–65. [PubMed: 12491153]
- 93.
- Gasnault J, Kahraman M, de de Herve Goer MG. et al. Critical role of JC virus-specific CD4 T-cell responses in preventing progressive multifocal leukoencephalopathy. AIDS. 2003;17(10):1443–1449. [PubMed: 12824781]
- 94.
- Amital H, Shoenfeld Y. Autoimmunity and autoimmune diseases such as Systemic lupus erythematosusIn: Lahita RG, ed.Systemic Lupus ErythematosusSan Diego, London, Boston, New York, Sidney, Tokio, Totonto: Academic Press,19991–16.
- 95.
- Reeves WH, Satoh M, Richards HB. Origins of antinuclear antibodiesIn: Lahita RG, ed.Systemic Lupus ErythematosusSan Diego, London, Boston, New York, Sidney, Tokio, Toronto: Academic Press,1999293–317.
- 96.
- Raz E, Brezis M, Rosenmann E. et al. Anti-DNA antibodies bind directly to renal antigens and induce kidney dysfunction in the isolated perfused rat kidney. J Immunol. 1989;142(9):3076–3082. [PubMed: 2785132]
- 97.
- Van BruggenMC, Kramers C, Berden JH. Autoimmunity against nucleosomes and lupus nephritis. Ann Med Interne Paris. 1996;147(7):485–489. [PubMed: 9092359]
- 98.
- Van BruggenMC, Walgreen B, Rijke TP. et al. Antigen specificity of anti-nuclear antibodies complexed to nucleosomes determines glomerular basement membrane binding in vivo. Eur J Immunol. 1997;27(6):1564–1569. [PubMed: 9209511]
- 99.
- Kramers C, Hylkema MN, Van Bruggen MC. et al. Anti-nucleosome antibodies complexed to nucleosomal antigens show anti-DNA reactivity and bind to rat glomerular basement membrane in vivo. J Clin Invest. 1994;94(2):568–577. [PMC free article: PMC296132] [PubMed: 8040312]
- 100.
- Berden JH, Licht R, Van Bruggen MC. et al. Role of nucleosomes for induction and glomerular binding of autoantibodies in lupus nephritis. Curr Opin Nephrol Hypertens. 1999;8(3):299–306. [PubMed: 10456260]
- 101.
- Vlahakos DV, Foster MH, Adams S. et al. Anti-DNA antibodies form immune deposits at distinct glomerular and vascular sites. Kidney Int. 1992;41(6):1690–1700. [PubMed: 1501424]
- 102.
- Madaio MP, Shlomchik MJ. Emerging concepts regarding B cells and autoantibodies in murine lupus nephritis. B cells have multiple. J Am Soc Nephrol. roles;1996;all autoantibodies are not equal [editorial]7(3):387–396. [PubMed: 8704103]
- 103.
- D'Andrea DM, Coupaye GerardB, Kleyman TR. et al. Lupus autoantibodies interact directly with distinct glomerular and vascular cell surface antigens. Kidney Int. 1996;49(5):1214–1221. [PubMed: 8731084]
- 104.
- Amoura Z, Piette JC, Bach JF. et al. The key role of nucleosomes in lupus. Arthritis Rheum. 1999;42(5):833–843. [PubMed: 10323438]
- 105.
- Amoura Z, Koutouzov S, Chabre H. et al. Presence of antinucleosome autoantibodies in a restricted set of connective tissue diseases: Antinucleosome antibodies of the IgG3 subclass are markers of renal pathogenicity in systemic lupus erythematosus. Arthritis Rheum. 2000;43(1):76–84. [PubMed: 10643702]
- 106.
- Cervera R, Vinas O, Ramos-Casals M. et al. Anti-chromatin antibodies in systemic lupus erythematosus: A useful marker for lupus nephropathy. Ann Rheum Dis. 2003;62(5):431–434. [PMC free article: PMC1754546] [PubMed: 12695155]
- 107.
- Benedek TG. Historical background of discoid and systemic lupus erythematosusIn: Wallace DJ, Hahn BH, eds.Dubois' Lupus erythematosusBaltimore, Philadelphia, London, Paris, Bangkok, Buenos Aires, Hong Kong, Munich, Sydney, Tokio, Wroclaw: Williams & Wilkins,19973–16.
- 108.
