

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

The keratin cytoskeleton of hepatocytes is affected in a variety of chronic liver diseases, such as alcoholic and nonalcoholic steatohepatitis (ASH, NASH), copper toxicosis, cholestasis and hepatocellular carcinoma. In these diseases hepatocytes reveal a derangement or even loss of the cytoplasmic keratin intermediate filament cytoskeleton and formation of cytoplasmic inclusions (Mallory bodies) consisting of misfolded and aggregated keratin as well as a variety of stress proteins. Keratin gene knock-out mice demonstrated that keratins fulfil besides a structural role providing mechanical stability to hepatocytes a role as target and modulator of toxic stress responses in that keratins interact with a variety of stress-related signaling pathways, are preferred targets of stress-induced protein misfolding and are substrates for caspases. Furthermore, the identification of mutations in keratin genes in patients with liver cirrhosis suggests that keratins act as genetic modifiers in liver diseases.
The Keratin Cytoskeleton of Hepatocytes and Bile Duct Epithelial Cells
The keratin intermediate filament (IF) cytoskeleton of hepatocytes is composed of the type I keratin 18 (K18) and type II keratin 8 (K8) in equimolar ratios.1-3 Bile duct epithelia have a more complex keratin composition and express in addition to K8 and K18 also K7 and K19. Since K7 is present in higher concentrations than K19 the major partner for K7 in bile duct epithelia is K18. This preferred partnership of K7 and K18 was demonstrated in K18 knock-out mice where in the absence of K18 there was also a loss of K7 in bile duct epithelia.4 In certain liver diseases, such as alcoholic steatohepatitis (ASH) and chronic cholestasis also single hepatocytes as well as small groups of hepatocytes express in addition to K8 and K18, K7 and, to a lesser extent, K19. This phenomenon was originally regarded as evidence for ductular metaplasia (i.e., hepatocytes acquire features of bile duct epithelial).5,6 A metaplastic transition of hepatocytes to bile duct epithelia, however, was so far not observed in mouse models for ASH or cholestasis.7 Another explanation for the occurrence of K7-positive hepatocytes is that in certain liver diseases regeneration of hepatocytes involves proliferation and differentiation of hepatocytic precursor cells (oval cells), which normally express K8, K18 as well as K7 and K19.8,9 Therefore, the presence of hepatocytes expressing K7 indicates that these hepatocytes originated from precursor cells and not by proliferation of differentiated hepatocytes, the latter of which is the classical mode of hepatocyte regeneration.9
The biological significance of the expression of different keratin pairs in bile duct epithelia and the expression of bile duct-type keratins in hepatocytes is unclear. Studies in keratin gene knock-out mice demonstrated that a regular liver can be formed in the absence either of K8, K18 or K19.4,10-12 However mice lacking K8 or K18 or expressing a mutated K18 gene were more sensitive to a variety of toxins and stress conditions.13 Furthermore, studies performed with epidermal keratins provided evidence that different keratin family members are associated with distinct cellular functions. In the epidermis keratinocytes replace the keratin pairs K5 / K14 by K1 / K10 during their course of differentiation.14,15 In situations of wound healing and in neoplasia keratinocytes start expressing different keratins, such as K16, which is associated with enhanced cell proliferation and migration.16,17 A relationship between specific keratin proteins and cell function was recently demonstrated in transfected keratinocytes, where expression of K16 resulted in higher cell proliferation rates whereas expression of K10 reduced proliferation.18,19
Alterations of the Keratin Cytoskeleton in Human Liver Diseases
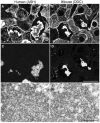
Alterations of the hepatocytic keratin IFs is a characteristic feature of ASH, which develops in approximately 20% of heavy drinkers and rapidly leads to the development of liver cirrhosis the major cause of death of alcoholics. In ASH hepatocytes on the one hand accumulate fat in the cytoplasm (steatosis), because of a general disturbance of lipid metabolism and, on the other hand, there are non steatotic hepatocytes, which are enlarged (ballooned) and may contain cytoplasmic protein aggregates termed Mallory bodies (MBs). Further morphologic alterations in ASH are a chicken wirelike fibrosis, an inflammatory reaction with predominantly polymorphonuclear granulocytes, and cholestasis.20-22 It has been demonstrated more than 20 years ago that the major cellar structure that is affected in ballooned hepatocytes is the keratin cytoskeleton. Ballooned hepatocytes reveal a derangement or even loss of the cytoplasmic keratin IFs and aggregation of keratin proteins as MBs23 (fig. 1). These alterations of the keratin cytoskeleton are characteristic but not specific of ASH since they are also seen in a variety of other liver diseases such as (i) nonalcoholic steatohepatitis (NASH), which occurs in patients with type II diabetes and obesity, after intestinal bypass surgery, and can be induce by certain drugs, (ii) chronic cholestasis, particularly primary biliary cirrhosis, (iii) copper intoxication as it occurs in patients with Wilson disease and Indian childhood cirrhosis, (iv) hepatocellular carcinoma24,25 (fig. 1).

