

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Methylenetetrahydrofolate reductase (MTHFR) is a key regulatory enzyme in folate and homocysteine metabolism. Research performed during the past decade has clarified our understanding of MTHFR deficiencies that cause hyperhomocysteinemia with homocystinuria, or mild hyperhomocysteinemia. The cloning of the MTHFR coding sequence was initially followed by the identification of the first deleterious mutations in MTHFR, in patients with homocystinuria. Shortly thereafter, the 677C→T variant was identified and shown to encode a thermolabile enzyme with reduced activity. Currently, a total of 34 rare but deleterious mutations in MTHFR, as well as a total of 9 common variants (polymorphisms) have been reported. The 677C→T (A222V) variant has been particularly noteworthy since it has become recognized as the most common genetic cause of hyperhomocysteinemia. The disruption of homocysteine metabolism by this polymorphism influences risk for several complex disorders, which are discussed in various chapters throughout this book. In this chapter, we describe the complex structure of the MTHFR gene, summarize the current state of knowledge on mutations/polymorphisms in MTHFR and discuss some initial findings in a newly generated mouse model for MTHFR deficiency.
Introduction
Methylenetetrahydrofolate reductase (MTHFR; EC 1.5.1.20) plays a central role in folate and homocysteine metabolism by catalyzing the conversion of 5,10-methylenetetrahydrofolate to 5-methyltetrahydrofolate, the primary circulatory form of folate which is utilized in homocysteine remethylation to methionine.1 The involvement of MTHFR in disease was first published by Mudd et al who identified a patient with homocystinuria due to a severe deficiency of the enzyme.2 This type of MTHFR deficiency, despite being the most common inborn error of folate metabolism, is still relatively rare. In 1988, a thermolabile variant of MTHFR was identified through enzymatic assays of lymphocyte extracts in patients with cardiovascular disease.3 This milder deficiency, which appeared to be more common, resulted in a mild to moderate elevation of plasma total homocysteine, an emerging risk factor for cardiovascular disease. The relationships, if any, between the severe and mild deficiencies were unclear, particularly since thermolabile MTHFR had also been observed in some families with homocystinuria.4,5 At that time, the investigations of MTHFR deficiency were limited to the laboratories with expertise in biochemical genetics methodologies. The isolation of the cDNA in 1994 opened up avenues for molecular genetic approaches to study MTHFR deficiencies.6 The cDNA isolation was quickly followed by identification of a common sequence variant at bp 677 (C→T) which encoded the thermolabile form of the enzyme.7 This variant has become recognized as the most common genetic cause of hyperhomocysteinemia and has been extensively investigated as a risk factor for several multifactorial disorders associated with disturbances in homocysteine metabolism. The experimental progression, from delineation of the rare, severe derangement of metabolism to the less deleterious consequences of the milder 677C→T mutation, was facilitated by the elucidation of molecular information on MTHFR. In this chapter, we review the molecular biology of the MTHFR cDNA and gene. We provide an overview of the genetic mutations in MTHFR, both rare and common sequence variants, and briefly describe our Mthfr-deficient mice, which serve as animal models for both the severe and mild forms of MTHFR deficiency. The reader is referred to other chapters in this book for additional discussions of severe MTHFR deficiency and of the biochemical and clinical consequences of the substitution at bp677.
DNA and Genomic Structure of MTHFR
Cloning the MTHFR Coding Sequence
We initially isolated a 90-bp MTHFR cDNA from pig liver RNA, using peptide sequences of the purified porcine liver enzyme that had been provided by Rowena Matthews and colleagues; degenerate oligonucleotides based on these peptide sequences were used for PCR amplification of porcine liver RNA.6 Sequence data from the porcine cDNA were then used to screen a human liver cDNA library by PCR for isolation of a partial human cDNA (1.3 kb), the predicted amino acid sequence of which showed strong homology to porcine MTHFR and to bacterial metF genes. Another human cDNA library was then screened by plaque hybridization with this 1.3 kb cDNA to obtain the missing C-terminal portion of the MTHFR coding sequence.7 A short 3' UTR sequence was included in the resulting cDNA clone, followed by a poly(A) tail (212 bp downstream of the stop codon).
The cDNA sequence deduced from this work was 2.2 kb in length and encompassed 11 exons.8 This cDNA sequence, available under GenBank GenInfo identifier (GI):6174884, was used as the reference sequence in subsequent reports on the identification of the numerous mutations in MTHFR (see below). When the deduced sequences of human and mouse MTHFR proteins are aligned, 90% of amino acids are identical.8 The location of intronic boundaries and most of the intron sizes are quite similar between the 2 species.
Porcine MTHFR was reported to be a dimer with a total molecular mass of about 150 kDa.9 Based on the structure of the porcine enzyme and initial Western blotting of human tissues, the major product of the human MTHFR gene appeared to be a 77 kDa protein; a second human isoform of approximately 70kDa was also observed on these Western blots. Our expression of the 2.2 kb cDNA in bacterial extracts resulted in a catalytically-active 70kDa protein, suggesting that additional coding sequences would be required to encode the 77kDa isoform.7 Complex alternative splicing in the 5' end of MTHFR was reported by our group10 and others.11 More recently, we identified the predicted upstream translational start site of MTHFR, generated from an alternatively-spliced mRNA, and expressed the cDNA; this cDNA encodes the larger 77 kDa isoform.12 This deduced N-terminal sequence is conserved in human, mouse and porcine MTHFR genes.
