

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Despite a common set of hallmark neuropathological lesions and clinical symptoms, Alzheimer's disease has an apparently complex etiology. The disease can be caused by autosomal dominant mutations in at least three genes (encoding the amyloid precursor protein (APP) and the two presenilins). In addition, it can be influenced by certain allelic variants of at least three “risk factor” genes (apolipoprotein E, antichymotrypsin, and interleukin-1), or may arise “sporadically” with no evident genetic component. In the end, as many as 30-40% of individuals over the age of 85 may have some symptoms of Alzheimer's—underscoring the fact that age itself is the strongest risk factor for the disease.
It has been known for almost twenty years that individuals with trisomy 21 (Down syndrome) exhibit Alzheimer neuropathology by the time they are 30-40 years old. Somewhat later, they also develop dementia, and eventually die of Alzheimer's disease. Because the gene for amyloid precursor protein (APP) resides on chromosome 21, its consequent overexpression in trisomy 21 cells presumably contributes to the development of Alzheimer's disease in Down syndrome individuals.
The connection between Down syndrome and Alzheimer's disease and the application of Occam's Razor led me to hypothesize that many cases of classical Alzheimer's disease—both of the genetic and late-onset, sporadic forms—might similarly be caused by chromosome mis-segregation leading to a small number of trisomy 21 cells developing during the life of the affected individual.
In this chapter, I will consider evidence from several laboratories that defects in mitosis, and particularly in chromosome segregation, may be a part of the Alzheimer disease process. In particular, mutations in the presenilin genes that cause Alzheimer's disease also cause chromosome instability. By generating a mosaic population of trisomy 21 and other aneuploid cells, such a mitotic defect could lead to Alzheimer pathology and dementia by inducing inflammation, apoptosis, and/or altered processing of the APP protein into the neurotoxic amyloid β-protein—all characteristic features of the disease. The possibility that many cases of Alzheimer's disease are mosaic for trisomy 21 suggests novel approaches to diagnosis and therapy.
Introduction
Alzheimer's disease arises when neurons in certain regions of the brain, particularly those involved in memory and cognition, are damaged and ultimately killed. A key step in this process is the polymerization of the Aβ peptide into neurotoxic protein filaments. Aggregates of these filaments accumulate in the brain as the characteristic neuropathological lesions termed “amyloid” and are thought to be an essential contributor to neuronal cell death in Alzheimer's disease.1-5 This hypothesis has been supported by many in vitro and in vivo experiments6-12 and has been recently strengthened greatly by our demonstration that amyloid formation catalyzed by the action of two amyloid-associated proteins, apolipoprotein E (apoE) and antichymotrypsin (ACT), on Aβ is required for neuronal disfunction and cognitive impairment in a mouse model of Alzheimer's disease.12 Together with the genetic evidence implicating the amyloid precursor protein (APP), apoE4, and ACT-A in the disease (for review, see reference 5), these results strongly point to either the process or product of amyloid formation as the key to Alzheimer's disease. The unanswered questions are how does amyloid formation arise in the majority of non-inherited Alzheimer's disease and what initiates neuronal cell death.
An important clue to the mechanism of Alzheimer's disease was the discovery that Down syndrome patients who live beyond the age of 30 or 40 develop brain neuropathology indistinguishable from that observed in classical Alzheimer's disease.13-15 Down syndrome is caused by the presence of three copies of chromosome 21, instead of the usual two, in every cell of the body from the moment of conception. The implication of this finding is that trisomy for chromosome 21 not only causes Down syndrome, but is also sufficient to cause Alzheimer's disease later in life.16
One possible explanation for the link between Down syndrome and Alzheimer's disease is that, in both disorders, a gene on chromosome 21 is over-expressed.due either to the 50% increased dosage of chromosome 21 genes in Down syndrome, or to potential somatic or inherited mutation or gene duplication in Alzheimer's disease.17,18 Indeed, the Aβ peptide, which the major component of the pathological amyloid deposits found in Alzheimer's disease brain, is encoded by a gene (amyloid precursor protein; APP) that resides on chromosome 21.19-22 APP is actually over-expressed in Down syndrome individuals somewhat more than the 50% expected from gene dosage alone.23-25 However, no known cases of Alzheimer's disease have resulted from a simple duplication of the APP gene or over expression of APP. It therefore seems to me more likely that trisomy 21 results in multiple abnormalities in gene expression that together result in Alzheimer's disease decades later.
One weekend, some years ago, I was deep in the New Hampshire woods attending a retreat for the MD-PhD students and faculty of The Harvard Medical School. Taking advantage of a relaxed atmosphere, I tried to concentrate on the curious relationship between Alzheimer's disease and Down syndrome. It occurred to me that if complete “trisomy 21” could lead to early Alzheimer's disease in Down syndrome, perhaps the slow development of some trisomy 21 cells over a lifetime could cause the Alzheimer's disease that affects elderly individuals.26 The more I thought about it the more it became clear that this model explained many, seemingly unrelated facts about Alzheimer's disease. It could also account for both the inherited and more common, non-familial form of the disorder, depending upon whether the defect in chromosome segregation that led to trisomy 21 mosaicism was the result of a genetic mutation or some environmental insult.
In order to be useful, a scientific model must be able to make testable predictions. The trisomy 21 model for Alzheimer's disease makes at least two major ones. The first is that Down syndrome and Alzheimer patients should share clinical features besides dementia that might be used as a diagnostic test for Alzheimer's disease.26 One such potential test is based on the finding that Down syndrome and Alzheimer's disease individuals have cholinergic deficits that, for instance make them hypersensitive to the pupil dilating effect of cholinergic antagonists.26,27 We and others are still in the process of testing this prediction. Increasingly promising results suggest that even individuals that are seemingly cognitively normal can dilate abnormally in response to the cholinergic antagonist tropicamide and can show cholinergic pathology and neuronal cell death not only in the cortex and hippocampus, but also in the Edinger-Westphal nucleus that controls the pupil.28-30 Such individuals are likely to be in the preclinical stage of Alzheimer's disease, which can last 10-30 years or more, and may be detected by their hypersensitive pupil dilation in response to tropicamide.
The second, and most important prediction of the trisomy 21 model of Alzheimer's disease is that Alzheimer patients should have developed a small number of trisomy 21 cells in their somatic tissues.26 Such trisomy 21 mosaicism would be produced over time by unequal chromosome segregation during mitosis. Ultimately, trisomy 21 cells in the mosaic individual could lead to Alzheimer's disease through the same (as yet unknown, and presumably multi-step) mechanism by which Down syndrome patients acquire the disease, but at a later age due to the modulating effect of the majority population of normal diploid cells.
Based on this suggestion that Alzheimer's disease may be a mosaic form of Down syndrome arising from defects in mitosis, certain further predictions could be made:26
- There should be alterations in the mitotic spindle apparatus or in mitosis-related proteins in Alzheimer's disease cells that could lead to chromosome mis-segregation and trisomy 21 mosaicism, and
- Mutations that cause Alzheimer's disease should occur in genes coding for proteins directly or indirectly involved in mitosis and chromosome segregation.
Each of these predictions can be tested, either by re-examination of the literature for findings supportive of the theory, or by new experimentation. In this chapter, we will first examine the published and unpublished evidence in favor of chromosome instability/mis-segregation and trisomy 21 mosaicism as a cause of Alzheimer's disease, then present evidence linking the Alzheimer presenilin mutations to mitosis and chromosome mis-segregation, and finally consider the resulting implications for Alzheimer diagnosis and therapy.
Trisomy 21 Mosaicism in Alzheimer's Disease
An early hint that chromosome mis-segregation and trisomy 21 mosaicism might be related to Alzheimer's disease was provided by epidemiological studies showing that women in some Alzheimer families in which the disease is inherited as an autosomal dominant mutation give birth to a significantly higher-than-normal number of Down syndrome children.31-33 Other researchers have failed to confirm the increased incidence of Down syndrome in families with inherited Alzheimer's disease, but they report that the number of relatives they analyzed was too few for the lack of Down syndrome to be statistically significant.34-36 Although the data in the largest studies were very suggestive, more work needs to be carried out to confirm the connection between a high frequency of Down syndrome and inherited Alzheimer's disease in the same family.
More recently, a strong association between Down syndrome and sporadic Alzheimer's disease was found. In a retrospective study, Schupf and her colleagues showed that young mothers of Down syndrome children have a five-fold greater risk of developing Alzheimer's disease later in life, when compared to either older Down syndrome mothers, or the general population, as though they suffered a novel form of “accelerated aging”.37 We interpret this result, instead, as indicating that the young Down syndrome mothers were most likely mosaic for trisomy 21 (as if they had a predisposition to chromosome mis-segregation) and that this mosaicism was reflected in their trisomy 21 offspring and their own increased risk for Alzheimer's disease.
Finally, trisomy 21 mosaicism was specifically found in two women who, though not mentally retarded and not characterized as having Down syndrome, developed Alzheimer-like dementia by age 40.38-40 One of the two women also had a Down syndrome child. An unusual family with an inherited aberrant chromosome 22-derived mini-chromosome was studied by Percy et al and found, in addition, to have a high frequency of Alzheimer's disease.41 The two living Alzheimer-affected members of the family carried the marker chromosome and one was also found to be mosaic for trisomy 21. The two patients reported by Schapiro38 and Rowe39 and their colleagues, and possibly the mosaic individual reported by Percy41 and her colleagues, demonstrate that it is not necessary for every cell of an individual to be trisomy 21 for the aberrant effects of this chromosome imbalance to result in early Alzheimer dementia. The often-later onset dementia of classic Alzheimer's disease—both genetic and sporadic—could thus result from an even smaller percentage of trisomy 21 cells that may go undetected.
It would be clearly advantageous to determine directly whether Alzheimer's disease patients are mosaic for trisomy 21. However, this apparently simple experiment is not straightforward because techniques must be used that can accurately determine low levels of mosaicism. Cytogenetic analysis of Alzheimer's disease patients has been carried out in a number of laboratories, with mixed results. Some investigators have reported small increases in aneuploidy or other abnormalities as measured by direct karyotyping, while others failed to confirm the finding, especially if they analyzed few or small families.42-50 Interestingly, the cytogenetic abnormality termed premature centromere division (PCD), which is a potential cause of improper chromosome segregation in vitro and in vivo, was found to be positively correlated with age and to be increased in women with familial Alzheimer's disease (3.6% vs. 0.6% in age-matched controls), particularly affecting the X chromosome.47,50,51 Furthermore, trisomy 21, 18, and X occurred in the lymphocytes and fibroblasts of a woman prone to PCD, who also had three trisomy 21 conceptuses.52
In contrast to metaphase karyotyping, fluorescence in situ hybridization (FISH) allows the number of copies of a particular chromosome to be determined regardless of the stage of the cell cycle (mitotic or interphase).53,54 The ability to examine interphase nuclei also greatly increases the number of cells that can be analyzed and, in principle, should allow easier detection of very low levels of aneuploidy. We therefore used FISH to determine the extent of aneuploidy, particularly chromosome 21 trisomy, in fibroblasts from Alzheimer and normal individuals.1,55,56 In analyzing thousands of cells from 27 Alzheimer and 13 control individuals, we found that fibroblasts from Alzheimer's disease patients exhibited more than twice the frequency of trisomy 21 as did cells from age-matched, normal individuals (fig. 1). The average percentage of trisomy 21 cells was 5.5% in Alzheimer's disease cultures and 2.5% in cultures from unaffected individuals. Using the same procedure, the number of trisomies in a Down syndrome culture was determined to be 98%. The greater frequency of trisomy 21 cells in Alzheimer's disease patients compared to controls was significant (p=0.007) and was not related to the age of the affected individuals. A small parallel study of chromosome 18 showed similar aneuploidy, indicating that the chromosome mis-segregation likely affected all chromosomes.