The common occurrence of alterations of the hepatocytic keratin cytoskeleton in such a variety of diseases poses the question whether there is a unique pathogenetic principle leading to this phenotype. Although the above mentioned diseases originate from different aetiologies, increased oxidative stress is a constant and common feature. For example, in ASH oxidation of ethanol results in the formation acetaldehyde, which depletes glutathione and disturbs mitochondrial function, both of which result in increased oxidative stress. Furthermore metabolism of alcohol by cytochrome P450 2E1 leads to generation of reactive oxygen species (ROS) as by-product.26 In NASH oxidative stress is an indirect consequence of mitochondrial overload with free fatty acids, and the increased release of TNF-α from adipose tissue.27 In chronic cholestasis there is accumulation of bile acids in hepatocytes, which also results in oxidative stress either directly by affecting signaling and mitochondrial function or indirectly by activation of polymorphonuclear granulocytes.28 In copper toxicosis copper leads to oxidative stress by mediating the formation of hydroxyl radicals by the Fenton reaction. The biological role of oxidative stress in copper storage-associated liver diseases was recently underlined by the common presence of MBs containing the stress-induced, ubiquitin binding protein p62 in livers with Wilson disease or Indian childhood cirrhosis.29 In liver cancer ballooned tumor cells containing MBs or cytoplasmic hyaline bodies, which share features with MBs and can be considered as precursors of MBs, are found in 10-20% of cancer cases30 (Denk et al, manuscript submitted). Comparative gene expression profiling of HCC with and without inclusions demonstrated increased expression of heat shock proteins, p62 and keratin in HCC containing inclusions, which supports the hypothesis that formation of cytoplasmic inclusions is associated with increased stress (Neumann et al, unpublished observations). This observation is in line with the emerging general evidence that cancer cells experience increased oxidative stress and reveal altered responses to reactive oxygen species.31
Animal Models to Study Hepatocytic Keratin Alterations
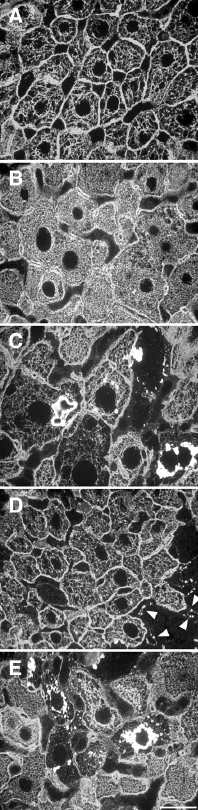
The alterations of the keratin cytoskeleton observed in human liver diseases can be reproduced in mice by feeding a diet containing griseofulvin (GF) or 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC)24,32,33 (fig. 2). Time course studies in GF- or DDC-fed mice, immunohistochemical and biochemical analyses of MBs as well as studies in K8 and K18 knock-out mice provided insights into the functional relevance of the keratin alterations in liver disease. DDC intoxication rapidly (within 2 days) leads to phosphorylation of keratin IFs at various phosphorylation sites.34 Some of these phosphorylation sites are targets for protein kinases typically activated in stress conditions indicating that DDC induces a stress response in hepatocytes.35 Furthermore mice respond to intoxication with marked overexpression of K8 and K18 resulting in a denser keratin IF network in enlarged hepatocytes34,36 (fig. 3). This overexpression of keratin apparently is an active (defence)response of hepatocytes to toxic injury since mice with one deleted K8 allele (K8+/-), which had limited capacities to increase keratin expression, were more sensitive to toxic injury than wild-type mice.37 The concept that keratin overexpression confers resistance to drug toxicity is not restricted to DCC intoxication since in a previous report marked induction of multidrug resistance in cultured mouse fibroblasts was achieved by transfection with K8 and K18 cDNAs.38 Interestingly, the overexpression of keratin in hepatocytes is a specific reaction to certain types of injury, such as ethanol, DDC, GF or cholestasis and not seen after intoxication with a series of cytostatic drugs7,34,36,39 (Lackner et al, unpublished observation). The mode of transcriptional control, which leads to these rather specific alterations in keratin expression in liver diseases, is unknown. Continuation of DDC or GF intoxication for up to two months leads to a reduction of the density to a loss of cytoplasmic keratin IFs in hepatocytes (fig. 3). The loosening of the keratin network is accompanied by the appearance small MBs at intersections of the keratin IF network at around 4-6 weeks of DDC intoxication. After 8 weeks larger MBs are found typically in the perinuclear region without association with keratin IFs (fig. 3). These alterations are reversible upon cessation of intoxication. After 4 weeks of recovery only granular remnants of MBs are found at the cell periphery preferentially in association with desmosomes of hepatocytes most of which are still devoid of a cytoplasmic keratin IF network (fig. 3). At this stage of recovery mice rapidly respond with MB formation upon reexposure to DDC.34 This enhanced reappearance of MBs is also seen in humans with ASH if patients are exposed to alcohol even after prolonged periods of abstinence and is reminiscent of a “toxic memory response”. Recent comparative studies in naïve mice and mice recovered from DDC intoxication, which were exposed to DDC for 3 days revealed marked differences in cytochrome P450 contents in these two groups of mice (Stumptner et al, unpublished observation). These findings indicate a relationship between DDC metabolism and cytoskeletal alterations. DDC is known to be metabolized by cytochrome P450 by N-demethylation, which leads to the generation of a methyl radical.40 This indicates that chronic radical injury is a major consequence of DDC intoxication and the putative common pathogenetic principle in human ASH, NASH and other diseases associated with MB formation.