With the increasing number of publications, sequences and other types of genetic information, the Internet is playing an increasingly larger role in the study of fundamental biomedical problems. Although GenBank was the first repository to contain sequence information on MTHFR, Table 1 provides a partial listing of the vast array of biological information available via the Internet about MTHFR. The reader is invited to consult these resources, as well as a few others that are cited throughout this chapter, to complement the text. However, objective analysis of the compiled data requires consultation of relevant publications.
Table 1
Description of MTHFR gene-related entries in current databases.
MTHFR/Mthfr Transcripts
Northern blot analysis has revealed MTHFR transcripts of approximately 2.8 and 7.2-7.7 kb in all tested tissues, and another of approximately 9.5 kb in brain, muscle, placenta, and stomach.8,12,13 For Mthfr, transcripts of 7.4, 6.3, 3.0 and 2.8 kb were observed.12 The different-sized transcripts result from alternate transcription start sites and multiple polyadenylation signals. The total abundance is low, and the proportion of each transcript differs among tissues. Overall expression is more intense in testis, intermediate in brain and kidney, and lower in other examined tissues.
Although difficult to achieve due to their lengths, isolation and analysis of the 5' and 3' ends of MTHFR/mthfr cDNAs12 has provided valuable information towards completion of the gene structure and analyses of regulatory sequences. Knowledge of the 3' UTR length (approximately 5 kb and 4 kb for the majority of MTHFR and Mthfr mRNA species, respectively) made it possible to predict the size of 5'UTR sequences. Since the coding sequence is about 2 kb and the main MTHFR transcripts on Northern blots do not exceed 9.5 kb (human) and 7.4 kb (mouse), this suggested that 5'UTR sequences were less than 2.5 kb and 1.4 kb for most of human and murine MTHFR mRNAs, respectively. Two clusters of transcription start sites of MTHFR/Mthfr mRNAs have been mapped by ribonuclease protection assays and are consistent with observations obtained by RT-PCR methodologies and Northern blotting.11 The identification of transcription start sites was critical for the prediction and the subsequent preliminary analysis of two promoters of the gene, that have to lie upstream of the transcribed sequences (Tran and Rozen, unpublished data).
Proximal transcription start sites in human MTHFR were identified 10 bp and 60 bp upstream of the ATG encoding the 77 kDa protein.12 These short 5'UTRs are expected to result in efficient translation of the 77-kDa enzyme. Some transcripts originating from an upstream region undergo splicing and do not contain the ATG for the long isoform (it is in a spliced-out segment); these transcripts are predicted to translate the 70 kDa protein, with a 5'UTR of approximately 50 nucleotides.12
Mapping
By fluorescence in situ hybridization (FISH), the human MTHFR gene was localized to 1p36.3.6 We also found close physical linkage between MTHFR and CLCN610 which was independently confirmed by Gaughan et al.13 Using RFLP analysis of 94 mice from an interspecific backcross panel, we localized the mouse Mthfr gene to the distal portion of mouse chromosome 4,14 which is the expected position for Mthfr based on known synteny between human and mouse genomes. This is also consistent with the observed linkage between murine Mthfr and Clcn6.12 For human MTHFR, we sequenced genomic clones that encompass 11 kb of sequences upstream of the MTHFR coding sequence.12 This DNA segment overlaps with GenBank entry AL02115, which shows that MTHFR is flanked by CLCN-6, NPPA, and NPPB at its 5' end. The location of these genes and the numerous STS markers in the genomic contig NT_004488.3 (which is the contig physically linked to MTHFR) concur with the result obtained by FISH.
Exploring the Introns
Although drafts of the sequence of human and mouse genomes are available and public databases contain a wealth of information about MTHFR-related sequences, the chromosome 1 segment encompassing the MTHFR gene is still a mosaic of sequences varying in data quality. Nonetheless, even in this incomplete state, the available information is useful. For example, several sequence contigs have made it possible to obtain the entire sequence of introns, although the relevant BAC clone has not been sequenced and/or assembled in its entirety. Figure 1 displays preliminary data from two BAC clones that contain a piece of human chromosome 1 DNA encompassing the MTHFR gene. The data reported as “clone 106H5 sequence” received an ambiguous annotation and an incorrect chromosomal location, which explains its “dead” status in NCBI databases. However, it can still be accessed using the NCBI ENTREZ retrieval tool with GI:15072573. The sequence of a segment of mouse chromosome 4 containing the entire Mthfr gene has been recently completed; the relevant BAC clone is also shown in Figure 1.