- Lahita RG. Systemic Lupus Erythematosus. 3th ed. New York: Academic Press. 1999
- 109.
- Rekvig OP. Polyoma induced autoimmunity to. Lupus. DNA;1997;experimental systems and clinical observations in human SLE6(3):325–326. [PubMed: 9296778]
- 110.
- Rekvig OP, Andreassen K, Moens U. Antibodies to DNA-towards an understanding of their origin and pathophysiological impact in systemic lupus erythematosus [editorial] Scand J Rheumatol. 1998;27(1):1–6. [PubMed: 9506871]
- 111.
- Max EE. Immunoglobulins: Molecular GeneticsIn: Paul WE, ed.Fundamental ImmunologyNew York: Lippincott, Williams & Wilkins,1999111–182.
- 112.
- Chen C, Nagy Z, Radic MZ. et al. The site and stage of anti-DNA B-cell deletion. Nature. 1995;373(6511):252–255. [PubMed: 7816141]
- 113.
- Chen C, Prak EL, Weigert M. Editing disease-associated autoantibodies. Immunity. 1997;6(1):97–105. [PubMed: 9052841]
- 114.
- Wagner SD, Neuberger MS. Somatic hypermutation of immunoglobulin genes. Annu Rev Immunol. 1996;14:441–457. [PubMed: 8962691]
- 115.
- Parker DC. T cell-dependent B cell activation. Annu Rev Immunol. 1993;11:331–360. [PubMed: 8476565]
- 116.
- Benacerraf B, Paul WE, Green I. Hapten-carrier relationships. Ann NY Acad Sci. 1970;169(1):93–104. [PubMed: 4244326]
- 117.
- Goodman JW. Antigenic determinants and antibody combining sitesIn: Sela M, ed.The antigensNew York: Academic Press,1975127–187.
- 118.
- Flaegstad T, Fredriksen K, Dahl B. et al. Inoculation with BK virus may break immunological tolerance to histone and DNA antigens. Proc Natl Acad Sci USA. 1988;85(21):8171–8175. [PMC free article: PMC282389] [PubMed: 2847152]
- 119.
- Aarden LA, de GrootER, Feltkamp TE. Immunology of DNA. III. Crithidia luciliae, a simple substrate for the determination of anti-dsDNA with the immunofluorescence technique. [PubMed: 52321]
- 120.
- Madaio MP, Hodder S, Schwartz RS. et al. Responsiveness of autoimmune and normal mice to nucleic acid antigens. J Immunol. 1984;132(2):872–876. [PubMed: 6690621]
- 121.
- Schwartz RS, Stollar BD. Origins of anti-DNA autoantibodies. J Clin Invest. 1985;75(2):321–327. [PMC free article: PMC423484] [PubMed: 2579097]
- 122.
- Stollar BD. Antibodies to DNA. CRC Crit Rev Biochem. 1986;20(1):1–36. [PubMed: 3514122]
- 123.
- Stollar BD. Immunochemistry of DNA. Int Rev Immunol. 1989;5(1):1–22. [PubMed: 2491157]
- 124.
- Rekvig OP, Flaegstad T, Fredriksen K. et al. Stimulation of clones specific for dsDNA or idiotypes of anti-dsDNA as a consequence of BK virus inoculation. Immunol Invest. 1989;18(5):657–669. [PubMed: 2544515]
- 125.
- Fredriksen K, Traavik T, Flaegstad T. et al. BK virus terminates tolerance to dsDNA and histone antigens in vivo. Immunol Invest. 1990;19(2):133–151. [PubMed: 2159950]
- 126.
- Fredriksen K, Traavik T, Rekvig OP. Anti-DNA antibodies induced by BK virus inoculations. Demonstration of the specificities for eukaryotic dsDNA and synthetic polynucleotides. Scand J Immunol. 1990;32(2):197–203. [PubMed: 2167511]
- 127.
- Fredriksen K, Brannsether B, Traavik T. et al. Antibodies to viral and mammalian native DNA in response to BK virus inoculation and subsequent immunization with calf thymus DNA. Scand J Immunol. 1991;34(1):109–119. [PubMed: 1648784]
- 128.
- Fredriksen K, Skogsholm A, Flaegstad T. et al. Antibodies to dsDNA are produced during primary BK virus infection in man, indicating that anti-dsDNA antibodies may be related to virus replication in vivo. Scand J Immunol. 1993;38(4):401–406. [PubMed: 8211002]
- 129.