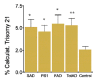
Figure 1
AD cells, including those with FAD mutations in PS-1 or 2 show increased trisomy 21 aneuploidy. Reprinted with permission from Neurobiology of Disease 1999; 6:167-179 ©1999 Elsevier.
When the chromosome study was initiated, we decided to examine all types of Alzheimer's disease with a particular focus on individuals with the early onset, familial form of the disease. The majority of these cases were, in the course of the investigation, shown to carry mutations in one of the two presenilin genes.57-62 We found that chromosome mis-segregation had occurred in all Alzheimer's disease individuals, including those with sporadic AD and those who are now know to carry a familial Alzheimer's disease (FAD)-causing mutation in either presenilin 1 or presenilin 2. Chromosome mis-segregation in sporadic AD patients has been elegantly confirmed in blood lymphocytes by Migliore et al63 and in the brains of sporadic AD patients by Yang and colleagues.64
Defects in Mitosis in Alzheimer's Disease
During the course of the studies described above, several lines of investigation provided independent evidence that defects in mitosis and/or in mitosis-specific proteins may be present in Alzheimer's disease patients—supporting another prediction of the trisomy 21 mosaicism model. Such defects could be expected to lead to chromosome mis-segregation, and thus could result in the trisomy 21 mosaicism and other aneuploidy observed in Alzheimer' disease cells and individuals, and, as will be discussed, may also lead to other features of the disease such as apoptosis and changes in amyloid precursor protein (APP) processing.
The most direct evidence for mitotic defects in Alzheimer's disease has been provided by experiments in which the mitotic spindles in dividing cells from Alzheimer patients were observed to be abnormal. For example, we briefly treated lymphocyte cell lines from presenilin mutant Alzheimer's disease and control patients with the microtubule-disrupting agent colchicine, and analyzing their karyotypes 40 hours later. This treatment causes many of the cells to exhibit separated chromatids in metaphase spreads. That is, in these karyotypes, the individual chromatids lay parallel to each other, separated by a clear gap, rather than being connected at their centromeres, as are chromosomes from untreated cultures. This metaphase chromosome pattern is similar to the spontaneous premature centromere division (PCD) that made the patients described above prone to chromosome mis-segregation and Alzheimer dementia. Comparison of colchicine-treated cells showed that those from Alzheimer's disease patients exhibited this karyotypic abnormality significantly more frequently than cells from normal individuals of similar age1 (Geller, Benjamin and Potter, in preparation). A similar Alzheimer-specific increase in PCD induced by microtubule-disrupting agents was reported by Migliori and colleagues.50
A related defect in mitosis (increased frequency of chromosomes displaced from the mitotic spindle after colchicine treatment) was found by Ford in cells derived from mothers of Down syndrome children.65 In addition, the rate of hyperdiploidy of all chromosomes was increased in cells from mothers with a Down syndrome child.66 These data indicate that such mothers may be generally prone to chromosome mis-segregation, which can lead to either gonadal trisomy 21 mosaicism or to meiotic non-disjunction, or both, and thus resulted in their trisomy 21 (Down syndrome) children. From the work of Schupf and colleagues37 discussed above, such mothers of Down syndrome children would also be expected to have an increased risk of developing Alzheimer's disease, again implicating chromosome mis-segregation in this disease process.
Further evidence for the potential involvement of cell cycle defects in Alzheimer's disease comes from the finding that both the amyloid precursor protein (APP) and the microtubule-associated protein tau found in Alzheimer paired helical filaments (PHF) become increasingly phosphorylated during mitosis67-69 (Geller and Potter, unpublished). Thus, the two proteins most intimately involved in forming the characteristic neuropathological lesions of Alzheimer's disease are altered in structure, and presumably function, at different times in the cell cycle. Furthermore, phospho-tau and other mitosis-specific phospho-proteins and enzymes are present in neurons of Alzheimer, but not normal, brains.70-77 Why these “post-mitotic” cells acquire mitosis-specific proteins is unknown. One possibility is that they have been stimulated to begin an aberrant mitosis. This, in turn, could have led to gain or loss of chromosomes, to changes in mitosis specific gene expression, or to apoptosis in response to an untenable physiological state—any or all of which could stimulate the development of Alzheimer pathology. 64,78,79
Potential Mitotic Function of the Presenilin Proteins
As mentioned above, many of the Alzheimer individuals whose fibroblasts we analyzed for trisomy 21 mosaicism belonged to large families that were subsequently shown to carry familial Alzheimer's disease (FAD) mutations in two related genes (AD3 and AD4), now called presenilin 1 and 2 (fig. 2). This result provided the first indication that familial Alzheimer's disease genes are likely to be involved in mitosis, and that their mutant forms predispose to chromosome mis-segregation—another of the predictions of the chromosome instability model of Alzheimer's disease.
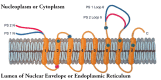
Figure 2
Probable transmembrane conformation of the two presenilin proteins (diagram modified by David Costa from ref. ).
The familial Alzheimer's disease genes encoding the presenilin proteins were identified on chromosome 14 (PS1) by St. George-Hyslop and colleagues based on previous genetic mapping57,58 and on chromosome 1 (PS2) by Schellenberg, Tanzi and colleagues59-61 and independently by us.62 Both presenilin proteins include ~8 transmembrane domains and are structurally similar.
Almost forty point mutations have been identified throughout the two presenilin genes that cause early onset familial Alzheimer's disease (FAD). The fact that all of the FAD mutations reside within the coding regions of the genes suggests that a dominant gain of function resulting for instance in changes in their proteins' function, localization, or structure, rather than quantitative changes in expression, are likely to underlie the genes' involvement in Alzheimer's disease.
The presence of multiple transmembrane domains and potential target sites for the mitosis-specific cdc-2 kinase in the presenilins, together with the chromosome mis-segregation results with presenilin-mutant human cells, led us to suggest that the PS-1 and PS-2 proteins might reside in the nuclear membrane and might be involved in chromosome organization and segregation during the cell cycle.62 We then tested this prediction by preparing a panel of antibodies to presenilin-derived peptides fused to glutathione-S-transferase and using immunocytochemical analysis to determine the normal intracellular location of the presenilin proteins.78
In the first approach to localizing the presenilins in the cell, PS-1- and PS-2-FLAG fusion proteins were expressed in transiently transfected cells and their intracellular location determined with anti-FLAG antibodies. Initial experiments indicated that the FLAG-tagged protein is partly localized in the endoplasmic reticulum and Golgi.78 This was not un-expected because these are the sites of synthesis and processing of transmembrane proteins and because other groups had also overexpressed the presenilin proteins in cells and found them primarily localized to the endoplasmic reticulum.81-83 However, because the localization of over-expressing proteins can easily become distorted, we sought to determine the subcellular location of the endogenous presenilin proteins. The affinity-purified presenilin antibodies were used to label the endogenous protein in cultured cells by Western blot analysis and immunocytochemistry.
Figure 3 shows an example of the intracellular localization of endogenous PS-2 in fibroblasts grown on cover slips. The labeling was primarily nuclear, which, in these flattened cells, was consistent with nuclear membrane localization. Putative PS-2 localization to the centrosome—identified as a small, strongly-labeled spot adjacent to the nucleus—was also apparent. These results, particularly the apparent centrosome labeling, provided additional evidence that the function of the PS-2 protein might be related to mitosis.

At higher magnification, the labeling of the nuclear membrane and the centrosome with the PS-2 loop antibody could be seen more clearly. In addition, a pronounced punctate pattern became apparent within the nucleus. This punctate nuclear membrane labeling pattern resembled the image generated by antisera directed against kinetochores—the protein complexes that bind the centromeres of the chromosomes and link them to the spindle microtubules during mitosis.84,85 By adjusting the plane of focus, the PS-2-labeled spots could be seen to be located justaposed to the inner membrane of the nuclear envelope, where the kinetochores reside in interphase cells.84,86
To confirm the localization of PS-2 to the nuclear membrane, centrosomes, and kinetochores, we performed a series of double-label experiments with both the rabbit PS-2 loop antibody and either a human anti-centrosome serum, a human anti-lamin serum, or a human anti-kinetochore serum, which could be distinguished from the PS-2 antibody with different fluorescent secondary antibodies.78 For example, a human auto-immune serum was obtained from a patient with the CREST variant of scleroderma, shown previously to specifically label kinetochores in both metaphase and interphase cells82,85 (F. McKeon, personal communication). The co-localization of the two signals (orange) identified the PS-2 protein as closely associated with the nuclear membrane, and with the kinetochores attached to the nuclear membrane during interphase (fig. 4). Perhaps the membrane-associated presenilins serve as anchors to attach the kinetochores to the inner nuclear membrane during interphase. Consistent with this hypothesis is the finding that the centrosome labeling remains during mitosis, but the nuclear envelope labeling becomes dispersed and the kinetochores are unlabelled.78

Colocalization of staining with presenilin and centrosome antibodies is shown in Figure 5. Because the presenilins are clearly transmembrane proteins, we assume that the centrosome localization reflects the proteins association with cytoplasmic vesicles that have accumulated next to the centrosome.

Figure 5
An example of a double immunolabeling of the centrosome with antibodies to presenilin 2 and to lamin.
Similar co-localization experiments were performed with an antibody to the N-terminus of PS-2 and also with two antibodies to the large hydrophilic loop and the N-terminus of PS-1.78 Identical results were obtained with all four antibodies, indicating that both presenilins are associated with the nuclear membrane, centrosome, and kinetochores of normal dividing cells.
Electron microscopy with gold-labelled secondary antibodies confirmed that the presenilins are associated with the inner nuclear membrane, the periphery of the centrosome, and punctate structures attached to the inner nuclear membrane.78 Several other Alzheimer researchers have confirmed that a major location for the presenilins in the cell is the nuclear envelope87,88 (B. Yankner, G. Schellenberg, T. Wisniewski and B. Frangione, B. De Strooper and G. Cole, personal communications). Furthermore, specific localization of presenilin protein to the centrosome and the kinetochore microtubules in early mouse embryos is induced by halting the cells in mitosis.89 Finally, Johnsingh et al90 and Pigino et al91 have found that the presenilins associate with the cytoskeleton-associate proteins CLIP 170 and restin and with microtubules, consistent with our localization findings.
Functional results are also consistent with the presenilins being involved in mitosis. The FAD presenilin mutations have been found to inhibit the cell cycle92 (Tanzi, personal communication; also see below), to increase sensitivity to apoptosis93-95 (Ma and Potter, unpublished), and to prevent the full-length proteins from being translocated to the nuclear envelope.88 Confirmation that the presenilin proteins are involved in chromosome segregation and Alzheimer's disease has recently been provided by the findings that a polymorphism in the presenilin 1 gene is associated with both an increased risk of developing Alzheimer's disease,96-114 and also with an increased risk of having a Down syndrome child via a meiosis II defect.115 Finally, when we examined the sequences of the PS1 and PS2, we found that in addition to the evident transmembrane domains, both proteins carry several S/TPXX amino acid motifs.62 Based on their function in other proteins, these motifs may be target sites for cdc2 kinase and suggest that PS1 and PS2 reside in the nuclear membrane and are involved in mitosis. In vitro experiments on one of these sites confirm that it is an effective target for cdc2 kinase but not, for example for CKII (Li and Potter, unpublished).