Mallory Bodies As a Product of the Cellular Response to Misfolded Keratin
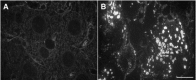
Occurrence of misfolded proteins is a major consequence of radical injury. Oxidation of amino acid residues results in conformational changes and exposure of hydrophobic residues at the protein surface. Such misfolded proteins are inactive, can disturb cellular functions and have the tendency to aggregate via hydrophobic interactions.41 Several cellular defence mechanisms against misfolded proteins exist. Misfolded proteins can either be refolded under consumption of ATP by heat shock protein 70 (HSP), preserved in a folding competent state by HSP 25/27, tagged by ubiquitin for degradation by the proteasome or aggregate in association with ubiquitin binding protein p62 as cytoplasmic inclusions recently designated in cultured cells as aggresomes or sequestosomes.41-50 Analysis of the protein composition of MBs by mass spectrometry revealed that MBs consist predominantly of keratin, ubiquitinated keratin, HSP 70, HSP 25 and p62.50 Immunohistochemical studies which confirmed the mass spectrometry data revealed in addition the presence α-B crystallin, another member of the small HSP family, and proteasomal subunits23,24,51-55 (Table 1). The protein composition of MBs indicates hat in these diseases all major cellular defence mechanism against misfolded proteins (i.e., misfolded keratin) are involved. Abnormal conformation of keratins in human ASH and experimentally induced MBs has been demonstrated by monoclonal antibodies recognizing conformation-dependent epitopes, the excess of keratin 8 over keratin 18 in MBs, which is not compatible with proper assembly of IFs, and the reduction in α-helically structured domains.34,56- 58 These data imply that keratin is the primary target for modification by radicals and misfolding, whereas all other MB components reflect the cellular response to misfolded proteins. The central role of keratins in MB formation has been demonstrated with keratin 8 and keratin 18 null mice. Keratin 18 null mice responded to DDC-intoxication with enhanced MB formation and formed MBs spontaneously at high age4 (Stumptner et al, manuscript submitted). In contrast no MBs were formed in the absence of keratin 837 (fig. 4). Surprisingly, keratin 8 null mice, which did not form MBs revealed markedly increased toxicity, whereas keratin 18 null mice tolerated DDC intoxication like wild type mice. This discrepancy in the phenotypes of DDC-treated keratin 8 null and keratin 18 null mice allows drawing several important conclusions: (i) Keratin 8 is the core component of MBs and all other MB components bind to or coassemble with keratin, (ii) MBs by themselves are not toxic to cells but MB occurrence is associated with better tolerance to toxic stress, (iii) since in both types of knock-out mice hepatocytes are devoid of a keratin IF cytoskeleton, the increased sensitivity of keratin 8 null mice cannot by related to the loss of keratin IFs but indicates that keratins fulfil in addition nonstructural roles in toxic stress situations.37,59
Table 1
Summary of major Mallory body components.
How Can Keratins Influence Toxic Cell Injury?