- Fredriksen K, Osei A, Sundsfjord A. et al. On the biological origin of anti-double-stranded (ds) DNA antibodies: Systemic lupus erythematosus-related anti-dsDNA antibodies are induced by polyomavirus BK in lupus-prone (NZBxNZW) F1 hybrids, but not in normal mice. Eur J Immunol. 1994;24(1):66–70. [PubMed: 8020573]
- 130.
- Gilkeson GS, Pippen AM, Pisetsky DS. Induction of cross-reactive anti-dsDNA antibodies in preautoimmune NZB/NZW mice by immunization with bacterial DNA. J Clin Invest. 1995;95(3):1398–1402. [PMC free article: PMC441482] [PubMed: 7883986]
- 131.
- Marion TN, Tillman DM, Jou NT. Interclonal and intraclonal diversity among anti-DNA antibodies from an (NZB x NZW)F1 mouse. J Immunol. 1990;145(7):2322–2332. [PubMed: 2118934]
- 132.
- Marion TN, Krishnan MR, Desai DD. et al. Monoclonal anti-DNA antibodies: Structure, specificity, and biology. Methods. 1997;11(1):3–11. [PubMed: 8990083]
- 133.
- Marion TN, Tillman DM, Jou NT. et al. Selection of immunoglobulin variable regions in autoimmunity to DNA. Immunol Rev. 1992;128:123–149. [PubMed: 1427921]
- 134.
- Krishnan MR, Marion TN. Structural similarity of antibody variable regions from immune and autoimmune anti-DNA antibodies. J Immunol. 1993;150(11):4948–4957. [PubMed: 8496596]
- 135.
- Krishnan MR, Marion TN. Comparison of the frequencies of arginines in heavy chain CDR3 of antibodies expressed in the primary B-cell repertoires of autoimmune-prone and normal mice. Scand J Immunol. 1998;48(3):223–232. [PubMed: 9743205]
- 136.
- Krishnan MR, Jou NT, Marion TN. Correlation between the amino acid position of arginine in VH- CDR3 and specificity for native DNA among autoimmune antibodies. J Immunol. 1996;157(6):2430–2439. [PubMed: 8805642]
- 137.
- Radic MZ, Mascelli MA, Erikson J. et al. Structural patterns in anti-DNA antibodies from MRL/ lpr mice. Cold Spring Harb Symp Quant Biol. 1989;54(Pt 2):933–946. [PubMed: 2640629]
- 138.
- Radic MZ, Weigert M. Genetic and structural evidence for antigen selection of anti- DNA antibodies. Annu Rev Immunol. 1994;12:487–520. [PubMed: 8011289]
- 139.
- Radic MZ, Mackle J, Erikson J. et al. Residues that mediate DNA binding of autoimmune antibodies. J Immunol. 1993;150(11):4966–4977. [PubMed: 8496598]
- 140.
- Shlomchik MJ, Marshak-Rothstein A, Wolfowicz CB. et al. The role of clonal selection and somatic mutation in autoimmunity. Nature. 1987;328(6133):805–811. [PubMed: 3498121]
- 141.
- Shlomchik MJ, Aucoin AH, Pisetsky DS. et al. Structure and function of anti-DNA autoantibodies derived from a single autoimmune mouse. Proc Natl Acad Sci USA. 1987;84(24):9150–9154. [PMC free article: PMC299710] [PubMed: 3480535]
- 142.
- Rekvig OP, Fredriksen K, Hokland K. et al. Molecular analyses of anti-DNA antibodies induced by polyomavirus BK in BALB/c mice Scand J Immunol 199541|(6):593–602 [published erratum appears in Scand J Immunol 1995 Aug42(2)286]. [PubMed: 7770729]
- 143.
- Bendiksen S, Rekvig OP, Van Ghelue M. et al. VP1 DNA sequences of JC and BK viruses detected in urine of systemic lupus erythematosus patients reveal no differences from strains expressed in normal individuals. J Gen Virol. 2000;81(Pt 11):2625–2633. [PubMed: 11038373]
- 144.