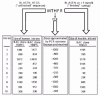
Detailed knowledge of MTHFR intronic sequences is critical for performing mutation screening across all exons and for the design of DNA diagnostic assays. It is also essential for the complete understanding of mutations that might involve activation of intronic cryptic splice sites. Although these types of mutations have not yet been reported for MTHFR, the availability of intronic data should facilitate their eventual identification and interpretation. Table 2 provides a list of primers used to amplify individual exons of the human gene, for mutation identification within exons and adjacent splice sites. The last base of the intronic primers is at least 20bp away from the exonic sequence, except for exon 9 primers which are 14 and 16 bp away from the exon. Two exons (exons 4 and 5) were amplified using two fragments.
Table 2
Primers used to amplify individual MTHFR exons.
MTHFR Pseudogene
We have recently shown that the mouse genome contains a pseudogene for Mthfr.15 The relevant locus (Mthfr-ps, 1259 bp) has been characterized. This nonfunctional pseudogene, which is similar to the Mthfr paralogous gene, arose by retrotransposition of a mis-spliced Mthfr transcript. The absence of intron 1, the partial retention of intron 2 and the location of this gene on chromosome 5 are features that are indicative of a partially-processed pseudogene. The lack of function was evident from a truncated coding sequence and from failure of reverse-transcription assays. These findings require consideration when designing PCR-based assays of the murine gene. Since some DNA is often present in a RNA sample, studies of the Mthfr genomic or mRNA sequences, including regulatory studies of Mthfr, need to distinguish between the functional transcript and the partially-processed nonfunctional pseudogene present in DNA.
Mutations in Severe MTHFR Deficiency
Cloning of the MTHFR cDNA/gene enabled the identification of disease-causing mutations in patients with severe MTHFR deficiency and homocystinuria. Although only 50 or so patients have been diagnosed worldwide with this type of deficiency, it is the most common inborn error of folate metabolism.16 Clinical symptoms displayed by MTHFR-deficient patients include developmental delay as well as various neurological and vascular problems, such as seizures, thromboses and vascular lesions. The chapter by MA Thomas and DS Rosenblatt provides a more comprehensive analysis of the clinical features in severe MTHFR deficiency.
To date, 34 mutations have been identified in MTHFR-deficient patients with homocystinuria (Table 3, fig. 2); detection mechanisms have included single strand conformation polymorphism (SSCP) analysis and direct sequencing of PCR or RT-PCR products.6,17-24 Primers for amplification of individual exons (Table 2) were designed on the basis of published genomic sequences.8 Identified mutations were predicted to be deleterious based on three criteria: 1. their absence from the general population, 2. the degree of conservation of the amino acid change, and 3. location of the mutated codon within a conserved region and/or predicted secondary structure. Whenever possible, mutations were expressed either in an in vitro bacterial expression system20,25 or in vivo in yeast,26 and assayed for enzyme activity (Table 4).
Table 3
Patient genotypes, age at onset of symptoms and enzyme activity in fibroblasts.

Table 4
In vitro assessment of MTHFR activity for different mutations.
Table 3 summarizes all the published mutations found in patients with severe MTHFR deficiency, along with their ethnic origin, age at onset of symptoms and residual activity of the enzyme. Twelve of the 34 mutations are present in the homozygous state in patients, while the remaining 22 are heterozygous. The 34 mutations can be classified into 8 nonsense mutations, 23 missense, 1 deletion and 2 splice variants. Exons 5 and 6 have the most mutations relative to their size. Since both exons are within the catalytic domain and may be involved in substrate binding,28 mutations in these exons may be more deleterious.
Nonsense Mutations
The eight nonsense mutations (28A→T, 559C→T, 1084C→T, 1134C→G, 1274G→A, 1420G→T, 1711C→T and 1762A→T) are scattered throughout the MTHFR polypeptide (fig. 2). Homozygous nonsense mutations can be very useful as they represent naturally occurring in vivo deletions of various portions of the C-terminus of the protein. Of the four homozygous mutant patients that have been reported, three mutations (559C→T in the catalytic domain, 1084C→T in the linker region, and 1762A→T in the regulatory domain) result in extreme enzyme deficiency (0-1%) in fibroblasts from these patients. It is not known whether the alleles harboring these mutations are translated or whether the mRNA is degraded by nonsense-mediated mRNA degradation (NMD);29 nonetheless, it seems that most of the polypeptide is required for enzyme activity. The fourth homozygous nonsense mutation (1711C→T in the regulatory region) was reported by Kluijtmans et al.22 The patient's specific enzyme activity was approximately 32%. This mutation is located in the C-terminus and does not appear to exert as dramatic an effect on enzyme activity, although the most distal nonsense mutation, at bp 1762, had been associated with very low activity.
The effects of the other four heterozygous nonsense mutations are unknown, but some impact may be predicted based on their location. The 5'-most mutation (28A→T; R6X) is at the beginning of the coding region and is unlikely to contribute to the patients' (II1 and II3, Table 3) residual enzyme activity. Mutation 1134C→G is just downstream of the linker region, leaving the catalytic domain intact but without potential inhibition from the SAM-binding site in the regulatory domain. Mutation 1274G→A is within the predicted SAM-binding site, while mutation 1420G→T is just downstream of it.