The most straightforward interpretation of the subcellular localization of PS-1 and PS-2 in dividing cells is that presenilin function is related to mitosis, chromosome organization and segregation, and, potentially, to mitosis-specific gene expression. More specifically, the nuclear membrane and kinetochore localization of the presenilins suggests that one function of these proteins may be to serve as kinetochore binding proteins or receptors (“kinetoceptors”) to anchor and organize the chromosomes on the nucleoplasmic surface of the inner nuclear membrane during interphase. In this role, the presenilins might bind DNA directly with special affinity for centromeric sequences, or they might interact with kinetochore proteins, possibly through microtubules. The point mutations and polymorphisms in the presenilin genes that cause or influence familial Alzheimer's disease may affect the ability of the presenilin proteins to link the chromosomes to the nuclear membrane and to release them at the appropriate time during mitosis, thus leading to chromosome mis-segregation and other consequent abnormalities seen in cells carrying mutant presenilin genes, such as inappropriate apoptosis. Interestingly, there are precedents for nuclear membrane proteins having a role in mitosis. Specifically, the yeast NDC-1 protein and the lamin B receptor both have seven-eight transmembrane domains, are located in the nuclear membrane, contain phosphorylatable S/TPXX motifs, and, in the case of NDC-1, cause chromosome mis-segregation when mutant.62,116-118
In order for a cell to leave G1 and enter mitosis, the kinetochores and their associated chromosomes must be released from the surface of the inner nuclear membrane and its associated proteins such as the presenilins. Two possible ways in which the putative presenilin binding to centromeres/kinetochores might be regulated during this process are by phosphorylation or by proteolytic cleavage. It is therefore interesting that PS-2 is phosphorylated in vivo by casein kinase 2,82 and in vitro by the cdc-2 kinase62 (Li and Potter, in preparation), and that both presenilin proteins, though found in full-length form, are also cleaved into two parts in normal and transfected cells.78,119,120
Mitotic Effects of the Normal and FAD Mutant Presenilin Proteins in Transfected Cells
The evidence that the presenilins are involved in mitosis and chromosome segregation is provided by their location within the cell and the finding that fibroblasts from individuals harboring an FAD mutation in PS-1 or PS-2 show chromosome mis-segregation. To further investigate possible mitotic functions of the PS proteins, the normal and FAD mutant versions of their genes have been cloned into expression vectors and introduced into mammalian cells with normal chromosome complements.
Preliminary evidence from transfected cells over-expressing the presenilin proteins are consistent with their involvement in chromosome segregation. AG09393 cells were transiently transfected with the pcDNA-3 vector either unmodified or expressing wild type PS-1 or PS-2. The combined level of aneuploidy in several experiments was statistically significantly higher in the cells receiving the PS-containing vectors compared to the cells receiving water or the vector (pcDNA3) alone (see Table 1). Overexpression of even normal presenilins clearly affects chromosome segregation. The chromosome changes included trisomies, monosomies, and translocations.
Table 1
Transient transfection experiments.
The fact that transient over-expression of even normal presenilin proteins causes massive chromosome instability and apoptosis,93,94 suggested that a more controlled expression system would be better able to show differences between the wild type and mutant presenilin genes. To this end, we have developed tetracycline activated expression vectors and are in the process of analyzing their effect on chromosome stability.
Chromosome and Cell Cycle Changes in Cells from Presenilin Transgenic and Knockout Mice
The demonstration that the presenilin proteins in mice were located in the same apparent structures as in human62 prompted some preliminary experiments in mouse cells to determine the effect of PS-1 deletion on the cell cycle. Shen et al developed a PS-1 knockout mouse which could be maintained in the heterozygous state and could generate homozygous knockout embryos.121 We prepared primary fibroblast cultures from such embryos and measured the cell division time and the number of in vitro passages needed to reach senescence compared to cells from a normal littermate. PS-1 knockout cells clearly grew more slowly than normal cells, and many of the knockout cells reached senescence as evidenced by flattening and cessation of mitosis, before any of the normal cells did.
We have also carried out a preliminary experiment to analyze the chromosome complement of fetal liver cells from a PS-1 knockout embryo in comparison to a normal littermate. The number of karyotypes that were abnormal was substantially (and statistically significant; P<0.05) higher in the E-15 PS-1 null liver cells than in cells obtained from a normal wild type littermate (see Table 2). In addition. the mitotic index was substantially smaller in the PS-1 KO cell cultures, also suggesting the presence of a defect in the cell cycle.
Table 2
Aneuploidy in PS1 knockout liver.
The results from the PS-1 knockout mouse cells have been complemented by similar experiments on cells derived from transgenic mice harboring and expressing the human PS-1 normal and PS-1-FAD mutant genes under the control of the PDGF promoter.122 Thus far, we have karyotype data from hundreds of spleen cells (in which the PDGF promoter is active) that reveal substantial aneuploidy in mice carrying a PS-1 gene harboring the M146L FAD mutation (Table 3). The number of aneuploid cells increases with the age of the transgenic mice. Flow cytometry confirms these results and also shows no aneuploidy in wt PS mice of 17 and 24 months of age.
Table 3
Aneuploidy in PS-1 mutant mice.
All of these results are consistent with PS-1 playing an important role in the normal physiology of dividing cells. The chromosome analysis specifically suggests that normal amounts of WT presenilins are essential for maintaining the fidelity of chromosome segregation. This result, if confirmed, could also explain the slower growth, the reduced lifespan in culture, and the tendency to undergo apoptosis of cells either knocked out or mutant for presenilins. The result also confirms the previous finding of chromosome mis-segregation and trisomy 21 in fibroblasts from presenilin mutant individuals with Alzheimer's disease.55
In Alzheimer's disease research, it has become almost axiomatic that the main cause of the disease is an increase in the relative amounts of the Alzheimer Aβ 1-42 and 1-40 peptides, with the Aβ 1-42 peptide being more prone to form amyloid deposits and induce neuronal damage.4 However, it is worth pointing out that the many, many fold increase in chromosome aneuploidy in cells carrying a presenilin mutation far exceeds the barely 2-fold effect of the mutations on the relative production of Aβ 1-42 and 1-40. Perhaps the main effect of the presenilin mutations is on the cell cycle and chromosome segregation, with a coincidental side effect being an alteration in Aβ production.
Mutation/Inactivation of the Tumor Suppressor p53 Causes Chromosome Instability and Regulates Expression of Presenilin
A completely independent result from the study of cancer supports the conclusion that the presenilins are involved in mitosis and chromosome segregation. p53 is a well-studied tumor suppressor gene whose mode of action is still not completely understood. Lack of p53 clearly predisposes to malignancy in humans and experimental animals. Indeed, p53 is the most common mutation in human cancers, and animals, humans, and cell lines mutant for p53 show abnormal centrosome duplication, chromosome mis-segregation, and aneuploidy.123-126 Thus p53 may exert its tumor suppressor function at least partly by promoting the orderly segregation of chromosomes during mitosis, with loss of p53 leading to aneuploidy. This conclusion is supported by two recent findings that together link p53 function to chromosome segregation and to the presenilins.
One approach to determining how a tumor suppressor gene acts to suppress unrestricted cell division is to identify other genes whose expression is either up-regulated or down-regulated by the tumor suppressor gene product and then to determine which are important downstream effectors of the pathway. One such study used a p53 mutant cell line into which a vector construct that expressed a temperature sensitive p53Val135 protein was stably transfected.127 Shifting the cells between the permissive and nonpermissive temperature and carrying out differential RNA display identified a series of genes that are up-regulated or down-regulated by wt p53. One such gene that was down-regulated is presenilin 1 (PS1), previously identified as carrying multiple mutations that cause autosomal dominant Alzheimer's disease.
Roperch and colleagues then assessed the importance of PS1 in p53 function by knocking out the PS1 protein by transfection of p53 mutant cells (which express large amounts of PS1) with an PS1 antisense RNA-expressing construct.127 The cells grew much more slowly, lost their ability to be cloned in soft agar, and generated few tumors when injected into nude mice. Those tumors that did develop had regained their presenilin expression, confirming the conclusion that PS1 is essential for the tumor-causing effect of the loss of p53 function.
The data from the Alzheimer's disease patients fibroblasts and the transgenic mice together with the suggestive involvement with p53 provide a strong indication that the presenilins may be involved in chromosome segregation, The location of the presenilins at the interphase kinetochore suggest a mechanism by which mutations in the presenilins could cause chromosome instability and aneuploidy. Appropriate chromosome segregation requires that very careful control of chromosome location and organization be maintained throughout the cell cycle, not merely during mitosis. This is presumably the reason that the kinetochores are attached to the inner surface of the nuclear membrane during interphase rather than being allowed to roam freely in the nucleoplasm. Therefore, it is likely that if this interphase location were to be disrupted by mutation in a protein such as a presenilin that seems to be connecting the interphase kinetochores with the inner nuclear membrane, then the end result could be a defect in chromosome segregation. For instance, if the chromosomes do not detach from the nuclear membrane at the right time and with the right speed and coordination, then they may not become available for attachment by the spindle microtubules with equal efficiency, and lagging chromosomes may become mis-segregated.
Presenilin Mutations May Effect Mitosis in both Neurons and Glia
The presence of trisomy 21 mosaicism in skin fibroblasts from Alzheimer's disease patients with presenilin mutations suggests that other cells from these individuals are also likely to be mosaic for trisomy 21. Which cells would, when aneuploid for chromosome 21, contribute most to Alzheimer neuropathology? The answer is not obvious. Neurons are the cells whose loss leads to the clinical symptoms of Alzheimer's disease and neurons produce large amounts of APP and of Aβ. Because mature neurons do not divide, it might be thought that they would be unaffected by mitotic proteins. There are two potential ways that FAD mutations in the presenilins could affect neurons directly. First, the presenilins may have an additional, non-nuclear, function in neurons, and, indeed, the majority of the presenilin protein in these cells is present in the somato-dendritic compartment. This localization has led to the reasonable hypothesis that FAD mutations in the presenilins change their interaction with the APP protein and consequently alter APP processing to produce the increased Aβ1-42 seen in presenilin mutation-carrying patients, cells, and transgenic mice. However, the results reviewed here force us to consider whether the apparent involvement of the presenilins in mitosis might also affect neuronal function and Aβ production. For example, it has become clear that new neurons are, in fact generated in the hippocampus of higher animals, including primates, not only during development, but thoughout adulthood as well.128-138 Specifically, the rate of new neuron formation in the dentate gyrus of the hippocampus is reported to be on the order of one to two thousand cells per day—more than enough to cause significant numbers of trisomy 21 or other aneuploid cells to accumulate over the course of 50 years under the influence of, for instance, a presenilin mutation. Alternatively, neurons may respond to mutations in FAD genes such as the presenilins by attempting to undergo inappropriate division, and perhaps becoming aneuploid and then undergoing apoptosis in the process. Indeed the aneuploid cells reported in AD patients by Yang and colleagues were brain neurons.64 Significantly, as mentioned above, phospho-epitopes and enzymes normally observed only in mitotic cells have been found in neurons in Alzheimer's disease brain. Furthermore, apoptotic neurons generate and release more Aβ than do healthy neurons.139,140
To an even greater extent than neurons, the glial cell population continues to divide in the adult primate brain141 and therefore could accumulate aneuploid cells throughout life. The involvement of glial cells in the neuropathology of Alzheimer's disease is indicated by increasing evidence that these cells mount an inflammatory reaction and an acute phase response that make an important contribution to the characteristic pathology and neuronal cell death of the Alzheimer's disease brain.7,142-149 Particularly interesting is the fact that an abnormally high number of microglia overexpress the lymphokine IL-1 in the areas of Alzheimer's disease and Down syndrome brain that exhibit, or will exhibit, Alzheimer's disease neuropathology.148,150,151 IL-1, in turn, induces astrocytes to express the amyloid-associated protein α1-antichymotrypsin (ACT). Both ACT and another amyloid-associated protein elicited by inflammation, apo E, can facilitate the polymerization of Aβ peptide into neurotoxic amyloid filaments.6-8,148,149,152 The essential function of apoE and ACT as amyloid promoting factors in vivo has been demonstrated recently in animal models of Alzheimer's disease.9-12 Indeed, we have recently shown that product or process of ACT and apoE-catalyzed amyloid formation in transgenic mice is responsible for their cognitive decline.12 In sum, a glial cell-led inflammatory cascade appears to play an essential role in Alzheimer amyloid formation, starting with the release of Il-1 from microglia. The constitutive activation of microglia in Down syndrome brain, even before birth, suggests the possibility that if microglia in a normal individual acquire an extra copy of chromosome 21, they may release Il-1 and initiate the inflammatory cascade that leads to the neuropathology of Alzheimer's disease.