- Bendiksen S, Van GhelueM, Rekvig OP. et al. A longitudinal study of human cytomegalovirus serology and viruria fails to detect active viral infection in 20 systemic lupus erythematosus patients. Lupus. 2000;9(2):120–126. [PubMed: 10787009]
- 145.
- Cerutti ML, Centeno JM, Goldbaum FA. et al. Generation of sequence-specific, high affinity anti-DNA antibodies. J Biol Chem. 2001;276(16):12769–12773. [PubMed: 11279040]
- 146.
- Sundar K, Jaques S, Spatz L. et al. Mice immunized with Epstein-Barr nuclear antigen-1 expressing plasmids secrete antibodies to EBNA-1, dsDNA and Sm. Faseb J. 2001;15:A 1058.
- 147.
- Verdolini R, Bugatti L, Giangiacomi M. et al. Systemic lupus erythematosus induced by Epstein-Barr virus infection. Br J Dermatol. 2002;146(5):877–881. [PubMed: 12000388]
- 148.
- Dong X, Hamilton KJ, Satoh M. et al. Initiation of autoimmunity to the p53 tumor suppressor protein by complexes of p53 and SV40 large T antigen. J Exp Med. 1994;179(4):1243–1252. [PMC free article: PMC2191430] [PubMed: 8145041]
- 149.
- Reeves WH, Dong X, Wang J. et al. Initiation of autoimmunity to self-proteins complexed with viral antigens. Ann NY Acad Sci. 1997;815:139–154. [PubMed: 9186651]
- 150.
- Mohan C, Adams S, Stanik V. et al. Nucleosome: A major immunogen for pathogenic autoantibody- inducing T cells of lupus. J Exp Med. 1993;177(5):1367–1381. [PMC free article: PMC2191002] [PubMed: 8478612]
- 151.
- Datta SK, Kaliyaperumal A. Nucleosome-driven autoimmune response in lupus. Pathogenic T helper cell epitopes and costimulatory signals. Ann NY Acad Sci. 1997;815:155–170. [PubMed: 9186652]
- 152.
- Voll RE, Roth EA, Girkontaite I. et al. Histone-specific Th0 and Th1 clones derived from systemic lupus erythematosus patients induce double-stranded DNA antibody production. Arthritis Rheum. 1997;40(12):2162–2171. [PubMed: 9416853]
- 153.
- Beverly B, Kang SM, Lenardo MJ. et al. Reversal of in vitro T cell clonal anergy by IL-2 stimulation. Int Immunol. 1992;4(6):661–671. [PubMed: 1616898]
- 154.
- Jenkins MK. The role of cell division in the induction of clonal anergy. Immunol Today. 1992;13(2):69–73. [PubMed: 1349483]
- 155.
- Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol. 1996;14:233–258. [PubMed: 8717514]
- 156.
- Gimmi CD, Freeman GJ, Gribben JG. et al. Human T-cell clonal anergy is induced by antigen presentation in the absence of B7 costimulation. Proc Natl Acad Sci USA. 1993;90(14):6586–6590. [PMC free article: PMC46977] [PubMed: 7688125]
- 157.
- Schwartz RH. Costimulation of T lymphocytes: The role of CD28, CTLA-4, and B7/BB1 in interleukin-2 production and immunotherapy. Cell. 1992;71(7):1065–1068. [PubMed: 1335362]
- 158.
- Mueller DL, Jenkins MK, Schwartz RH. Clonal expansion versus functional clonal inactivation: A costimulatory signalling pathway determines the outcome of T cell antigen receptor occupancy. Annu Rev Immunol. 1989;7:445–480. [PubMed: 2653373]
- 159.
- Schwartz RH. T cell anergy. Annu Rev Immunol. 2003;21:305–334. [PubMed: 12471050]
- 160.
- Mamula MJ, Lin RH, Janeway Jr CA. et al. Breaking T cell tolerance with foreign and self coimmunogens. A study of autoimmune B and T cell epitopes of cytochromec. J Immunol. 1992;149(3):789–795. [PubMed: 1321851]
- 161.
- Lin RH, Mamula MJ, Hardin JA. et al. Induction of autoreactive B cells allows priming of autoreactive T cells. J Exp Med. 1991;173(6):1433–1439. [PMC free article: PMC2190847] [PubMed: 1851798]
- 162.