Deletion of the C-terminal domain of human MTHFR was carried out by our group, in a bacterial expression system,20 and by another group that expressed the deleted cDNA in yeast lacking endogenous MTHFR (met11-/-).26 When protein levels were normalized, the bacterial expression system yielded a 4-fold increase in enzyme activity relative to the wild type enzyme (Table 4). Yeast met11-/- cells expressing only the N-terminus of MTHFR grew more slowly than their parental cells, with an observed residual activity of 24%. Thus, in vitro expression experiments suggest that the presence of the C-terminus has an inhibitory effect on MTHFR activity; this finding is consistent with the localization of the SAM binding (inhibitory) domain in the C-terminal region. In vivo data in yeast suggest that the C-terminal domain is critical for cell growth. The C-terminus may stabilize the protein in vivo, since the truncation leads to reduced MTHFR protein levels.26
Missense Mutations
Most of the missense mutations are found in the catalytic N-terminal domain of MTHFR (16 out of 23, fig. 2). Mutations 1141C→T, 1172G→A, and 1274G→C all lie within the predicted SAM-binding site and may influence SAM binding. Mutations 1615C→T, 1727C→T, 1755G→A and 1768G→A are downstream of the linker, while mutation 1081C→T resides within it. With the exception of mutation 1727C→T, the other three mutations are found in patients diagnosed in their second decade of life, possibly indicating a mild effect on enzyme activity. Furthermore, in vitro expression of the 1081C→T mutation indicated it had no detectable effect on enzyme activity (Table 4); we therefore suggested that perhaps it had an effect on enzyme stability.
Seven N-terminal missense mutations exist in the homozygous state in patients: 458G→T, 692C→T, 764C→T, 983A→G, 985C→T, 1010T→C and 1027T→G. All of these mutations, except for 985C→T, are predicted to have a severe deleterious effect on enzyme activity since the patients were diagnosed within the first decade of life. The detrimental effect of mutations 458G→T, 692C→T, 983A→G, and 1027T→G was confirmed by various mutagenesis experiments (Table 4). The enzyme carrying a mutation at bp 983 has been shown to be stabilized by FAD in vitro, similarly to the enzyme carrying the polymorphism at bp 677. Whether the patient with this mutation would benefit from riboflavin supplementation remains to be determined. Mutation 985C→T is interesting in that the homozygous mutant patient has 20% enzyme activity, but when the mutation was expressed in vitro, it resulted in an approximate 5-fold increase in enzyme activity. Again, this may indicate that the mutation affects enzyme stability rather than activity in vivo.20
Site-directed mutagenesis was used to examine the effect of some of the heterozygous mutations on enzyme activity. Mutations 164G→C, 980T→C, 1015C→T, 1025T→C all had low enzyme activity (10%, Table 4). Mutations 167G→A and 1141C→T did not have considerable effects on activity. Interestingly, mutation 1025T→C, present in exon 5, caused retention of intron 5 in vivo. Although there is no clear explanation for this observation, it may allude to the presence of an exon splicing enhancer in that region.
Other Mutations
Two splice site mutations have been identified. The first, 249-1G→T, is located within the splice-acceptor dinucleotide (AG) in intron 1.18 The second, 792+1G→A, mutates a 5' splice donor site, resulting in activation of a cryptic splice site and deletion of 57 nucleotides.17
Only one deletion has been reported in severe MTHFR deficiency - patient 1569 (Table 3).20 This is a two-nucleotide deletion of AG at positions 1553 and 1554, with a predicted frameshift.
Severe Mutations and Mild Polymorphisms
In vitro and in vivo data suggest that the 677C→T polymorphism may modulate enzyme activity even in patients with severe MTHFR deficiency. This has been demonstrated in in vitro studies where the presence of the valine allele of the 677C→T polymorphism decreased enzyme activity by approximately 50%, both on its own and in cis with severe mutations (Table 4; refs. 20 and 25). A similar phenomenon has been observed in vivo where the 677 polymorphism appears to contribute to the thermolability of MTHFR in patients with severe MTHFR deficiency,18,20,21 although the initial reports4 had assumed that the thermolability was due to the deleterious mutation (since the polymorphism had not yet been identified). Although the presence of known deleterious mutations may be a good predictor of enzyme activity, the effect of mild polymorphisms can add to the complexity of genotype-phenotype analysis.