Although it would seem reasonable that amyloid should develop in the regions immediately surrounding aberrant cells (for instance trisomy 21 neurons or glial cells), the precedent provided by other amyloidoses suggests that this need not be the case. For instance, the autosomal-dominantly inherited diseases Familial Amyloidotic Polyneuropathy and Hereditary Cerebral Hemorrhage with Amyloidosis of both the Dutch and Icelandic types have very specific regions of amyloid deposition despite the fact that all cells in the body carry the point mutation in the affected amyloid gene (transthyretin, cystatin C, or APP respectively), and that these genes are expressed in many parts of the body where the amyloid does not deposit.153 Thus, by analogy, the trisomy 21 cells that are relevant for the formation of amyloid pathology in Down syndrome (and, according to the hypothesis presented here, Alzheimer's disease) need not reside in the brain at all. Indeed, some researchers have suggested that the Aβ peptide is transported to the brain by the circulation, after having been generated elsewhere.154
How Aneuploidy May Lead to Alzheimer's Disease
Several potential mechanisms could explain how the generation of trisomy 21 or other aneuploid cells by FAD-mutant presenilin proteins could lead to Alzheimer's disease. For example, aneuploid cells might be prone to apoptosis. Indeed, cortical neurons from Down syndrome fetuses undergo spontaneous apoptosis in vitro.155 Such apoptosis would lead to neurodegeneration directly, but it could also indirectly affect APP processing and the production of the Aβ peptide. Support for this latter hypothesis is provided by the findings that Down syndrome fetal brains and adult sera contain a higher ratio of the pathogenic Aβ1-42 compared to Aβ1-40156 and that inducing apoptosis in normal human neurons by serum starvation or other treatments increases their secretion of the Aβ peptide.139,140 A more direct connection between the presenilin genes and apoptosis is indicated by the finding that the overexpression of even normal presenilin genes in transfected cells (which we would expect to disrupt orderly chromosome segregation) results in increased sensitivity of the cells to apoptotic stimuli95,157,158 (Ma and Potter, unpublished). Furthermore, expression of a C-terminal fragment of PS-2 in cells was shown to inhibit induced apoptosis, which was then restored by overexpression of full-length PS-2.93,94
How could presenilin-induced apoptosis lead to the other commonly-reported effect of expressing the FAD mutant presenilins in human patients, tranfected cells, or trangenic animals—an increase in the ratio of secreted Aβ1-42 over Aβ1-40.122,159-162 One possibility is that the mutant presenilins induce changes in mitosis and chromosome segregation that affect the level of mitosis-specific phosphorylation of various proteins. In as much as altering the phosphorylation of APP alters the production and secretion of Aβ, and the phosphorylation of the APP protein naturally changes during the cell cycle,67 a cell cycle-altering mutation in a protein such as one of the presenilins could affect APP processing indirectly through its phosphorylation state and thus lead to the observed increase in Aβ1-42.
Potential Involvement of Other Genes in Chromosome Mis-Segregation in Alzheimer's Disease
Thus far, it is clear that trisomy 21 mosaicism can cause some sporadic cases of Alzheimer's disease, and there is strong circumstantial evidence favoring such a mechanism as one explanation for how the FAD mutations in the presenilin genes exert their Alzheimer-causing effect. Thus a key prediction of the trisomy 21 mosaicism model for Alzheimer's disease has been, at least partly, fulfilled. Is it possible that other forms of Alzheimer's disease also involve trisomy 21 mosaicism? Here the evidence is intriguing. For example, FAD mutations in the APP gene result both in apoptosis and in increased production of Aβ1-42 over Aβ1-40, which is also characteristic of trisomy 21 and of the presenilin mutations.163-166 Then there is the finding that the APP protein not only becomes hyperphosphorylated during mitosis,67 but that APP phosphorylation also modulates APP processing.167-171 Furthermore, endogenous APP localizes to the centrosome and the nuclear membrane in dividing cells just as we have found for the presenilins172,173 (Li and Potter, unpublished), and has also been found to interact with the presenilins by co-immunoprecipitation from extracts of APP and PS2 double-overexpressing cells.174 Also, in our study of aneuploidy in Alzheimer's disease cell lines, the two lines harboring an APP mutation also exhibited increased trisomy 21.56 Perhaps the APP mutations act not directly, but indirectly, on APP processing, by altering the cell cycle and its pattern of phosphorylation of proteins including APP itself. For example, the cytoplasmic tail of APP binds to a protein that drives cells though the S-M checkpoint and causes apoptosis in neurons.175 Finally, knocking a gene that is highly homologous to APP, termed APLP2, in mice is lethal because the cells of the very early embryo undergo massive chromosome mis-segregation.176,177
The other major genetic risk factor for Alzheimer's disease is the inheritance of the ε4 allele of apolipoprotein E. Although the best evidence indicates that the apoE4 protein promotes Alzheimer's disease by promoting the polymerization of the Aβ peptide into neurotoxic amyloid filaments (discussed above), it is possible that apoE4 has additional functions related to mitosis and chromosome segregation. Such a conclusion was reached, for example, by Avramopoulos and colleagues who showed that young mothers of Down syndrome children are significantly more likely to carry an apo ε4/ε4 genotype.178 The authors suggested that the demonstrated inability of the apoE4 protein (compared to apoE3) to bind to the microtubule-associated protein tau179 disrupts the balance of microtubule polymerization/depolymerization, and thus affects the mechanics of chromosome segregation. This explanation is interesting in light of the facts that a similar group of Down syndrome mothers was shown by Schupf et al to have an increased risk of developing Alzheimer's disease37 and that the highest level of trisomy 21 mosaicism in our sample of Alzheimer's disease patients was found in an apo ε4/ε4 individual.56 It is also interesting that exogenously-added apoE halts lymphocyte proliferation in the G1A phase of the cell cycle.180 Further biochemical and chromosome segregation studies are under way to test directly whether apoE4 affects chromosome segregation.
Although improper chromosome segregation can result from a genetic mutation, it can also be caused by environmental agents. Of the many exogenous factors that influence chromosome segregation, microtubule-disrupting agents such as colchicine and low doses of radiation are perhaps the best studied.181 Aluminum, the consumption of which some studies show to have a weak but significant association with the development of Alzheimer's disease, also binds to microtubules and, in the form of aluminum silicate, causes chromosome nondisjunction in cultured cells.182-184 Thus, the large proportion of Alzheimer's disease cases that arise in a sporadic manner not directly attributable to the inheritance of a genetic mutation can also be understood in the light of the chromosome 21 trisomy model.
Implications of Trisomy 21 Mosaicism in Alzheimer's Disease for Future Diagnosis and Therapy
The mechanistic implication of the findings discussed above is that an early step in the pathogenic pathway to Alzheimer's disease may involve a defect in chromosome segregation that leads to trisomy 21 mosaicism. This conclusion can be exploited in our search for more effective diagnoses and treatments for Alzheimers disease.
As already discussed, the similar hypersensitivity of Alzheimer and Down syndrome individuals to cholinergic agonists or antagonists may serve as an Alzheimer diagnostic test. Another, very straightforward potential diagnostic test based on our finding of trisomy 21 cells among Alzheimer patients fibroblasts would be to use chromosome analysis to directly assess the level of trisomy 21 mosaicism in an individual. Although we have found that cell lines from Alzheimer individuals as a group have increased trisomy 21 mosaicism, there is substantial overlap in the data between the Alzheimer patients and the control individuals. Clearly, the test must be made more sensitive to be useful. One approach, which we have initiated, is to use primary cells directly derived from the patients rather than fibroblast cell lines. This would reduce the influence of culture conditions on the chromosome analysis and potentially provide more rapid results. Specifically, buccal cells, which the individual scrapes with a soft brush from the inside of their cheek, can be assayed for trisomy 21 and other aneuploidy by modification of the standard fluorescence in situ hybridization (FISH) technique. This scraping method also has the advantage of sampling cells from a larger area than does a needle skin biopsy, thus reducing the possibility of missing a small clone of trisomy 21 or other aneuploid cells that might be important. It can also be repeated a number of times to strengthen the confidence of the analysis. The data thus far are too preliminary to make strong conclusions, but it is reasonable to be concerned that an individual who is clearly and reproducibly found to be mosaic for trisomy 21 may be at risk for developing Alzheimer's disease later in life. Similarly, as discussed above, women who have a trisomy 21 fetus at an early age might consider undergoing chromosome analysis of not only their blood lymphocytes, but also their fibroblasts, to assess whether or not trisomy 21 mosaicism is present.
The development in the last ten years of a clearer understanding of the pathogenic pathway that leads to Alzheimer's disease has identified key steps in this pathway that might serve as targets for therapeutic intervention. For instance, the APP processing enzymes that generate the Aβ peptide, the process of Aβ polymerization itself, the amyloid-inducing interaction of ACT or apoE with Aβ, the production of IL-1 by microglial cells, or its recognition by the receptors on the surfaces of astrocytes are all potential steps in the pathway that our current knowledge suggests would be effective Alzheimer therapeutic targets, and for which drug screening assays can be designed. The implication of the data presented in this review that trisomy 21 mosaicism may be one of the very first steps in the Alzheimer pathogenic pathway suggests a new set of approaches to therapy that may be particularly valuable because they are targeted at the very earliest step in the pathway. In some situations it may be possible to determine the mechanism of chromosome mis-segregation or defective mitosis-specific gene expression. Drugs that would rectify the defect could then be selected for. For example, if the familial Alzheimer's disease (FAD) mutations in the presenilins result in a kinetochore receptor that fails to bind the kinetochore sufficiently tightly to prevent chromosomes from becoming disorganized at the start of mitosis, a drug that improved presenilin kinetochore binding could be developed. Similarly, if apoE4 interaction with microtubules is defective and leads to chromosome mis-segregation, a drug could be developed to increase the effectiveness of apoE4 in individuals carrying this allele.
Alternatively, it may be possible to strengthen the fidelity of the chromosome segregation process prophylactically. Several approaches are possible. It may be possible to identify and eliminate environmental toxins that cause chromosome mis-segregation, or to develop agents that counteract the effect of the toxins. Such treatments might include heavy metal chelaters, antioxidants, and promoters of microtubule assembly. Drugs that improve chromosome segregation might also include those that affect DNA topoisomerase II or centromere binding proteins such as CBF1 or DIS1.185-187
A more difficult, but potentially equally effective approach to therapy, would be to determine whether it is the aneuploid—for instance, trisomy 21—cells per se that are contributing to the development of Alzheimer's disease, as appears to be the case in Down's syndrome, or whether it is the secondary effects of aberrant mitosis or attempted mitosis that are important. If the latter is the case, then the drugs discussed above would be appropriate. If the trisomy 21 cells themselves are the problem, then it may be possible to target them for removal from the body by exploiting unusual features of their cell biology. Specifically, several genes are overexpressed in trisomy 21 cells to a much greater extent than would be expected by the 50% increase in gene dosage. An example is the APP protein itself; another is the interferon α-2 receptor. Antibodies that recognize such cell surface antigens that are overexpressed to a great extent in trisomy 21 cells could be coupled to cellular toxins and used to selectively eliminate these cells. Similarly, if the preliminary evidence indicating that trisomy 21 microglial cells are constitutively active and expressing the lymphokine IL-1 is confirmed, it may be possible to eliminate IL-1 positive microglial cells at a single point in time by microglia-targeted gene therapy with a toxin gene under the control of a regulatable IL-1 promoter. Trisomy 21 microglial cells that receive the gene would self-destruct, but only if another co-inducting signal were also present—for instance, tetracycline. Once the trisomy 21 cells were eliminated, the co-inducer could be removed, and the remaining, diploid, microglial cells (though still containing the toxin gene under the control of the IL-1 promoter and the tetracycline enhancer) would still be able to carry out their normal function during infection or other demands for brain inflammation in which the IL-1 cytokine must be produced under proper regulatory control. Because the toxin gene would not be activated, the normal microglial cells could be induced to express IL-1 without being killed.