Keratins were shown to undergo extensive modifications in various cell culture and in vivo conditions. They include phosphorylation, ubiquitination, proteolytic cleavage, and cross linking, which modulate the status of the keratin IF cytoskeleton and the interaction of keratins with several cellular proteins, particularly signaling molecules.35,54,60,61 K8 and 18 are substrates of a variety of protein kinases involved in mitosis, apoptosis and stress.35 Antibodies directed against specific phosphoepitopes on keratin revealed that K8 and 18 become phosphorylated at many sites in human ASH as well as in DDC-fed mice.62 Some of these phosphorylation sites were targets of stress-induced protein kinases and were phosphorylated already within one day of intoxication. This indicates that phosphorylation of keratin might be one of the initial mechanisms of involving keratins in toxic stress responses. A direct role of keratin phosphorylation as modulator of cell toxicity was demonstrated in transgenic mice expressing a K18 with a mutated phosphorylation site. These mice were more sensitive to treatment with griseofulvin or microcystin than wild-type mice or transgenic mice expressing nonmutated K18.63 The functional relationship between keratin phosphorylation and toxicity could be based on the fact that phosphorylation status of keratins regulates IF assembly/disassembly as well as interaction of keratin with various signaling proteins. For example, a phosphorylation-dependent interaction of K18 with 14-3-3 was demonstrated recently.64 14-3-3 occurs in various isoforms and is an important integrator of major signaling pathways regulating cell proliferation and apoptosis.65 The biological significance of the interaction of keratin with 14-3-3 was show in mice with an impaired keratin cytoskeleton, in which an association between alterations of keratin, 14-3-3 and cell proliferation was observed.66 Furthermore, keratin is able to modulateTNF-α signaling. Association of keratin to tumor necrosis factor receptor 2 (TNFR2) influences TNF-α-induced activation of Jun NH2-terminal kinase (JNK) as well as NFκB.67 The interaction of keratin with TNF-α signaling could play a central role in ASH and NASH where enhanced production of TNF-α and activation of JNK and NFκB are constant findings.27,68-70 Furthermore keratins were shown to modulate induction and execution of apoptosis by interfering at various levels with the apoptotic program. In K8-deficient mice there is enhanced targeting of Fas to the plasma membrane, which results in three- to four-fold increased sensitivity to Fas-mediated apoptosis.71 A similar observation of increased Fas-mediated apoptosis was made in mice expressing mutated K18.72 Furthermore K8- as well as in K18- deficient mice were up to 100-fold more sensitive to TNF-α-induced cell death, which was explained by the interaction of K8 and K18 with TNFR2.67 Other studies, however, did only demonstrate a relationship between K8 or K18 and Fas but not TNF-α- induced apoptosis.71,72 A further mechanism by which keratin modulates Fas or TNF-α mediated apoptosis was recently demonstrated in keratin 8 null mice. These mice showed marked reduction of c-Flip and did not respond with Erk1/2 activation upon stimulation of Fas, TNF-α receptor or TNF-α-related apoptosis-inducing ligand receptor.73 A direct role of keratin in this pathway was suggested by the fact that keratin was found in association with a complex consisting of c-Flip, Raf, and Erk1/2.
Keratins are also targets in and modulators of the execution of apoptosis since keratins are cleaved by and associate with caspases. Cleavage of keratins by caspases leads to a disruption of the keratin IF network and the formation of small globular cytoplasmic inclusions containing cleaved and hyperphosphorylated keratins as well as activated caspases.74 The association of activated caspases with keratin inclusions is a characteristic feature of epithelial cell apoptosis and is expected to influence the activity of caspases in the execution of the apoptotic program.
Mutations of Keratin Genes and Liver Diseases
Heterozygous mutations in the K8 and K18 genes were found in 17 out of 467 (3.6%) explanted livers from patients with end-stages of various acute and chronic liver diseases.75-77 75% of all mutations identified were at K8 Y53H, K8 G61C and K18 H127L, suggesting existence of mutation hot spots. The keratin mutations were shown to lead to reorganization and partial collapse of the keratin IF network in patient livers.77 Furthermore the mutations affected keratin assembly in transfected cells if they were exposed to oxidative stress.76 The preference of keratin mutations in patients with cryptogenic liver cirrhosis indicates that the presence of a keratin mutation might predispose to the development of liver cirrhosis, in particular in patients with NASH since NASH is regarded as the major cause of cryptogenic liver cirrhosis.
Since keratin mutations were found also in a healthy cohort in a frequency of 0.6%, heterozygous keratin mutations per se do not lead to liver disease but could rather act as a genetic modifier. Interestingly, keratin mutations were not found in patients with earlier stages of liver diseases.78 This discrepancy to the studies performed by B. Omary in end-stage liver disease75-77 underlines that heterozygous mutations of K8 and K18 are not the cause of liver disease in humans but could characterize patients with risk for enhanced progression of liver disease to cirrhosis requiring liver transplantation.
References
- 1.
- Franke WW, Denk H, Kalt R. et al. Biochemical and immunological identification of cytokeratin proteins present in hepatocytes of mammalian liver tissue. Exp Cell Res. 1981;131:299–318. [PubMed: 6162655]
- 2.
- Moll R, Franke WW, Schiller D. et al. The catalog of human cytokeratins: Patterns of expression in normal epithelia, tumors and cultured cells. Cell. 1982;31:11–24. [PubMed: 6186379]
- 3.
- Fuchs E, Weber K. Intermediate filaments: Structure, dynamics, function, and disease. Annu Rev Biochem. 1994;63:345–382. [PubMed: 7979242]
- 4.
- Magin TM, Schröder R, Leitgeb S. et al. Lessons from keratin 18 knockout mice; formation of novel keratin filaments, secondary loss of keratin 7 and accumulation of liver specific keratin 8-positive aggregates. J Cell Biol. 1998;140:1441–1451. [PMC free article: PMC2132680] [PubMed: 9508776]
- 5.