- Mamula MJ, Fatenejad S, Craft J. B cells process and present lupus autoantigens that initiate autoimmune T cell responses. J Immunol. 1994;152(3):1453–1461. [PubMed: 8301145]
- 163.
- Desai DD, Krishnan MR, Swindle JT. et al. Antigen-specific induction of antibodies against native mammalian DNA in nonautoimmune mice. J Immunol. 1993;151(3):1614–1626. [PubMed: 8393048]
- 164.
- Kisielow P, von BoehmerH. Development and selection of T cells: Facts and puzzles. Adv Immunol. 1995;58:87–209. [PubMed: 7741032]
- 165.
- Sprent J, Kishimoto H, Cai Z. et al. The thymus and T cell death. Adv Exp Med Biol. 1996;406:191–198. [PubMed: 8910685]
- 166.
- Surh CD, Sprent J. T-cell apoptosis detected in situ during positive and negative selection in the thymus. Nature. 1994;372(6501):100–103. [PubMed: 7969401]
- 167.
- Liu Y, Janeway JrCA. Microbial induction of costimulatory activity for CD4 T-cell growth. Int Immunol. 1991;3(4):323–332. [PubMed: 1831651]
- 168.
- Collins T, Korman AJ, Wake CT. et al. Immune interferon activates multiple class II major histocompatibility complex genes and the associated invariant chain gene in human endothelial cells and dermal fibroblasts. Proc Natl Acad Sci USA. 1984;81(15):4917–4921. [PMC free article: PMC391603] [PubMed: 6431411]
- 169.
- Page C, Rose M, Yacoub M. et al. Antigenic heterogeneity of vascular endothelium. Am J Pathol. 1992;141(3):673–683. [PMC free article: PMC1886681] [PubMed: 1519671]
- 170.
- Marelli BergFM, Hargreaves RE, Carmichael P. et al. Major histocompatibility complex class II-expressing endothelial cells induce allospecific nonresponsiveness in naïve. T cells J Exp Med. 1996;183(4):1603–1612. [PMC free article: PMC2192539] [PubMed: 8666918]
- 171.
- Marelli-Berg FM, Frasca L, Imami N. et al. Lack of T cell proliferation without induction of nonresponsiveness after antigen presentation by endothelial cells. Transplantation. 1999;68(2):280–287. [PubMed: 10440402]
- 172.
- Corrigall VM, Solau-Gervais E, Panayi GS. Lack of CD80 expression by fibroblast-like synoviocytes leading to anergy in T lymphocytes. Arthritis Rheum. 2000;43(7):1606–1615. [PubMed: 10902766]
- 173.
- Byrne B, Madrigal-Estebas L, McEvoy A. et al. Human duodenal epithelial cells constitutively express molecular components of antigen presentation but not costimulatory molecules. Hum Immunol. 2002;63(11):977–986. [PubMed: 12392850]
- 174.
- Lohse AW, Knolle PA, Bilo K. et al. Antigen-presenting function and B7 expression of murine sinusoidal endothelial cells and Kupffer cells. Gastroenterology. 1996;110(4):1175–1181. [PubMed: 8613007]
- 175.
- Murray AG, Khodadoust MM, Pober JS. et al. Porcine aortic endothelial cells activate human T cells: Direct presentation of MHC antigens and costimulation by ligands for human CD2 and CD28. Immunity. 1994;1(1):57–63. [PubMed: 7889399]
- 176.
- Denton MD, Geehan CS, Alexander SI. et al. Endothelial cells modify the costimulatory capacity of transmigrating leukocytes and promote CD28-mediated CD4(+) T cell alloactivation. J Exp Med. 1999;190(4):555–566. [PMC free article: PMC2195607] [PubMed: 10449526]
- 177.
- June CH, Ledbetter JA, Linsley PS. et al. Role of the CD28 receptor in T-cell activation. Immunol Today. 1990;11i(6):211–216. [PubMed: 2162180]
- 178.
- Marelli-Berg FM, Lechler RI. Antigen presentation by parenchymal cells: A route to peripheral tolerance? Immunol Rev. 1999;172:297–314. [PubMed: 10631955]
- 179.
- Napirei M, Karsunky H, Zevnik B. et al. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat Genet. 2000;25(2):177–181. [PubMed: 10835632]
- 180.