In summary, some Alzheimer's disease patients are clearly mosaic for trisomy 21, and many others may have low levels of trisomy 21 mosaicism. The mechanism(s) by which these abnormal cells arise and whether/how they contribute to the pathogenesis of the disease are areas of active investigation. Further exploration of these novel findings has the potential to contribute to the development of diagnoses and therapies for Alzheimer's disease and to increase our understanding of basic cellular processes.
References
- 1.
- Potter H, Ma J, Das S. et al. Beyond β-protein: new steps in the pathogenic pathway to Alzheimer's diseaseIn: Iqbal K, Mortimer JA, Winblad B et al, eds.Research Advances in Alzheimer's Disease and Related DisordersNew York: John Wiley and Sons,1995643–654.
- 2.
- Yankner BA. Mechanisms of neuronal degeneration in Alzheimer's disease. Neuron. 1996;16:921–932. [PubMed: 8630250]
- 3.
- Potter H. Beyond beta protein—The essential role of inflammation in Alzheimer amyloid formationIn: Bondy SC, Campell A, eds.Inflammatory Events in NeurodegenerationProminent Press,2001 .
- 4.
- Selkoe DJ. Physiol Rev. Alzheimer's disease Genes proteins and therapy;2001;81:741–766. [PubMed: 11274343]
- 5.
- Potter H, Wefes IM, Nilsson LNG. The inflammation-induced pathological chaperones ACT and apoE are necessary catalysts of Alzheimer amyloid formation Neurobiol Aging 2001. in press . [PubMed: 11755000]
- 6.
- Ma J, Yee A, Brewer HB. et al. The Alzheimer amyloid-associated proteins a1-antichymotrypsin and apolipoprotein E promote the assembly of the Alzheimer β-protein into filaments Nature 199437292–94.(see also “News and Views” same issue) [PubMed: 7969426]
- 7.
- Wisniewski T, Castaño EM, Golabek A. et al. Acceleration of Alzheimer's fibril formation by apolipoprotein E in vitro. Am J Pathol. 1994;145:1030–1035. [PMC free article: PMC1887417] [PubMed: 7977635]
- 8.
- Bales KR, Verina T, Dodel RC. et al. Lack of apolipoprotein E dramatically reduces amyloid β-peptide deposition. Nature Genetics. 1997;17:263–264. [PubMed: 9354781]
- 9.
- Holzman DM, Bales KR, Tenkova T. et al. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2000;97:2892–2897. [PMC free article: PMC16026] [PubMed: 10694577]
- 10.
- Mucke L, Yu G-Q, McConlogue L. et al. Astroglial expression of human α1-antichymotrypsin enhances Alzheimer-like pathology in amyloid protein precursor transgenic mice. Am J Pathol. 2000;157:2003–2010. [PMC free article: PMC1885780] [PubMed: 11106573]
- 11.
- Nilsson LNG, Bales KR, DiCarlo D. et al. α-1-antichymotrypsin promotes beta-sheet amyloid plaque deposition in transgenic mouse model of Alzheimer's disease. J Neurosci. 2001;21:1444–1451. [PMC free article: PMC6762932] [PubMed: 11222634]
- 12.
- Nilsson LNG, Arendash GW, Low MA. et al. Cognitive impairment depends on ApoE and ACT-catalyzed amyloid formation in an Alzheimer's mouse model. (submitted). [PubMed: 15312961]
- 13.
- Olson MI, Shaw CM. Presenile dementia and. Brain. Alzheimer's disease in mongolism;1969;92:147–156. [PubMed: 4237656]
- 14.
- Glenner G, Wong CW. Alzheimer disease and Down's syndrome: Sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun. 1984;122:1131–1135. [PubMed: 6236805]
- 15.
- Wisniewski HM, Rabe A, Wisniewski KE. Neuropathology and dementia in people with Down's syndromeIn: Davies P, Finch C, eds.Molecular Neuropathology of AgingNew York: Cold Spring Harbor Laboratory,1988399–413.
- 16.
- Epstein CJ. The consequences of chromosome imbalance. Am J Med Genet Supp. 1990;7:31–37. [PubMed: 2149968]
- 17.
- Schweber M. A possible unitary genetic hypothesis for Alzheimer's disease and Down syndrome. Ann NY Acad Sci. 1985;450:223–238. [PubMed: 3160290]
- 18.
- Schweber MS. Alzheimer's Disease Related DisordersSan Diego: Alan R Liss, Inc.1989247–267.
- 19.
- Goldgaber D, Lerman MJ, McBride OW. et al. Characterization and chromosomal localization of a cDNA encoding brain amyloid of Alzheimer's disease. Science. 1987;235:877–880. [PubMed: 3810169]
- 20.
- Kang J, Lemaire HG, Unterback A. et al. The precursor of Alzheimer disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–736. [PubMed: 2881207]
- 21.
- Robakis NK, Ramakrishna N, Wolfe G. et al. Molecular cloning and characterization of a cDNA encoding the cerebrovascular and the neuritic plaque amyloid peptides. Proc Natl Acad Sci USA. 1987;84:4190–4194. [PMC free article: PMC305050] [PubMed: 3035574]
- 22.
- Tanzi RE, Gusella JF, Watkins PC. et al. Amyloid β-protein gene; cDNA, mRNA distributions, and genetic linkage near the Alzheimer locus. Science. 1987;235:880–884. [PubMed: 2949367]
- 23.
- Neve RL, Finch EA, Dawes LR. Expression of the Alzheimer amyloid precursor gene transcripts in the human brain. Neuron. 1988;1:669–677. [PubMed: 2908447]
- 24.
- Rumble B, Retallack R, Hilbich C. et al. Amyloid A4 protein and its precursor in Down's syndrome and Alzheimer's disease. NEJM. 1989;320(22):1446–1452. [PubMed: 2566117]
- 25.
- Oyama F, Cairns NJ, Shimada H. et al. Down's syndrome: Up-regulation of beta-amyloid protein precursor and tau mRNAs and their defective coordination. J Neurochem. 1994;62(3):1062–1066. [PubMed: 8113792]
- 26.
- Potter H. Review and hypothesis: Alzheimer disease and Down syndrome—Chromosome 21 nondisjunction may underlie both disorders. Am J Hum Genet. 1991;48:1192–1200. [PMC free article: PMC1683102] [PubMed: 1827946]
- 27.
- Scinto LFM, Daffner KR, Dressler D. et al. A potential non-invasive neurobiological test for Alzheimer's disease. Science. 1994;266:1051–1054. [PubMed: 7973660]
- 28.
- Scinto LFM, Rentz DM, Potter H. et al. Pupil assay and Alzheimer's disease: A critical analysis. Neurology. 1999;52:673. [PubMed: 10025819]
- 29.
- Scinto LMF, Wu CK, Firla KM. et al. Focal Pathology in the Edinger-Westphal nucleus explains pupillar hypersensitivity in Alzheimer's disease. Acta Neuropath. 1999;97:557–564. [PubMed: 10378374]
- 30.
- Scinto LF, Frosch M, Wu CK. et al. Selective cell loss in Edinger-Westphal in asymptomatic elders and Alzheimer's patients. Neurobiol Aging. 2001;22:729–36. [PubMed: 11705632]
- 31.
- Heston LL, Mastri AR. The genetics of Alzheimer's disease. Arch Gen Psychiatry. 1977;34:976–981. [PubMed: 142459]
- 32.
- Heston LL, Mastri AR, Anderson VE. et al. Dementia of the Alzheimer type. Arch Gen Psychiat. 1981;38:1084–1090. [PubMed: 6457579]
- 33.
- Heyman A, Wilkinson W, Hurwitz B. et al. Alzheimer's disease: Genetic aspects and associated clinical disorders. Ann Neurol. 1983;14:507–515. [PubMed: 6228188]
- 34.
- Whalley LJ, Carothers AD, Collyer S. et al. A study of familial factors in Alzheimer's disease. Brit J Psychiat. 1982;140:249–256. [PubMed: 6212094]
- 35.
- Amaducci LA, Fratiglioni L, Rocca WA. et al. Risk factors for clinically diagnosed Alzheimer's disease: A case-control study of an Italian population. Neurology. 1986;36:922–931. [PubMed: 3714054]
- 36.
- Chandra V, Philipose V, Bell PA. et al. Case-control study of late onset “probable Alzheimer's disease.” Neurology. 1987;37:1295–1300. [PubMed: 3614650]
- 37.
- Schupf N, Kapell D, Lee JH. et al. Increased risk of Alzheimer's disease in mothers of adults with Down's syndrome. The Lancet. 1994;344:353–356. [PubMed: 7914304]
- 38.
- Schapiro MB, Kumar A, White B. et al. Alzheimer's disease (AD) in mosaic/translocation Down's syndrome (Ds) without mental retardation. Neurology. 1989;39:169.
- 39.
- Rowe IF, Ridler MAC, Gibberd FB. Presenile dementia associated with mosaic trisomy 21 in a patient with a Down syndrome child. Lancet. 1989;July:229. [PubMed: 2568565]
- 40.
- Hardy J, Goate A, Owen M. et al. Presenile dementia associated with mosaic trisomy 21 in a patient with a Down syndrome child. Lancet. 1989;September:743. [PubMed: 2570989]
- 41.
- Percy ME, Markovic VD, McLachlan DRC. et al. Family with 22-derived marker chromosome and late-onset dementia of the Alzheimer type: I. Application of a new model for estimation of the risk of disease associated with the marker. Am J Med Genet. 1991;39:307–313. [PubMed: 1867282]
- 42.
- Jarvik LF, Yen F-S, Goldstein F. Chromosomes and mental status. A study of women residing in institutions for the elderly. Arch Gen Psychiat. 1974;30:186–190. [PubMed: 4809925]
- 43.
- Ward BE, Cook RH, Robinson A. et al. Increased aneuploidy in Alzheimer disease. Am J Med Genet. 1979;3:137–144. [PubMed: 474626]
- 44.
- Nordenson I, Adolfsson R, Beckman G. et al. Chromosomal abnormality in dementia of Alzheimer type. Lancet. 1980;1:481–482. [PubMed: 6102203]
- 45.
- White BJ, Crandall C, Goudsmit J. et al. Cytogenetic studies of familial and sporadic Alzheimer disease. Am J Med Genet. 1981;10:77–89. [PubMed: 7294063]
- 46.
- Buckton KE, Whalley LJ, Lee M. Chromosome changes in Alzheimer's presenile dementia. J Med Genet. 1983;20:46–51. [PMC free article: PMC1048985] [PubMed: 6842534]
- 47.
- Moorhead PS, Heyman A. Chromosome studies of patients with Alzheimer's disease. Am J Med Genet. 1983;14:545–556. [PubMed: 6859105]
- 48.
- Kormann-Bortolotto MH, Smith MAC, Neto JT. Alzheimer's disease and ageing: A chromosomal approach. Gerontol. 1993;39:1–6. [PubMed: 8440486]
- 49.