- Van EykenP, Sciot R, Desmet VL. A cytokeratin immunohistochemical study of alcoholic liver disease: Evidence that hepatocytes can express “bile duct-type” cytokeratins. Histopathology. 1988;13:605–617. [PubMed: 2466751]
- 6.
- Van EykenP, Sciot R, Desmet VL. A cytokeratin immunohistochemical study of cholestatic liver disease: Evidence that hepatocytes can express “bile duct-type” cytokeratins. Histopathology. 1989;15:125–135. [PubMed: 2476370]
- 7.
- Fickert P, Trauner M, Fuchsbichler A. et al. Cytokeratins as targets for bile acid induced toxicity. Am J Pathol. 2002;160:491–499. [PMC free article: PMC1850630] [PubMed: 11839569]
- 8.
- Van EykenP, Sciot R, Callea F. et al. The development of the intrahepatic bile ducts in man: A keratin-immunohistochemical study. Hepatology. 1988;8:1586–1595. [PubMed: 2461337]
- 9.
- Roskams TA, Libbrecht L, Desmet VJ. Progenitor cells in diseased human liver. Semin Liver Dis. 2003;23:385–396. [PubMed: 14722815]
- 10.
- Baribault H, Price H, Miyai K. et al. Mid-gestational lethality in mice lacking keratin 8. Genes Dev. 1993;7:1191–1202. [PubMed: 7686525]
- 11.
- Baribault H, Penner J, Iozzo RV. et al. Colorectal hyperplasia and inflammation in keratin 8-deficient FVB/N mice. Genes Dev. 1994;8:2964–2974. [PubMed: 7528156]
- 12.
- Hesse M, Franz T, Tamai Y. et al. Targeted deletion of keratins 18 and 19 leads to trophoblast fragility and early embryonic lethality. EMBO J. 2000;19:5060–5070. [PMC free article: PMC302090] [PubMed: 11013209]
- 13.
- Omary BM, Ku NO, Toivola DM. Keratins: Guardians of the liver. Hepatology. 2002;35:251–257. [PubMed: 11826396]
- 14.
- Herrmann H, Hesse M, Reichenzeller M. et al. Functional complexity of intermediate filament cytoskeletons: From structure to assembly to gene ablation. Int Rev Cytol. 2003;223:83–175. [PubMed: 12641211]
- 15.
- Fuchs E. Epidermal differentiation: The bare essentials. J Cell Biol. 1990;111:2807–2814. [PMC free article: PMC2116387] [PubMed: 2269655]
- 16.
- Paladini RD, Takahashi K, Bravo NS. et al. Onset of reepithelialization after skin injury correlates with a reorganization of keratin filaments in wound edge keratinocytes: Defining a potential role for keratin 16. J Cell Biol. 1996;132:381–397. [PMC free article: PMC2120730] [PubMed: 8636216]
- 17.
- McGowan K, Coulombe PA. The wound repair-associated keratins 6, 16, and 17. Insightsinto the role of intermediate filaments in specifying keratinocyte cytoarchitecture. Subcell Biochem. 1998;31:173–204. [PubMed: 9932493]
- 18.
- Paramio JM, Casanova ML, Segrelles C. et al. Modulation of cell proliferation by cytokeratins K10 and K16. Mol Cell Biol. 1999;19:3086–3094. [PMC free article: PMC84102] [PubMed: 10082575]
- 19.
- Paramio JM, Segrelles C, Ruiz S. et al. Inhibition of protein kinase B (PKB) and PKCzeta mediates keratin K10-induced cell cycle arrest. Mol Cell Biol. 2001;21:7449–7459. [PMC free article: PMC99917] [PubMed: 11585925]
- 20.
- Burt AD, Mutton A, Day CP. Diagnosis and interpretation of steatosis and steatohepatitis. Sem Diagnostic Pathol. 1998;15:246–258. [PubMed: 9845426]
- 21.
- Hall P. Pathological spectrum of alcoholic liver diseaseIn: Hall P, ed.Pathology and Pathogenesis: Alcoholic Liver DiseaseLondon, Boston, Melbourne, Auckland: Edward Arnold,199541–68.
- 22.
- Brunt E. Nonalcoholic Steatohepatitis. Semin Liver Dis. 2004;24:3–20. [PubMed: 15085483]
- 23.
- Denk H, Franke WW, Eckerstorfer R. et al. Formation and involution of Mallory bodies (alcoholic hyalin) in murine and human liver revealed by immunofluorescence microscopy with antibodies to prekeratin. Proc Natl Acad Sci USA. 1979;76:4112–4116. [PMC free article: PMC383988] [PubMed: 386356]
- 24.
- Denk H, Stumptner C, Zatloukal K. Mallory body revisited. J Hepatol. 2000;32:689–702. [PubMed: 10782920]
- 25.