Polymorphisms in MTHFR
677C→T Variant
Historically, investigations of MTHFR genetic defects had focussed on the characterization of rare inborn errors leading to severe hyperhomocysteinemia and homocystinuria. With the identification of a MTHFR polymorphism (i.e., a common mutation) that results in mild hyperhomocysteinemia, it became clear that some diseases of adulthood, such as cardiovascular disease, reflect milder versions of the fulminant biochemical lesions present in the newborn or child with severe MTHFR deficiency.7
Biochemical and Molecular Studies
In an attempt to identify the molecular basis of severe and mild MTHFR deficiency, we had performed SSCP analyses on several types of patients. Ironically, we first identified the C-to-T substitution at bp677, that converts an alanine to a valine residue (A222V), in a patient with severe MTHFR deficiency and homocystinuria.7 This substitution was found to be equally common in homocystinuric patients and in a control population. Consequently, homocystinuric patients often have 3 or 4 mutations in the MTHFR gene (two distinct severe mutations and one or two copies of the A to V change, see ref. 18). To confirm that the substitution at bp 677 altered enzyme function, we performed site-directed mutagenesis of the cDNA and expressed the mutation in bacterial extracts; the mutagenized cDNA encoded an enzyme with reduced activity and increased thermolability.7 Furthermore, there was excellent correlation between reduced specific MTHFR activity in lymphocytes at 37°C, increased thermolability at 46°C and the presence of the A to V mutation in 96 patients with CAD.30
The initial report by Frosst et al7 also demonstrated the association of the homozygous mutant genotype (677TT) with mild hyperhomocysteinemia. However, it soon became evident that this association was present only in individuals with low folate status.31 In that study of 365 individuals from the NHLBI Family Heart Study, we found that the homozygous mutant genotype was associated with higher levels of plasma homocysteine.31 However, when the group was subdivided on the basis of plasma folate, there was a more dramatic increase in plasma homocysteine in individuals who were homozygous mutant and had plasma folate values below the median. There was no effect of the genotype on homocysteine levels in individuals with plasma folate values above the median. These findings suggested that folate supplementation should be effective in treating hyperhomocysteinemia in individuals with the mutation. A comprehensive discussion of this topic (mild MTHFR deficiency, hyperhomocysteinemia and interaction with folate status) is provided in Chapter 5.
Biochemical studies of wild type and mutant enzymes have provided a rationale for the protective effect of folate on hyperhomocysteinemia in mutant individuals. Lymphocyte extracts, heated in the presence and absence of 5-methylTHF, retain higher residual MTHFR activity after heating with increasing amounts of folate; the protective effect is greater on the mutant than on the wild type enzyme.28 This phenomenon has been reproduced with the recombinant wild type and mutant human enzymes expressed in heated bacterial extracts. The A222V polymorphism in human MTHFR has been mimicked in the E. coli enzyme by introducing the homologous mutation A177V. The biochemical properties of these bacterial enzymes, as well as more recent studies on purified human MTHFR,32 have addressed the mechanisms of this protective effect and are discussed more thoroughly in Chapter 3.
The aforementioned studies also demonstrated that the MTHFR cofactor, FAD, could protect the mutant enzyme from destabilization, suggesting that riboflavin, the precursor of FAD, should be considered as a modifier of enzyme activity and consequently of hyperhomocysteinemia (see Chapter 6). The mutation in bacterial MTHFR (A177V) increases the propensity for bacterial MTHFR to lose its essential flavin cofactor, and folate may protect the E. coli enzymes against flavin loss.
Clinical Impact
Since hyperhomocysteinemia had emerged as a risk factor for cardiovascular disease, the 677C→T variant became an excellent candidate for risk modification of this complex trait; the initial studies33,34 supported this concept while subsequent studies reached different conclusions. A couple of reports35,36 had also demonstrated elevated plasma total homocysteine in families with neural tube defects. Consequently, soon after its initial identification, the MTHFR variant was reported to be the first genetic risk factor for neural tube defects.37 The number of clinical conditions influenced by the 677 variant has grown considerably; the majority of the studies have used the initial HinfI digestion protocol7 for diagnosis of the variant. This book devotes individual chapters to the first two disorders (cardiovascular disease and neural tube defects) as well as to many of the more recent clinical conditions reported to be influenced by mild MTHFR deficiency. Although the mechanisms by which this variant influences disease progression may be unique to the specific condition, the various possibilities include the following:
- disruption in methionine or S-adenosylmethionine synthesis, with consequent effects on methylation. A disruption in methylation may also occur due to the conversion of homocysteine to S-adenosylhomocysteine, an inhibitor of several methyltransferases. Individuals with the TT genotype have decreased methylation in lymphocytes; this disturbance may be folate dependent.40Since altered DNA methylation is associated with changes in gene expression, mild MTHFR deficiency could influence developmental or oncogenic processes through this mechanism.
- altered distribution of folate metabolites—decreased MTHFR activity should result in a decrease in methyltetrahydrofolate and an increase in other folate forms, such as methylenetetrahydrofolate and other nonmethyl forms. This has been demonstrated in lymphocytes of TT individuals.41The redistribution of folates could affect thymidine or purine synthesis, with consequent effects on DNA synthesis or repair.