- Lao JI, Beyer K, Fernandez-Novoa L. et al. Cytogenetic signs of genomic instability in Alzheimer's disease. Neurobio Aging. 1996;17:S62 (supplement).
- 50.
- Migliore L, Testa A, Scarpato R. et al. Spontaneous and induced aneuploidy in peripheral blood lymphocytes of patients with Alzheimer's disease. Hum Genet. 1997;101:299–305. [PubMed: 9439658]
- 51.
- Fitzgerald PH, Pickering AF, Mercer JM. et al. Premature centromere division: A mechanism of non-disjunction causing X chromosome aneuploidy in somatic cells of man. Ann Hum Genet. 1975;38:417–428. [PubMed: 1190734]
- 52.
- Fitzgerald PH, Archer SA, Morris CM. Evidence for the repeated primary non-disjunction of chromosome 21 as a result of premature centromere division (PCD). Hum Gene. 1986;72:58–62. [PubMed: 2935477]
- 53.
- Lichter P, Cremer T, Tang CJC. et al. Rapid detection of human chromosome 21 aberrations by in situ hybridization. Proc Natl Acad Sci USA. 1988;85:9664–9668. [PMC free article: PMC282830] [PubMed: 2974158]
- 54.
- Fuscoe JC, Collins CC, Pinkel D. et al. An efficient method for selecting unique-sequence clones from DNA libraries and its application to fluorescent staining of human chromosome 21 using in situ hybridization. Genomics. 1989;5:100–109. [PubMed: 2527800]
- 55.
- Potter H, Geller LN. Alzheimer's disease, Down's syndrome, and chromosome segregation. The Lancet. 1996;348:66. [PubMed: 8691961]
- 56.
- Geller LN, Potter H. Chromosome mis-segregation and trisomy 21 mosaicism in Alzheimer's disease. Neurobiol Disease. 1999;6:167–179. [PubMed: 10408806]
- 57.
- Schellenberg GD, Bird TD, Wijsman EM. et al. Genetic linkage evidence for a familial Alzheimer's disease locus on chromosome 14. Science. 1993;258:668–671. [PubMed: 1411576]
- 58.
- Sherrington R, Rogaev EI, Liang Y. et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995;375:754–760. [PubMed: 7596406]
- 59.
- Levy-Lahad E, Wijsman EM, Nemens E. et al. A familial Alzheimer's disease locus on chromosome I. Science. 1995a;269:970–973. [PubMed: 7638621]
- 60.
- Levy-Lahad E, Wasco W, Poorkaj P. et al. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science. 1995b;269:973–977. [PubMed: 7638622]
- 61.
- Rogaev EI, Sherrington R, Rogaeva EA. et al. Familial Alzheimer's disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene. Nature. 1995;376:775–778. [PubMed: 7651536]
- 62.
- Li J, Ma J, Potter H. Identification and expression analysis of a potential familial Alzheimer's disease gene on chromosome 1 related to AD3. Proc Natl Acad Sci USA. 1995;92:12180–12184. [PMC free article: PMC40320] [PubMed: 8618867]
- 63.
- Migliore L, Botto N, Scarpato R. et al. Preferential occurrence of chromosome 21 malsegregation in peripheral blood lymphocytes of Alzheimer's disease patients. Cytogenet Cell Genet. 1999;87:41–46. [PubMed: 10640809]
- 64.
- Yang Y, Geldmacher DS, Herrup K. DNA replication precedes neuronal cell death in Alzheimer's disease. J Neurosci. 2001;21:2661–2668. [PMC free article: PMC6762514] [PubMed: 11306619]
- 65.
- Ford JH. Spindle microtubular dysfunction in mothers of Down syndrome children. Hum Genet. 1984;68:295–298. [PubMed: 6239816]
- 66.
- Staessen C, Maes AM, Kirsch-Volders M. et al. Is there a predisposition for meiotic non-disjunction that may be detected by mitotic hyperploidy? Clinical Genetics. 1983;24:184–190. [PubMed: 6226461]
- 67.
- Suzuki T, Oishi M, Marshak DR. et al. Cell cycle-dependent regulation of the phosphorylation and metabolism of the Alzheimer amyloid precursor protein. EMBO J. 1994;13:1114–1122. [PMC free article: PMC394920] [PubMed: 8131745]
- 68.
- Pope WB, Lambert MP, Leypold B. et al. Microtubule-associated protein tau is hyperphosphorylated during mitosis in the human neuroblastoma cell line SH-SY5Y. Exper Neurol. 1994;126:185–194. [PubMed: 7925819]
- 69.
- Preuss U, DÖring F, Illenberger S. et al. Cell cycle-dependent phosphorylation and microtubule binding of tau protein stably transfected in Chinese hamster ovary cells. Mol Biol Cell. 1995;6:1397–1410. [PMC free article: PMC301295] [PubMed: 8573794]
- 70.
- Arendt T, Holzer M, Grossmann A. et al. Increased expression and subcellular translocation of the mitogen activated protein kinase and mitogen-activated protein kinase in Alzheimer's disease. Neuroscience. 1995;68:5–18. [PubMed: 7477934]
- 71.
- Arendt T, Rodel L, Gartner U. et al. Expression of the cyclin-dependent kinase inhibitor p16 in Alzheimer's disease. NeuroReport. 1996;7:3047–3049. [PubMed: 9116237]
- 72.
- Smith TW, Lippa CF. TI Ki-67 immunoreactivity in Alzheimer's disease and other neurodegenerative disorders. J Neuropathol Exp Neurol. 1995;54:297–303. [PubMed: 7745428]
- 73.
- Vincent I, Rosado M, Davies P. Mitotic mechanisms in Alzheimer's disease? J Cell Biol. 1996;132:413–425. [PMC free article: PMC2120731] [PubMed: 8636218]
- 74.
- Vincent I, Jicha G, Rosado M. et al. Aberrant expression of mitotic Cdc2/cyclin B1 kinase in degenerating neurons of Alzheimer's disease brain. J Neurosci. 1997;17:3588–3598. [PMC free article: PMC6573674] [PubMed: 9133382]
- 75.
- Kondratick CM, Vandre DD. Alzheimer's disease neurofibrillary tangles contain mitosis-specific phosphoepitopes. J Neurochem. 1996;67:2405–2416. [PubMed: 8931473]
- 76.
- Nagy Z, Esiri MM, Cato AM. et al. Cell cycle markers in the hippocampus in Alzheimer's disease. Acta Neuropathologica. 1997;94:6–15. [PubMed: 9224524]
- 77.
- McShea A, Harris PLR, Webster KR. et al. Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer's disease. Amer J Pathol. 1997;150:1933–1939. [PMC free article: PMC1858317] [PubMed: 9176387]
- 78.
- Li J, Xu M, Zhou H. et al. Alzheimer presenilins in the nuclear membrane, interphase kinetochores, and centrosomes suggest a role in chromosome segregation. Cell. 1997;90:917–927. [PubMed: 9298903]
- 79.
- McShea A, Wahl AF, Smith MA. Re-entry into the cell cycle: A mechanism for neurodegeneration in Alzheimer disease Medical Hypotheses 1998. in press . [PubMed: 10459833]
- 80.
- Schellenberg GD, Bird TD, Wijsman EM. et al. Genetic linkage evidence for a familial Alzheimer's disease locus on chromosome 14. Science. 1992;258:668–71. [PubMed: 1411576]
- 81.
- Kovacs DM, Fausett HJ, Page KJ. et al. Alzheimer-associated presenilins 1 and 2: Neuronal expression in brain and localization to intracellular membranes in mammalian cells. Nature Med. 1996;2:224–229. [PubMed: 8574969]
- 82.
- Walter J, Capell A, Grünberg J. et al. The Alzheimer's disease-associated presenilins are differentially phosphorylated proteins located predominantly within the endoplasmic reticulum. Molec Med. 1996;2:673–91. [PMC free article: PMC2230134] [PubMed: 8972483]
- 83.
- Strooper BDe, Beullens M, Contreras B. et al. Phosphorylation, subcellular localization, and membrane orientation of the Alzheimer's disease-associated presenilins. J Biol Chem. 1997;272:3590–3598. [PubMed: 9013610]
- 84.
- Moroi Y, Hartman AL, Nakane PK. et al. Distribution of kinetochore (centromere) antigen in mammalian cell nuclei. J Cell Biol. 1981;90:254–259. [PMC free article: PMC2111823] [PubMed: 7019222]
- 85.
- Brenner S, Pepper D, Berns MW. et al. Kinetochore structure, duplication, and distribution in mammalian cells: analysis by human autoantibodies from scleroderma patients. J Cell Biol. 1981;91:95–102. [PMC free article: PMC2111947] [PubMed: 7298727]
- 86.
- Weimer R, Haaf T, Kruger J. et al. Characterization of centromere arrangements and test for random distribution in G0, G1, S, G2, G1, and early S' phase in human lymphocytes. Hum Genet. 1992;88:673–682. [PubMed: 1551672]
- 87.
- Annaert WG, Levesque L, Craessaerts K. et al. Presenilin 1 controls gamma-secretase processing of amyloid precursor protein in pre-golgi compartments of hippocampal neurons. J Cell Biol. 1999;147:277–294. [PMC free article: PMC2174229] [PubMed: 10525535]
- 88.
- Honda T, Nihonmatsu N, Yasutake K. et al. Familial Alzheimer's disease-associated mutations block translocation of full-length presenilin 1 to the nuclear envelope. Neurosci Res. 2000;37:101–111. [PubMed: 10867173]
- 89.
- Jeong SJ, Kim HS, Chang KA. et al. Subcellular localization of presenilins during mouse preimplantationdevelopment. FASEB J. 2000;14(14):2171–2176. [PubMed: 11053237]
- 90.
- Johnsingh AA, Johnston JM, Merz G. et al. Altered binding of mutated presenilin with cytoskeleton-interacting proteins. FEBS Lett. 2000;465(1):53–58. [PubMed: 10620705]
- 91.
- Pigino G, Pelsman A, Mori H. et al. Presenilin-1 mutations reduce cytoskeletal association, deregulate neurite growth, and potentiate neuronal dystrophy and tau phosphorylation. J Neurosci. 2001;21:834–842. [PMC free article: PMC6762317] [PubMed: 11157069]
- 92.
- Janicki SM, Monteiro MJ. Presenilin overexpression arrests cells in the G1 phase of the cell cycle: arrest potentiated by the Alzheimer's disease PS2 (N141I) Mutant. Am J Pathol. 1999;155:135–144. [PMC free article: PMC1866651] [PubMed: 10393846]
- 93.
- Vito P, Lacana E, D'Adamio L. Interfering with apoptosis: Ca(2+)-binding protein ALG-2 and Alzheimer's disease gene ALG-3. Science. 1996;271:521–165. [PubMed: 8560270]
- 94.
- Vito P, Wolozin B, Ganjei JK. et al. Requirement of the familial Alzheimers disease gene PS2 for apoptosis. Opposing effect of ALG-3. J Biol Chem. 1996;271: 31025–31028. [PubMed: 8940094]
- 95.
- Wolozin B, Iwasaki K, Vito P. et al. Participation of presenilin 2 in apoptosis: Enhanced basal activity conferred by an Alzheimer mutation. Science. 1996;274:1710–1713. [PubMed: 8939861]
- 96.
- Wragg M, Hutton M, Talbot C. Genetic association between intronic polymorphism in presenilin-1 gene and late-onset Alzheimer's disease. Alzheimer's Disease Collaborative Group. Lancet. 1996;347(9000):509–512. [PubMed: 8596269]
- 97.
- Higuchi S, Muramatsu T, Matsushita S. et al. Presenilin-1 polymorphism and Alzheimer's disease. Lancet. 1996;347:1186. [PubMed: 8609783]
- 98.