- Zatloukal K, Stumptner C, Fuchsbichler A. et al. The keratin cytoskeleton in liver disease. J Pathol. 2004;204:367–376. [PubMed: 15495250]
- 26.
- Lieber CS. Alcoholic liver disease: New insights in pathogenesis lead to new treatments. J Hepatol. 2000;32:113–128. [PubMed: 10728799]
- 27.
- Angulo P. Nonalcoholic fatty liver disease. N Engl J Med. 2002;16:1221–1231. [PubMed: 11961152]
- 28.
- Fang Y, Han SI, Mitchell C. et al. Bile acids induce mitochondrial ROS, which promote activation of receptor tyrosine kinases and signaling pathways in rat hepatocytes. Hepatology. 2004;40:961–971. [PubMed: 15382121]
- 29.
- Muller T, Langner C, Fuchsbichler A. et al. Immunohistochemical analysis of Mallory bodies in Wilsonian and non hepatic copper toxicosis. Hepatology. 2004;39:963–969. [PubMed: 15057900]
- 30.
- Stumptner C, Heid H, Fuchsbichler A. et al. Analysis of intracytoplasmic hyaline bodies in a hepatocellular carcinoma. Demonstration of p62 as major constituent. Am J Pathol. 1999;154:1701–1710. [PMC free article: PMC1866621] [PubMed: 10362795]
- 31.
- Benhar M, Engelberg D, Levitzki A. ROS, stress activated kinases and stress signaling in cancer. EMBO Rep. 2002;3:420–425. [PMC free article: PMC1084107] [PubMed: 11991946]
- 32.
- Denk H, Gschnait F, Wolff K. Hepatocellular hyalin (Mallory bodies) in long term griseofulvin-treated mice: A new experimental model for the study of hyalin formation. Lab Invest. 1975;32:773–776. [PubMed: 50498]
- 33.
- Tsunoo C, Harwood TR, Arak S. et al. Cytoskeletal alterations leading to Mallory body formation in livers of mice fed 3,5-diethoxycarbonyl-1,4-dihydrocollidine. J Hepatol. 1987;5:85–97. [PubMed: 2443554]
- 34.
- Stumptner C, Fuchsbichler A, Lehner K. et al. Sequence of events in the assembly of Mallory body components in mouse liver: Clues to the pathogenesis and significance of Mallory body formation. J Hepatol. 2001;34:665–675. [PubMed: 11434612]
- 35.
- Omary MB, Ku NO, Liao J. et al. Keratin modifications and solubility properties in epithelial cells and in vitro. Subcell Biochem. 1998;31:105–140. [PubMed: 9932491]
- 36.
- Cadrin M, Hovington H, Marceau N. et al. Early perturbation in keratin and actin gene expression and fibrillar organisation in griseofulvin-fed mouse liver. J Hepatol. 2000;33:199–207. [PubMed: 10952237]
- 37.
- Zatloukal K, Stumptner C, Lehner M. et al. Cytokeratin 8 protects from hepatotoxicity, and its ratio to cytokeratin 18 determines the ability of hepatocytes to form Mallory bodies. Am J Pathol. 2000;156:1263–1274. [PMC free article: PMC1876873] [PubMed: 10751352]
- 38.
- Bauman PA, Dalton WS, Anderson JM. et al. Expression of cytokeratin confers multiple drug resistance. Proc Natl Acad Sci USA. 1994;91:5311–5314. [PMC free article: PMC43984] [PubMed: 7515497]
- 39.
- Fickert P, Trauner M, Fuchsbichler A. et al. Bile acid-induced Mallory body formation in drug-primed mouse liver. Am J Pathol. 2002;161:2019–2026. [PMC free article: PMC1850910] [PubMed: 12466118]
- 40.
- Tephly TR, Coffman BL, Ingall G. et al. Identification of N-methylprotophorphyrin IX in livers of untreated mice and mice treated with 3,5-diethoxycarbonyl-1,4-dihydrocollidine: Source of the methyl group. Arch Biochem Biophys. 1981;212:120–126. [PubMed: 6895449]
- 41.
- Grune T, Reinheckel T, Davies KJA. Degradation of oxidized proteins in mammalian cells. FASEB J. 1997;11:526–534. [PubMed: 9212076]
- 42.
- Ehrnsperger M, Gräber S, Gaeste M. et al. Binding of nonnative protein to Hsp25 during heat shock creates a reservoir of folding intermediates for reactivation. EMBO J. 1997;16:221–229. [PMC free article: PMC1169629] [PubMed: 9029143]
- 43.
- Hartl FU. Molecular chaperones in cellular protein folding. Nature. 1996;381:571–580. [PubMed: 8637592]
- 44.
- Vadlamudi RK, Joung I, Strominger J. et al. p62, a phosphotyrosine-independent ligand of the SH2 domain of p56lck, belongs to a new class of ubiquitin-binding proteins. J Bio Chem. 1996;271:20235–20237. [PubMed: 8702753]
- 45.