- Yoh K, Itoh K, Enomoto A. et al. Nrf2-deficient female mice develop lupus-like autoimmune nephritis. Kidney Int. 2001;60(4):1343–1353. [PubMed: 11576348]
- 181.
- Herrmann M, Zoller OM, Hagenhofer M. What triggers anti-dsDNA antibodies? Mol Biol Rep. 1996. pp. 265–267. [PubMed: 9112239]
- 182.
- Berden JH, Grootscholten C, Jurgen WC. et al. Lupus nephritis: A nucleosome waste disposal defect? J Nephrol. 2002;15(Suppl 6):S1–10. [PubMed: 12515368]
- 183.
- Meurman OH, Mantyjarvi RA, Salmi AA. et al. Prevalence of antibodies to a human papova virus (BK virus) in subacute sclerosing panencephalitis and multiple sclerosis patients. Z Neurol. 1972;203(2):191–194. [PubMed: 4119029]
- 184.
- Arthur RR, Dagostin S, Shah KV. Detection of BK virus and JC virus in urine and brain tissue by the polymerase chain reaction. J Clin Microbiol. 1989;27(6):1174–1179. [PMC free article: PMC267522] [PubMed: 2546971]
- 185.
- Arthur RR, Shah KV. Occurrence and significance of papovaviruses BK and JC in the urine. Prog Med Virol. 1989;36:42–61. [PubMed: 2555837]
- 186.
- Azzi A, Cesaro S, Laszlo D. et al. Human polyomavirus BK (BKV) load and haemorrhagic cystitis in bone marrow transplantation patients. J Clin Virol. 1999;14(2):79–86. [PubMed: 10588450]
- 187.
- Azzi A, De SantisR, Ciappi S. et al. Human polyomaviruses DNA detection in peripheral blood leukocytes from immunocompetent and immunocompromised individuals. J Neurovirol. 1996;2(6):411–416. [PubMed: 8972423]
- 188.
- Biel SS, Held TK, Landt O. et al. Rapid quantification and differentiation of human polyomavirus DNA in undiluted urine from patients after bone marrow transplantation. J Clin Microbiol. 2000;38(10):3689–3695. [PMC free article: PMC87458] [PubMed: 11015385]
- 189.
- Degener AM, Pietropaolo V, Di Taranto C. et al. Identification of a new control region in the genome of the DDP strain of BK virus isolated from PBMC. J Med Virol. 1999;58(4):413–419. [PubMed: 10421410]
- 190.
- Degener AM, Pietropaolo V, Di Taranto C. et al. Detection of JC and BK viral genome in specimens of HIV-1 infected subjects. New Microbiol. 1997;20(2):115–122. [PubMed: 9208421]
- 191.
- Di TarantoC, Pietropaolo V, Orsi GB. et al. Detection of BK polyomavirus genotypes in healthy and HIV-positive children. Eur J Epidemiol. 1997;13(6):653–657. [PubMed: 9324211]
- 192.
- Itoh S, Irie K, Nakamura Y. et al. Cytologic and genetic study of polyomavirus-infected or polyomavirus-activated cells in human urine. Arch Pathol Lab Med. 1998;122(4):333–337. [PubMed: 9648901]
- 193.
- Jin L, Pietropaolo V, Booth JC. et al. Prevalence and distribution of BK virus subtypes in healthy people and immunocompromized patients detected by PCR-restriction enzyme analysis. Clin Diagn Virol. 1995;3:285–295. [PubMed: 15566809]
- 194.
- Jin L. Rapid genomic typing of BK virus directly from clinical specimens. Mol Cell probes. 1993;7:331–334. [PubMed: 8232350]
- 195.
- Leung AY, Suen CK, Lie AK. et al. Quantification of polyoma BK viruria in hemorrhagic cystitis complicating bone marrow transplantation. Blood. 2001;98(6):1971–1978. [PubMed: 11535537]
- 196.
- Reese JM, Reissing M, Daniel RW. et al. Occurrence of BK virus and BK virus-specific antibodies in the urine of patients receiving chemotherapy for malignancy. Infect Immun. 1975;11(6):1375–1381. [PMC free article: PMC415225] [PubMed: 166919]
- 197.
- Shah KV, Daniel RW, Strickler HD. et al. Investigation of human urine for genomic sequences of the primate polyomaviruses simian virus 40, BK virus, and JC virus. J Infect Dis. 1997;176(6):1618–1621. [PubMed: 9395377]
- 198.