- Isoe K, Urakami K, Ji Y. et al. Presenilin-1 polymorphism in patients with Alzheimer's disease, vascular dementia and alcohol-associated dementia in Japanese population. Acta Neurol Scand. 1996;94:326–328. [PubMed: 8947284]
- 99.
- Kehoe P, Williams J, Holmans P. et al. Association between a PS-1 intronic polymorphism and late onset Alzheimer's disease. NeuroReport. 1996a;7:2155–2158. [PubMed: 8930979]
- 100.
- Kehoe P, Williams J, Lovestone S. et al. Presenilin-1 polymorphism and Alzheimer's disease. Lancet. 1996b;347:1185. [PubMed: 8609781]
- 101.
- Brookes AJ, Howell WM, Woodburn K. et al. Presenilin-I, presenilin-II, and VLDL-R associations in early onset Alzheimer's disease. Lancet. 1997;350:336–337. [PubMed: 9251642]
- 102.
- Ezquerra M, Blesa R, Tolosa E. et al. The genotype 2/2 of the presenilin-1 polymorphism is decreased in Spanish early-onset Alzheimer's disease. Neurosci Lett. 1997;227:201–204. [PubMed: 9185685]
- 103.
- Nishiwaki Y, Kamino K, Yoshiiwa A. et al. T/G polymorphism at intron 9 of presenilin 1 gene is associated with, but not responsible for sporadic late-onset Alzheimer's disease in Japanese population. Neurosci Lett. 1997;227:123–126. [PubMed: 9180219]
- 104.
- Tilley L, Morgan K, Grainger J. et al. Evaluation of polymorphisms in the presenilin-1 gene and the butyrylcholinesterase gene as risk factors in sporadic Alzheimer's disease. Eur J Hum Genet. 1999;7:659–663. [PubMed: 10482954]
- 105.
- Perez-Tur J, Froelich S, Prihar G. et al. A mutation in Alzheimer's disease destroying a splice acceptor site in the presenilin-1 gene. Neuroreport. 1995;7:297–301. [PubMed: 8742474]
- 106.
- Scott WK, Growdon JH, Roses AD. et al. Presenilin-1 polymorphism and Alzheimer's disease. Lancet. 1996;347:1186–1187. [PubMed: 8609784]
- 107.
- Scott WK, Yamaoka LH, Locke PA. et al. No association or linkage between an intronic polymorphism of presenilin-1 and sporadic or late-onset familial Alzheimer disease. Genet Epidemiol. 1997;14:307–315. [PubMed: 9181359]
- 108.
- Cai X, Stanton J, Fallin D. et al. No association between the intronic presenilin-1 polymorphism and Alzheimer's disease in clinic and population-based samples. Am J Med Genet. 1997;74:202–203. [PubMed: 9129725]
- 109.
- Lendon CL, Myers A, Cumming A. et al. A polymorphism in the presenilin 1 gene does not modify risk for Alzheimer's disease in a cohort with sporadic early onset. Neurosci Lett. 1997;228:212–214. [PubMed: 9218645]
- 110.
- Singleton AB, Lamb H, Leake A. et al. No association between an intronic polymorphism in the presenilin-1 gene and Alzheimer's disease. Neurosci Lett. 1997;234:19–22. [PubMed: 9347936]
- 111.
- Sorbi S, Nacmias B, Tedde A. et al. Presenilin-1 gene intronic polymorphism in sporadic and familial Alzheimer's disease. Neurosci Lett. 1997;222:132–134. [PubMed: 9111746]
- 112.
- Tysoe C, Galinsky D, Robinson D. et al. Analysis of α-1 antichymotrypsin, presenilin-1, angiotensin-converting enzyme, and methylenetetrahydrofolate reductase loci as candidates for dementia. Am J Med Genet. 1997;74:207–212. [PubMed: 9129727]
- 113.
- Tysoe C, Whittaker J, Cairns NJ. et al. Presenilin-1 intron 8 polymorphism is not associated with autopsy-confirmed late-onset Alzheimer's disease. Neurosci Lett. 1997b;222:68–69. [PubMed: 9121726]
- 114.
- Wu X, Jiang S, Lin S. et al. No association between the intronic presenilin 1 polymorphism and Alzheimer's disease in the Chinese population. Am J Med Genet. 1999;88:1–3. [PubMed: 10050959]
- 115.
- Petersen MB, Karadima G, Samaritaki M. et al. Association between presenilin-1 polymorphism and maternal meiosis II errors in Down syndrome. Am J Med Genet. 2000;93:366–72. [PubMed: 10951459]
- 116.
- Courvalin JC, Segil N, Blobel G. et al. The lamin B receptor of the inner nuclear membrane undergoes mitosis-specific phosphorylation and is a substrate for p34cdc2-type protein kinase. J Biol Chem. 1992;267:19035–19038. [PubMed: 1326541]
- 117.
- Winey M, Hoyt MA, Chan C. et al. NDC1: A nuclear periphery component required for yeasts spindle pole body duplication. J Cell Biol. 1993;122:743–751. [PMC free article: PMC2119589] [PubMed: 8349727]
- 118.
- Ye Q, Worman HJ. Primary structure analysis and lamin B and DNA binding of human LBR, an integral protein of the nuclear envelope inner membrane. J Biol Chem. 1994;269:11306–11311. [PubMed: 8157662]
- 119.
- Thinakaran G, Borchelt DR, Lee MK. et al. Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron. 1996;17:181–190. [PubMed: 8755489]
- 120.
- Mercken M, Takahashi H, Honda T. et al. Characterization of human presenilin 1 using N-terminal specific monoclonal antibodies: evidence that Alzheimer mutations affect proteolytic processing. FEBS Letters. 1996;389:297–303. [PubMed: 8766720]
- 121.
- Shen J, Bronson RT, Chen DF. et al. Skeletal and CNS defects in Presenilin-1-deficient mice. Cell. 1997;89:629–639. [PubMed: 9160754]
- 122.
- Duff K, Eckman C, Zher C. et al. Increased amyloid-β42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383:710–713. [PubMed: 8878479]
- 123.
- Donehower LA, Godley LA, Aldaz CM. et al. Deficiency of p53 accelerates mammary tumorigenesis in Wnt-1 transgenic mice and promotes chromosomal instability. Dev. 1995;9:882–895. [PubMed: 7705663]
- 124.
- Fukasawa K, Choi T, Kuriyama R. et al. Abnormal centrosome amplification in the absence of p53. Science. 1996;271:1744–1747. [PubMed: 8596939]
- 125.
- Donehower LA. Genetic instability in animal tumorigenesis models. Cancer Surv. 1997;29:329–352. [PubMed: 9338107]
- 126.
- Boyle JM, Mitchell ELD, Graves MJ. et al. Chromosome instability is a predominant trait of fibroblasts from Li-Fraumeni families. Br J Cancer. 1998;77:2181–2192. [PMC free article: PMC2150396] [PubMed: 9649131]
- 127.
- Roperch JP, Alvaro V, Prieur S. et al. Inhibition of presenilin 1 expression is promoted by p53 and p21WAF-1 and results in apoptosis and tumor suppression. Nat Med. 1998;14:835–838. [PubMed: 9662377]
- 128.
- Altman J, Das GD. Autoradiographic and histologic evidence of postnatal neurogenesis in rats. J Comp Neurol. 1965;124:319–335. [PubMed: 5861717]
- 129.
- Kaplan MS, Hinds JW. Neurogenesis in the adult rat: electron microscopic analysis of light radioautographs. Science. 1977;197:1092–1094. [PubMed: 887941]
- 130.
- Bayer SA, Yackel JW, Puri PS. Neurons in the rat dentate gyrus granular layer substantially increase during juvenile and adult life. Science. 1982;216:890–892. [PubMed: 7079742]
- 131.
- Cameron HA, Woolley CS, McEwen BS. et al. Differentiation of newly born neurons and glia in the dentate gyrus of the adult rat. Neuroscience. 1993;56:337–344. [PubMed: 8247264]
- 132.
- Kaplan MS, Bell DH. Mitotic neuroblasts in the 9-day-old and 11-month-old rodent hippocampus. J Neurosci. 1984;4:1429–1441. [PMC free article: PMC6564978] [PubMed: 6726341]
- 133.
- Gage FH, Ray J, Fisher LJ. Isolation, characterization, and use of stem cells from the CNS. Annu Rev Neurosci. 1995;18:159–192. [PubMed: 7605059]
- 134.
- Kuhn GH, Dickinson-Anson H, Gage FH. Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J Neurosci. 1996;16(6):2027–2033. [PMC free article: PMC6578509] [PubMed: 8604047]
- 135.
- Gould E, Tanapat P, McEwen BS. et al. Neurogenesis in the dentate gyrus of adult primates can be suppressed by stress. Proc Natl Acad Sci USA. 1998;95:3168–3171. [PMC free article: PMC19713] [PubMed: 9501234]
- 136.
- Gould E, Reeves AJ, Graziano MS. et al. Neurogenesis in the neocortex of adult primates. Science. 1999a;286:548–552. [PubMed: 10521353]
- 137.
- Gould E, Reeves AJ, Fallah M. et al. Hippocampal neurogenesis in adult Old World primates. Proc Natl Acad Sci USA. 1999b;96:5263–5267. [PMC free article: PMC21852] [PubMed: 10220454]
- 138.
- Eriksson PS, Perfilieva E, Bjork-Eriksson T. et al. Neurogenesis in the adult human hippocampus. Nature Med. 1998;4:1313–1317. [PubMed: 9809557]
- 139.
- LeBlank A. Increased production of 4KDa amyloid beta peptide in serum-deprived human primary neuron cultures: possible involvement of apoptosis. J Neurosci. 1995;15:7837–7846. [PMC free article: PMC6577962] [PubMed: 8613723]
- 140.
- Galli C, Piccini A, Ciotti MT. et al. Increased amyloidogenic secretion in cerebellar granule cells undergoing apoptosis. Proc Natl Acad Sci USA. 1998;95:1247–1252. [PMC free article: PMC18734] [PubMed: 9448317]
- 141.
- Rakic P. Limits of neurogenesis in primates. Science. 1985;227:1054–1056. [PubMed: 3975601]
- 142.
- McGeer PL, Itagaki S, Boyes E. et al. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci Lett. 1987;79:195–200. [PubMed: 3670729]
- 143.
- Abraham CR, Selkoe DJ, Potter H. Immunochemical identification of the serine protease inhibitor α1-antichymotrypsin in the brain amyloid deposits of Alzheimer's disease. Cell. 1988;52:487–501. [PubMed: 3257719]
- 144.
- Abraham CR, Potter H. Alzheimer's disease: Recent advances in understanding the brain amyloid deposits. Biotechnology. 1998;7:147–153.
- 145.
- Potter H. The involvement of astrocytes and an acute phase response in the amyloid deposition of Alzheimer's diseaseIn: Yu ACH, Hertz LMD, eds.Neuronal-astrocytic interactions: Implication for normal and pathological CNS function. Prog Brain Res 199294447–458. [PubMed: 1287729]
- 146.
- McGeer PL, Kawamata T, Walker DG. et al. Microglia in degenerative neurological disease. Glia. 1993;7:84–92. [PubMed: 8423066]
- 147.
- Eikelenboom P, Zhan SS, van Gool WA. et al. Inflammatory mechanisms in Alzheimer's disease. TIPS. 1994;15:447–450. [PubMed: 7886816]
- 148.
- Das S, Potter H. Expression of the Alzheimer amyloid-promoting factor antichymotrypsin is induced in human astrocytes by IL-1. Neuron. 1995;14:447–456. [PubMed: 7857652]
- 149.
- Ma J, Brewer JrHB, Potter H. Alzheimer Aβ neurotoxicity: promotion by antichymotrypsin, ApoE4; inhibition by Aβ-related peptides. Neurobiol Aging. 1996;17:773–780. [PubMed: 8892351]
- 150.