- Shin J. P62 and the sequestosome, a novel mechanism for protein metabolism. Arch Pharm. 1998;21:629–633. [PubMed: 9868528]
- 46.
- Wickner S, Maurizi MR, Gottesman S. Posttranslational quality control: Folding, refolding, and degrading proteins. Science. 1999;286:1888–1893. [PubMed: 10583944]
- 47.
- Johnston JA, Ward CL, Kapito RR. Aggresomes: A cellular response to misfolded proteins. J Cell Biol. 1998;143:1883–1898. [PMC free article: PMC2175217] [PubMed: 9864362]
- 48.
- Stumptner C, Fuchsbichler A, Heid H. et al. Mallory body—A disease associated type of sequestosome. Hepatology. 2002;35:1053–1062. [PubMed: 11981755]
- 49.
- Ciechanover A, Schwartz AL. Ubiquitin-mediated degradation of cellular proteins in health and disease. Hepatology. 2002;35:3–6. [PubMed: 11786953]
- 50.
- Zatloukal K, Stumptner C, Fuchsbichler A. et al. p62 a common component of the cellular response to aggregated proteins. Am J Pathol. 2002;160:255–263. [PMC free article: PMC1867135] [PubMed: 11786419]
- 51.
- Franke WW, Denk H, Schmid E. et al. Ultrastructural, biochemical and immunologic characterization of Mallory bodies in livers of griseofulvin-treated mice. Fimbriated rods of filaments containing prekeratin-like polypeptides. Lab Invest. 1979;40:207–220. [PubMed: 219290]
- 52.
- Lowe J, Blanchard A, Morell K. et al. Ubiquitin is a common factor in intermediate filament inclusion bodies of diverse type in man, including those of Parkinson's disease, Pick's disease, and Alzheimer's disease, as well as Rosenthal fibers in cerebellar astrocytomas, cytoplasmic bodies in muscle, and Mallory bodies in alcoholic liver disease. J Pathol. 1988;155:9–15. [PubMed: 2837558]
- 53.
- Lowe J, McDermott H, Pike I. et al. Alpha B crystallin expression in nonlenticular tissues and selective presence in ubiquitinated inclusion bodies in human disease. J Pathol. 1992;166:61–68. [PubMed: 1311375]
- 54.
- Ohta M, Marceau N, Perry G. et al. Ubiquitin is present on the cytokeratin intermediate filaments and Mallory bodies of hepatocytes. Lab Invest. 1988;59:848–856. [PubMed: 2462130]
- 55.
- Riley NE, Li J, McPhaul LW. et al. Heat shock proteins are present in Mallory bodies (cytokeratin aggresomes) in human liver biopsy specimens. Exp Mol Pathol. 2003;74:168–172. [PubMed: 12710948]
- 56.
- Hazan R, Denk H, Franke WW. et al. Change of cytokeratin organization during development of Mallory bodies as revealed by a monoclonal antibody. Lab Invest. 1986;54:543–553. [PubMed: 2422438]
- 57.
- Zatloukal K, Fesus L, Denk H. et al. High amount of ε - (γ-glutamyl) lysine cross-links in Mallory bodies. Lab Invest. 1992;66:774–777. [PubMed: 1351114]
- 58.
- Cadrin M, French SW, Wong TT. Alteration in molecular structure of cytoskeleton proteins in griseofulvin-treated mouse liver: A pressure tuning infrared spectroscopy study. Exp Mol Pathol. 1991;55:170–179. [PubMed: 1936212]
- 59.
- Coulombe PA, Omary MB. ‘Hard’ and ‘soft’ principles defining the structure, function and regulation of keratin intermediate filaments. Curr Opin Cell Biol. 2002;14:110–122. [PubMed: 11792552]
- 60.
- Zatloukal K, Denk H, Lackinger E. et al. Hepatocellular cytokeratins as substrates of transglutaminases. Lab Invest. 1989;61:603–608. [PubMed: 2481149]
- 61.
- Ku N-O, Omary B. Keratins turn over by ubiquitination in a phosphorylation-modulated fashion. J Cell Biol. 2000;149:547–552. [PMC free article: PMC2174842] [PubMed: 10791969]
- 62.
- Stumptner C, Omary MB, Fickert P. et al. Hepatocyte cytokeratins are hyperphosphorylated at multiple sites in human alcoholic hepatitis and in a Mallory body mouse model. Am J Pathol. 2000;156:77–90. [PMC free article: PMC1868635] [PubMed: 10623656]
- 63.
- Ku NO, Michie SA, Soetikno RM. et al. Mutation of a major keratin phosphorylation site predisposes to hepatotoxic injury in transgenic mice. J Cell Biol. 1998;143:2023–2032. [PMC free article: PMC2175212] [PubMed: 9864372]
- 64.