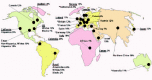
As discussed in subsequent chapters, the 677C→T polymorphism may only be a modest risk factor for some disorders. However, from a world population standpoint, it may represent a considerable burden since the variant is so common. The initial report identified homozygosity in 10%-15% of Canadian controls.7 Subsequent studies by many groups have revealed regional and ethnic variations of the frequency of the 677C→T homozygous mutant genotype. Figure 3 shows the prevalence of homozygosity by geographical and racial/ethnic group. The prevalence of the variant is relatively high in the general population, with homozygosity (TT) of 6-14% in several White populations. The 677T allele is less common in African Blacks and in Blacks living outside of Africa (Brazil, United States), with homozygosity frequencies of less than 2%. The 677T allele appears to be very common among Hispanics. Studies in Hispanic populations in California and Colombia have shown %TT of 21% and 18%, respectively; this is likely due to the equally high frequencies reported in their southern Mediterranean ancestral population.42-44 Although there is considerable variation in population frequencies, the mutation may have only occurred once, on a founder haplotype.45 This finding alludes to a selective advantage of the 677T allele for maintenance of this high frequency haplotype in many populations. The variant has been shown to be protective in some neoplasias (see Chapter 14), but this phenomenon would not explain a significant reproductive advantage. The aforementioned effects of mild MTHFR deficiency on methylation or on DNA synthesis and repair could have beneficial effects on early development, but experimental studies in this regard have not been reported.
Other Polymorphisms in MTHFR
Table 5 lists all reported variants in MTHFR. Only the 677 and 1298 substitutions have been expressed and confirmed to affect enzyme activity. The functional impact, if any, of the other polymorphisms remains unknown.
Table 5
Polymorphic mutations in 5,10-methylenetetrahydrofolate reductase.
A point mutation in exon 7 (1298A→C) results in a glutamate to alanine substitution (E429A). Site-directed mutagenesis of the MTHFR cDNA and expression in bacterial extracts have shown that the activity of the encoded enzyme is decreased (to 68% of the wild type enzyme), but not as dramatically as that for the 677T allele (for which residual activity is 45% of wild type).48 The enzyme mutated at bp 1298 is not thermolabile. Activities in lymphocyte studies of individuals with this variant closely parallel the results with the recombinant enzyme. Homozygotes represent approximately 8% of individuals in the tested populations, largely European (range is from 4% to 12% for most tested populations).50 These homozygotes do not appear to have higher serum homocysteine levels than controls. However, individuals who are compound heterozygotes for the 1298C and 677T alleles tend to have a biochemical profile closer to that seen among 677C→T homozygotes, with increased serum homocysteine levels.48
The 1298A→C mutation was initially examined by PCR and MboII digestion. However, a silent mutation (1317T→C) also creates a MboII site and results in a digestion pattern extremely similar to that of the 1298A→C variant. We reported a Fnu4HI diagnostic assay that makes it possible to detect the mutation at position 1298 without interference by the genotype at position 1317.48 We occasionally experienced difficulties with the Fnu4HI diagnostic assay. For this reason, we designed a new ACRS assay (Artificially Created Restriction Site) based on a MwoI digestion (unpublished), involving the sense primer: 5'-TGGGGGGCGGAGCTGGCCAGTGA-3'. The two underlined letters are intentional mismatches creating a MwoI site (GCNNNNNNNGC) that constitutes an internal digestion control, in addition to introducing a second MwoI site if the 1298C allele is amplified. Following amplification (35 cycles; 94°C 1 min, 64°C 1 min, 72°C 2 min) with the antisense primer (5'-AGGCCAGGGGCAGGGGATGAA-3'), the 137 bp amplicon is digested to generate fragments of 127 bp (1298A allele) or 119bp (1298C variant).
Most studies have reported no or few cases with 677T and 1298C alleles in the cis configuration. 43,48,51,52 It is likely that these mutations arose independently on different alleles and recombination has not occurred frequently enough, within the requisite small interval, to place the two mutations on the same chromosome. Furthermore, a recombinant enzyme containing both the 677T and 1298C substitutions has the same activity as the recombinant enzyme containing only the 677T allele,27 suggesting that 677T/1298C homozygotes do not have decreased survival (compared to 677TT homozygotes).
A third variant, R594Q, results from the change 1793G→A.49 It is less common than the 677C→T or 1298A→C polymorphism, with allele frequencies of 6.9% among Caucasians, 5.8% among Hispanics and 3.1% among African-Americans. Homozygosity was observed only in Caucasians (a single individual in 159 tested subjects).
The 1068T→C allele (reported as 1059T→C)53 is silent and appeared to be in linkage disequilibrium with the 1298A→C mutation. Trembath et al52 referred to it as 1059T→C instead of 1068T→C because they used numbering that had been reported in an old version (GI:499223) of U09806. In this entry, GenBank staff had removed the portion corresponding to the synthetic linker that was present in the original reference sequence (published as fig. 1 in ref. 6).