- Griffin WST, Stanley LC, Ling C. et al. Brain interleukin-1 and S100 immunoreactivity elevated in Down's syndrome and Alzheimer's disease. Proc Natl Acad Sci USA. 1989;86:7611–7615. [PMC free article: PMC298116] [PubMed: 2529544]
- 151.
- Sheng JG, Mrak RE, Griffin WS. Microglial interleukin-1 alpha expression in brain regions in Alzheimer's disease: correlation with neuritic plaque distribution. Neuropathol Appl Neurobiol. 1995;21:290–301. [PubMed: 7494597]
- 152.
- Sanan DA. et al. Apolipoprotein E associates with beta amyloid peptide of Alzheimer's disease to form novel monofibrils. Isoform apoE4 associates more efficiently than apoE3. J Clin Invest. 1994;94:860–869. [PMC free article: PMC296168] [PubMed: 8040342]
- 153.
- Castano EM, Frangione B. Biology of disease: Human amyloidosis, Alzheimer disease and related disorders. Lab Invest. 1988;58:122–132. [PubMed: 3276958]
- 154.
- Selkoe DJ. Molecular pathology of amyloidogenic proteins and the role of vascular amyloidosis in Alzheimer's disease. Neurobiol Aging. 1989b;10:387–3395. [PubMed: 2510035]
- 155.
- Busciglio J, Yankner BA. Apoptosis and increased generation of reactive oxygen species in Down's syndrome neurons in vitro. Nature. 1997;378:776–779. [PubMed: 8524410]
- 156.
- Teller JK, Russo C, DeBusk LM. et al. Presence of soluble amyloid β-peptide precedes amyloid plaque formation in Down's syndrome. Nature Medicine. 1996;2:93–95. [PubMed: 8564851]
- 157.
- Deng G, Pike CJ, Cotman CW. Alzheimer-associated presenilin-2 confers increased sensitivity to apoptosis in PC12 cells. FEBS Letters. 1996;397:50–54. [PubMed: 8941712]
- 158.
- Janicki S, Monteiro MJ. Increased apoptosis arising from increased expression of the Alzheimer's disease-associated presenilin-2 mutation (N141I). J Cell Biol. 1997;139:485–495. [PMC free article: PMC2139804] [PubMed: 9334350]
- 159.
- Martins RN, Turner BA, Carroll RT. et al. High levels of amyloid-β protein from S182 (Glu246) familial Alzheimer's cells. NeuroReport. 1995;7:217–220. [PubMed: 8742455]
- 160.
- Scheuner D, Eckman C, Jensen M. et al. Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nature Med. 1996;2:864–870. [PubMed: 8705854]
- 161.
- Borchelt DR, Thinakaran G, Eckman CB. et al. Familial Alzheimer's disease-linked presenilin 1 variants elevate Aβ1-42/1-40 ratio in vitro and in vivo. Neuron. 1996;17:1005–1013. [PubMed: 8938131]
- 162.
- Citron M, Westaway D, Xia W. et al. Mutant presenilins of Alzheimer's disease increase production of 42-residue amyloid β-protein in both transfected cells and transgenic mice. Nature Med. 1997;3:67–72. [PubMed: 8986743]
- 163.
- Suzuki N, Cheung TT, Cai XD. et al. An increased percentage of long amyloid β protein secreted by familial amyloid β protein precursor (βAPP717) mutants. Science. 1994b;264:1336–1340. [PubMed: 8191290]
- 164.
- Yamatsuji T, Matsui T, Okamoto T. et al. G protein-mediated neuronal DNA fragmentation induced by familial Alzheimer's disease-associated mutants of APP. Science. 1996;272:1349–1352. [PubMed: 8650548]
- 165.
- Yamatsuji T, Okamoto T, Takeda S. et al. Expression of V642 APP mutant causes cellular apoptosis as Alzheimer trait-linked phenotype. EMBO J. 1996;15:498–509. [PMC free article: PMC449968] [PubMed: 8599933]
- 166.
- Zhao B, Chrest FJ, Horton Jr WE. et al. Expression of mutant amyloid precursor proteins induces apoptosis in PC12 cells. J Neurosci Res. 1997;47:253–263. [PubMed: 9039647]
- 167.
- Gandy S, Czernik AJ, Greengard P. Phosphorylation of Alzheimer disease amyloid precursor protein peptide by protein kinase C and Ca2+/calmodulin-dependent protein kinase II. Proc Natl Acad Sci USA. 1988;85:6218–6221. [PMC free article: PMC281937] [PubMed: 3137567]
- 168.
- Buxbaum JD, Gandy SE, Cicchetti P. et al. Processing of Alzheimer b/A4 amyloid precursor protein: modulation by agents that regulate protein phosphorylation. Proc Natl Acad Sci USA. 1990;87:6003–6006. [PMC free article: PMC54458] [PubMed: 2116015]
- 169.
- Demaerschlack I, Delvaux A, Octave JN. Activation of protein kinase C increases the extracellular release of the transmembrane amyloid protein precursor of Alzheimer's disease. Biochimica et Biophysica Acta. 1993;1181:214–218. [PubMed: 8100450]
- 170.
- da Cruz e Silva EF, da Cruz e Silva OAB, Zaia CTBV. et al. Inhibition of protein phosphatase 1 stimulates secretion of Alzheimer amyloid precursor protein. Mol Med. 1995;1:535–541. [PMC free article: PMC2229959] [PubMed: 8529119]
- 171.
- Xu H, Sweeney D, Greengard P. et al. Metabolism of Alzheimer b-amyloid precursor protein: regulation by protein kinase A in intact cells and in a cell-free system. Proc Natl Acad Sci USA. 1996;93:4081–4084. [PMC free article: PMC39490] [PubMed: 8633020]
- 172.
- Autilio-Gambetti L, Morandi A, Tabaton M. et al. The amyloid percursor protein of Alzheimer disease is expressed as a 130 kDa polypeptide in various cultured cell types. FEBS Lett. 1988;241:94–98. [PubMed: 2904381]
- 173.
- Dranovsky A, Oxberry W, Lyubsky S. et al. APP is associated with mitotic centrosomes and a distinct cytoplasmic network which colocalizes with depolymerized tubulin Neurobiol Aging 199617S196(abs788)
- 174.
- Weidemann A, Paliga K, Durrwang U. et al. Formation of stable complexes between two Alzheimer's disease gene products: presenilin-2 and beta-amyloid precursor protein. Nature Med. 1997;3:328–323. [PubMed: 9055862]
- 175.
- Chen Y, McPhie DL, Hirschberg J. et al. The amyloid precursor protein-binding protein APP-BP1 drives the cell cycle through the S-M checkpoint and causes apoptosis. J Biol Chem. 2000;275:8929–8935. [PubMed: 10722740]
- 176.
- von der Kammer H, Hanes J, Klaudiny J. et al. A human amyloid precursor-like protein is highly homologous to a mouse sequence-specific DNA-binding protein. DNA Cell Biol. 1994;13:1137–1143. [PubMed: 7702756]
- 177.
- Rassoulzadegan M, Yang Y, Cuzin F. APLP2, a member of the Alzheimer precursor protein family, is required for correct genomic segregation in dividing mouse cells. EMBO J. 1998;17:4647–4656. [PMC free article: PMC1170794] [PubMed: 9707424]
- 178.
- Avramopoulos D, Mikkelsen M, Vassilopoulos D. et al. Apolipoprotein E allele distribution in parents of Down's syndrome children. Lancet. 1996;347:862–865. [PubMed: 8622392]
- 179.
- Strittmatter WJ, Roses D. Apolipoprotein E and Alzheimer disease. Proc Natl Acad Sci USA. 1995;92:4725–4727. [PMC free article: PMC41779] [PubMed: 7761390]
- 180.
- Mistry MJ, Clay MA, Kelly ME. et al. Apolipoprotein E restricts interleukin-dependent T lymphocyte proliferation at the G1A/G1B boundary. Cell Immunol. 1995;160:14–23. [PubMed: 7842480]
- 181.
- Uchida IA, Lee CPV, Byrnes EM. Chromosome aberrations in vitro by low doses of radiation: nondisjunction in lymphocytes of young adults. Am J Hum Genet. 1975;27:419–429. [PMC free article: PMC1762864] [PubMed: 803021]
- 182.
- Martyn CN, Osmond C, Edwardson JA. et al. Geographical relation between Alzheimer's disease and aluminium in drinking water. Lancet. 1989;January:59–62. [PubMed: 2562879]
- 183.
- Palekar LD, Eyre JF, Most BM. et al. Metaphase and anaphase analysis of V79 cells exposed to erionite, UICC chrysotile, and UICC crocidolite. Carcinogen. 1987;8:553–560. [PubMed: 3030579]
- 184.
- Ganrot PO. Metabolism and possible health effects of aluminum. Environ Health Persp. 1986;65:363–441. [PMC free article: PMC1474689] [PubMed: 2940082]
- 185.
- Holm C, Stearns T, Botstein D. DNA topoisomerase II must act at mitosis to prevent nondisjunction and chromosome breakage. Mol Cell Biol. 1898;9:159–168. [PMC free article: PMC362157] [PubMed: 2538717]
- 186.
- Cai M, Davis RW. Yeast centromere binding protein CBF1, of the helix-loop-helix protein family, is required for chromosome stability and methionine prototrophy. Cell. 1990;61:437–446. [PubMed: 2185892]
- 187.
- Rockmill B, Fogel S. DISI: A yeast gene required for proper meiotic chromosome disjunction. Genetics. 1988;119:261–272. [PMC free article: PMC1203410] [PubMed: 3294101]
- 188.
- Barinaga M. Missing Alzheimer's gene found. Science. 269:917–918z. [PubMed: 7638610]
- Introduction
- Trisomy 21 Mosaicism in Alzheimer's Disease
- Defects in Mitosis in Alzheimer's Disease
- Potential Mitotic Function of the Presenilin Proteins
- Mitotic Effects of the Normal and FAD Mutant Presenilin Proteins in Transfected Cells
- Chromosome and Cell Cycle Changes in Cells from Presenilin Transgenic and Knockout Mice
- Mutation/Inactivation of the Tumor Suppressor p53 Causes Chromosome Instability and Regulates Expression of Presenilin
- Presenilin Mutations May Effect Mitosis in both Neurons and Glia
- How Aneuploidy May Lead to Alzheimer's Disease
- Potential Involvement of Other Genes in Chromosome Mis-Segregation in Alzheimer's Disease
- Implications of Trisomy 21 Mosaicism in Alzheimer's Disease for Future Diagnosis and Therapy
- References
- Cell Cycle and Chromosome Segregation Defects in Alzheimer's Disease - Madame Cu...Cell Cycle and Chromosome Segregation Defects in Alzheimer's Disease - Madame Curie Bioscience Database
- Genetic Susceptibility to Infectious Diseases Linked to NRAMP1 Gene in Farm Anim...Genetic Susceptibility to Infectious Diseases Linked to NRAMP1 Gene in Farm Animals - Madame Curie Bioscience Database
- Conformational Dynamics within the Ribosome - Madame Curie Bioscience DatabaseConformational Dynamics within the Ribosome - Madame Curie Bioscience Database
- Neuroprotective Strategies in Animal and in Vitro Models of Neuronal Damage: Isc...Neuroprotective Strategies in Animal and in Vitro Models of Neuronal Damage: Ischemia and Stroke - Madame Curie Bioscience Database
- Regulation of Voltage-Sensitive Ca2+ Channels in Bipolar Cells by Divalent Catio...Regulation of Voltage-Sensitive Ca2+ Channels in Bipolar Cells by Divalent Cations and Polyamines - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...