- Ku NO, Michie S, Resurreccion EZ. et al. Keratin binding to 14-3-3 proteins modulates keratin filaments and hepatocyte mitotic progression. Proc Natl Acad Sci USA. 2002;99:4373–4378. [PMC free article: PMC123655] [PubMed: 11917136]
- 65.
- Hermeking H. The 14-3-3 cancer connection. Nat Rev Cancer. 2003;3:931–943. [PubMed: 14737123]
- 66.
- Toivola DM, Nieminen MI, Hesse M. et al. Disturbances in hepatic cell-cycle regulation in mice with assembly-deficient keratins 8/18. Hepatology. 2001;34:1174–1183. [PubMed: 11732007]
- 67.
- Caulin C, Ware CF, Magin TM. et al. Keratin-dependent, epithelial resistance to tumor necrosis factor-induced apoptosis. J Cell Biol. 2000;149:17–22. [PMC free article: PMC2175089] [PubMed: 10747083]
- 68.
- Deaciuc IV. Alcohol and cytokine networks. Alcohol. 1997;14:421–430. [PubMed: 9305456]
- 69.
- Tsukamoto H, Lu SC. Current concepts in the pathogenetic of alcoholic liver injury. FASEB J. 2001;15:1335–1349. [PubMed: 11387231]
- 70.
- Diehl AM. Cytokine regulation of liver injury and repair. Immunol Rev. 2000;174:160–71. [PubMed: 10807515]
- 71.
- Gilbert S, Loranger A, Daigle N. et al. Simple epithelium keratins 8 and 18 provide resistance to Fas-mediated apoptosis. The protection occurs through a receptor- targeting modulation. J Cell Biol. 2001;154:763–773. [PMC free article: PMC2196458] [PubMed: 11514590]
- 72.
- Ku NO, Soetikno RM, Omary MB. Keratin mutation in transgenic mice predisposes to fas but not TNF-Induced apoptosis and massive liver injury. Hepatology. 2003;37:1006–1014. [PubMed: 12717381]
- 73.
- Gilbert S, Loranger A, Marceau N. Keratins modulate c-Flip/extracellular signal-regulated kinase 1 and 2 antiapoptotic signaling in simple epithelial cells. Mol Cell Biol. 2004;24:7072–7081. [PMC free article: PMC479742] [PubMed: 15282307]
- 74.
- MacFarlane M, Merrison W, Dinsdale D. et al. Active caspases and cleaved cytokeratins are sequestered into cytoplasmic inclusions in TRAIL-induced apoptosis. J Cell Biol. 2000;148:1239–1254. [PMC free article: PMC2174305] [PubMed: 10725337]
- 75.
- Ku NO, Wright TL, Terrault NA. et al. Mutation of human keratin 18 in association with cryptogenic cirrhosis. J Clin Invest. 1997;99:19–23. [PMC free article: PMC507762] [PubMed: 9011570]
- 76.
- Ku NO, Gish R, Wright TL. et al. Keratin 8 mutations in patients with cryptogenic liver disease. N Engl J Med. 2001;344:1580–1587. [PubMed: 11372009]
- 77.
- Ku NO, Darling JM, Krams SM. et al. Keratin 8 and 18 mutations are risk factors for developing liver disease of multiple etiologies. PNAS. 2003;100:6063–6068. [PMC free article: PMC156326] [PubMed: 12724528]
- 78.
- Hesse M, Berg T, Wiedenmann B. et al. A frequent keratin 8 p.L227L polymorphism, but no point mutations in keratin 8 and 18 genes, in patients with various liver disorders. J Med Gen. 2004;41:e42. [PMC free article: PMC1735733] [PubMed: 15060118]
- The Keratin Cytoskeleton of Hepatocytes and Bile Duct Epithelial Cells
- Alterations of the Keratin Cytoskeleton in Human Liver Diseases
- Animal Models to Study Hepatocytic Keratin Alterations
- Mallory Bodies As a Product of the Cellular Response to Misfolded Keratin
- How Can Keratins Influence Toxic Cell Injury?
- Mutations of Keratin Genes and Liver Diseases
- References
- Keratins As Targets in and Modulators of Liver Diseases - Madame Curie Bioscienc...Keratins As Targets in and Modulators of Liver Diseases - Madame Curie Bioscience Database
- Embryonic Salivary Gland Branching Morphogenesis - Madame Curie Bioscience Datab...Embryonic Salivary Gland Branching Morphogenesis - Madame Curie Bioscience Database
- Earthworm Immunity - Madame Curie Bioscience DatabaseEarthworm Immunity - Madame Curie Bioscience Database
- Overview of Intracellular Compartments and Trafficking Pathways - Madame Curie B...Overview of Intracellular Compartments and Trafficking Pathways - Madame Curie Bioscience Database
- Subcellular Compartmentalization of Insulin Signaling Processes and GLUT4 Traffi...Subcellular Compartmentalization of Insulin Signaling Processes and GLUT4 Trafficking Events - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...