Recommendations for Nomenclature of MTHFR Mutations and Numbering of Bases
This chapter would not be complete without a discussion of ambiguities related to MTHFR reference sequences and nomenclature. The common C-to-T change at position 677 is often referred to as “C677T”. Based on the official nomenclature system for human gene mutations,54 this would correspond to the format for an amino acid codon change i.e., C677T should stand for “Cysteine at codon 677 substituted by Threonine”. The designation “677C→Trdquo; follows the nomenclature rules for a change in the nucleotide sequence. The reference sequence was first shown in Figure 1 of ref. 6 for the initial report of MTHFR mutations. Because it contained linkers used to generate the cDNA library, it was a “synthetic construct” based on GenBank classification (see GI:6174884 for GenBank accession no U09806). As mentioned in ref. 6, this sequence was predicted to be missing an upstream ATG and therefore the numbering started from the synthetic linker. The same numbering principle was used for describing the thermolabile variant,7 and this numbering method was conserved through virtually all subsequent publications. Ideally, the A of the ATG initiator methionine codon should be designated as nucleotide 1.54 Nucleotide 677 of the reference sequence corresponds to nucleotide 665 of the open reading frame for the 70 kDa isoform (GenBank accession nos XM_030156 and NM_005957, where a second upstream ATG had not yet been identified). Given the large number of scientific publications in which MTHFR bases were numbered according to the original report, it would become very confusing to establish a new reference sequence to conform to the guidelines suggested by Antonarakis et al54 even though the upstream ATG has now been identified.12 Consequently, we suggest maintaining GI:6174884-based numbering, as long as investigators clearly state the reference sequence in their reports. Because several versions of the same accession number (U09806) are now present in the GenBank database, it would be appropriate to mention GI:6174884, since older versions of U09806 have a different 5' end. GI: 6174884 was created specifically to match the numbering used in the initial report.6
The GenBank GI:6174884 is appropriate for designation of bases in the short MTHFR isoform sequence. There are, however, some specific cases that cannot use this reference sequence. If mutations are identified in the segment encompassing the coding sequences specific to the 77 kDa MTHFR isoform, and are absent in GI:6174884, it would then be appropriate to use negative numbering based on the initially- identified ATG initiator Met codon (for the 70 kDa isoform), with the nucleotide 5' to this ATG labelled “-1”. This would not create any ambiguities as long as the proper reference sequence is cited, and it would also be appropriate and unambiguous for designating bases in the 5'UTR sequences. In these situations, given the alternative splicing and the alternative usage of two close splice acceptor sites (A2 or A3)12 located between the 2 ATG initiator Met codons, it may become confusing to choose a cDNA reference sequence. Using a genomic DNA reference sequence would then be important (for example, such human genomic sequences were submitted to GenBank and are cited in ref. 12). The specific reference sequence (whether cDNA or genomic) needs to be quoted in all reports of MTHFR mutations or gene structure.
For amino acid numbering, the protein sequence of the short isoform (associated with GI:6174884) should be used, since this is already in common usage in the literature. However, for designation of amino acids that are specific to the 77 kDa isoform, it would be appropriate to specify which acceptor site was used to generate the mRNA in question (usage of A2 or A3 acceptor sites in ref. 12).
Animal Model of MTHFR Deficiency
To investigate the in vivo pathogenetic mechanisms of MTHFR deficiency, we generated Mthfr knockout mice.55 Plasma total homocysteine levels in heterozygous and homozygous knockout mice were 1.6- and 10-fold higher than those in wild type littermates, respectively. Both heterozygous and homozygous knockouts had either significantly decreased S-adenosylmethionine levels or significantly increased S-adenosylhomocysteine levels, or both, with global DNA hypomethylation. Heterozygosity for the knockout allele does not yield an abnormal phenotype. The homozygotes are smaller, with developmental retardation and cerebellar pathology. Abnormal lipid deposition in the proximal aorta was observed in older heterozygotes and homozygotes, alluding to an atherogenic effect of hyperhomocysteinemia in these mice.
Based on these initial observations, the homozygous knockout mice appear to be a good animal model for homocystinuria due to severe MTHFR deficiency, based on the complete enzymatic deficiency and the dramatic elevation in plasma total homocysteine. The heterozygous knockout mice, with approximately 60% residual enzyme activity and a moderate elevation in plasma total homocysteine, appear to be a good animal model for individuals that are homozygous for the 677T variant; these individuals have approximately 40% of the activity of 677C wild type homozygotes.30
Microarray analysis of brain RNA from knockout Mthfr mice revealed altered expression of several genes.56 RT-PCR and, in some cases, Western blots were used as a validation method to confirm a representative set of observations obtained from the analysis of the high-density oligonucleotide arrays. Interestingly, Mthfd2 expression was increased in Mthfr-/- mice. This gene encodes a bifunctional enzyme which can generate the MTHFR substrate 5,10-methyleneTHF; the altered expression is likely to be part of a cellular response triggered by deficient MTHFR activity. Among other differentially-expressed genes, the decreased expression of Itpr1 (inositol 1,4,5-triphosphate receptor, type 1) may reflect homocysteine-induced calcium influx, which is one of the proposed routes for compromised neuronal homeostasis.57
These mice have already proven useful in assessing the impact of betaine in MTHFR deficiency. 58 Although many studies had reported the homocysteine lowering effect of betaine in homocystinuria, betaine administration in moderate hyperhomocysteinemic states had not been extensively investigated. Betaine supplementation reduced plasma homocysteine in mice of all 3 Mthfr genotypes (+/+, +/-, and -/-), restored liver betaine and phosphocholine levels in the deficient mice, and prevented severe steatosis in the homozygous knockout animals. These observations highlighted the importance of betaine as an alternate methyl donor when folate-dependent remethylation is compromised and prompted us to examine this phenomenon in a human study, where we found a significant negative correlation between plasma homocysteine and plasma betaine in patients with cardiovascular disease.58
Conclusion
Since the isolation of the cDNA in 1994, the work on the mammalian MTHFR gene has resulted in significant advances in our understanding of its genomic organization, genetic variations and involvement in human disorders. Several important issues, however, remain to be addressed. Little is known about the regulation of this gene despite the fact that the enzyme links folate and homocysteine metabolism, and is involved in such critical cellular processes as DNA synthesis and DNA methylation. Investigations are required to understand the regulatory regions and their modulation, as well as the factors that affect alternative splicing and synthesis of the two protein isoforms. These are not easy tasks given the unusually complex MTHFR gene structure.