- Tsai RT, Wang M, Ou WC. et al. Incidence of JC viruria is higher than that of BK viruria in Taiwan. J Med Virol. 1997;52(3):253–257. [PubMed: 9210032]
- 199.
- Ling PD, Lednicky JA, Keitel WA. et al. The dynamics of herpesvirus and polyomavirus reactivation and shedding in healthy adults: A 14-month longitudinal study. J Infect Dis. 2003;187(10):1571–1580. [PubMed: 12721937]
- 200.
- Ding R, Medeiros M, Dadhania D. et al. Noninvasive diagnosis of BK virus nephritis by measurement of messenger RNA for BK virus VP1 in urine. Transplantation. 2002;74(7):987–994. [PubMed: 12394843]
- 201.
- Hogan TF, Padgett BL, Walker DL. et al. Survey of human polyomavirus (JCV, BKV) infections in 139 patients with lung cancer, breast cancer, melanoma, or lymphoma. Prog Clin Biol Res. 1983;105:311–324. [PubMed: 6304768]
- 202.
- Stoner GL, Agostini HT, Ryschkewitsch CF. et al. JC virus excreted by multiple sclerosis patients and paired controls from Hungary. Mult Scler. 1998;4(2):45–48. [PubMed: 9599332]
- 203.
- Pagani E, Delbue S, Mancuso R. et al. Molecular analysis of JC virus genotypes circulating among the Italian healthy population. J Neurovirol. 2003;9(5):559–566. [PubMed: 13129770]
- 204.
- Chima SC, Ryschkewitsch CF, Stoner GL. Molecular epidemiology of human polyomavirus JC in the Biaka Pygmies and Bantu of Central Africa. Mem Inst Oswaldo Cruz. 1998;93(5):615–623. [PubMed: 9830527]
- 205.
- Ryschkewitsch CF, Friedlaender JS, Mgone CS. et al. Human polyomavirus JC variants in Papua New Guinea and Guam reflect ancient population settlement and viral evolution. Microbes Infect. 2000;2(9):987–996. [PubMed: 10967279]
- 206.
- Miranda JJ, Sugimoto C, Paraguison R. et al. Genetic diversity of JC virus in the modern Filipino population: Implications for the peopling of the Philippines. Am J Phys Anthropol. 2003;120(2):125–132. [PubMed: 12541330]
- 207.
- Koralnik IJ, Boden D, Mai VX. et al. JC virus DNA load in patients with and without progressive multifocal leukoencephalopathy Neurology 199952|(2):253–260. [PubMed: 9932940]
- 208.
- Li RM, Branton MH, Tanawattanacharoen S. et al. Molecular identification of SV40 infection in human subjects and possible association with kidney disease. J Am Soc Nephrol. 2002;13(9):2320–2330. [PubMed: 12191976]
- 209.
- Markowitz RB, Dynan WS. Binding of cellular proteins to the regulatory region of BK virus DNA. J Virol. 1988;62(9):3388–3398. [PMC free article: PMC253462] [PubMed: 2841492]
- 210.
- Moens U, Johansen T, Johnsen JI. et al. Noncoding control region of naturally occurring BK virus variants: Sequence comparison and functional analysis. Virus Genes. 1995;10(3):261–275. [PubMed: 8560788]
- Immunity and Autoimmunity Induced by Polyomaviruses: Clinical, Experimental and ...Immunity and Autoimmunity Induced by Polyomaviruses: Clinical, Experimental and Theoretical Aspects - Madame Curie Bioscience Database
- Methylenetetrahydrofolate Reductase Polymorphisms: Pharmacogenetic Effects - Mad...Methylenetetrahydrofolate Reductase Polymorphisms: Pharmacogenetic Effects - Madame Curie Bioscience Database
- NMD and Human Disease - Madame Curie Bioscience DatabaseNMD and Human Disease - Madame Curie Bioscience Database
- Telomeres: Guardians of Genomic Integrity or Double Agents of Evolution? - Madam...Telomeres: Guardians of Genomic Integrity or Double Agents of Evolution? - Madame Curie Bioscience Database
- Preprotein Translocation through the Sec Translocon in Bacteria - Madame Curie B...Preprotein Translocation through the Sec Translocon in Bacteria - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...