Although numerous clinical association studies have been performed on MTHFR variants, conclusions have been contradictory in some cases, due to the multifactorial nature of the disorders and our inability to identify the multiple genetic and environmental factors that can interact with MTHFR polymorphisms to impact disease risk. The biologic and tissue-specific impact of MTHFR deficiency has also not been adequately addressed since these types of investigations cannot be readily performed in human subjects; the availability of an animal model may be useful in this regard.
References
- 1.
- Rosenblatt DS. Inherited disorders of folate transport and metabolism. In: Scriver CR, Beaudet AL, Sly S, Valle D, eds. The Metabolic and Molecular Bases of Inherited Disease. Seventh ed. New-York: McGraw-Hill. 1995:3111–3128.
- 2.
- Mudd SH, Uhlendorf BW, Freeman JM. et al. Homocystinuria associated with decreased methylenetetrahydrofolate reductase activity. Biochem Biophys Res Commun. 1972;46:905–912. [PubMed: 5057914]
- 3.
- Kang SS, Zhou J, Wong PW. et al. Intermediate homocysteinemia: A thermolabile variant of methylenetetrahydrofolate reductase. Am J Hum Genet. 1988;43:414–421. [PMC free article: PMC1715503] [PubMed: 3177384]
- 4.
- Rosenblatt DS, Erbe RW. Methylenetetrahydrofolate reductase in cultured human cells. II. Genetic and biochemical studies of methylenetetrahydrofolate reductase deficiency. Pediat Res. 1977;11:1141–1143. [PubMed: 917613]
- 5.
- Kang SS, Wong PWK, Bock HGO. et al. Intermediate hyperhomocysteinemia resulting from compound heterozygosity of methylenetetrahydrofolate reductase mutations. Am J Hum Genet. 1991;48:546–551. [PMC free article: PMC1682989] [PubMed: 1998340]
- 6.
- Goyette P, Sumner JS, Milos R. et al. Human methylenetetrahydrofolate reductase: Isolation of cDNA, mapping and mutation identification. Nature Genet. 1994;7:195–200. [PubMed: 7920641]
- 7.
- Frosst P, Blom HJ, Milos R. et al. A candidate genetic risk factor for vascular disease: A common mutation in methylenetetrahydrofolate reductase. Nature Genet. 1995;10:111–113. [PubMed: 7647779]
- 8.
- Goyette P, Pai A, Milos R. et al. Gene structure of human and mouse methylenetetrahydrofolate reductase (MTHFR). Mamm Genome. 1998;9:652–656. [PubMed: 9680386]
- 9.
- Daubner SC, Matthews RG. Purification and properties of methylenetetrahydrofolate reductase from pig liver. J Biol Chem. 1982;10:140–145. [PubMed: 6975779]
- 10.
- Chan M, Tran P, Goyette P. et al. Analysis of the 5' region of the methylenetetrahydrofolate reductase (MTHFR) gene reveals multiple exons with alternative splicing, and an overlapping gene. FASEB J. 1999;13:A1375.
- 11.
- Homberger A, Linnebank M, Winter C. et al. Genomic structure and transcript variants of the human methylenetetrahydrofolate reductase gene. Eur J Human Genet. 2000;8:725–729. [PubMed: 10980581]
- 12.
- Tran P, Leclerc D, Chan M. et al. Multiple transcription start sites and alternative splicing in the methylenetetrahydrofolate reductase gene result in two enzyme isoforms. Mamm Genome. 2002;13:483–492. [PubMed: 12370778]
- 13.
- Gaughan DJ, Barbaux S, Kluijtmans LAJ. et al. The human and mouse methylenetetrahydrofolate reductase (MTHFR) genes: Genomic organization, mRNA structure and linkage to the CLCN6 gene. Gene. 2000;257:279–289. [PubMed: 11080594]
- 14.
- Frosst P, Zhang Z-X, Pai A. et al. The methylenetetrahydrofolate reductase (Mthfr) gene maps to distal mouse Chromosome 4. Mamm Genome. 1996;7:864–869. [PubMed: 8875901]
- 15.
- Leclerc D, Darwich-Codore H, Rozen R. Characterization of a pseudogene for murine methylenetetrahydrofolate reductase. Mol Cell Biochem. 2003;252:391–395. [PubMed: 14577615]
- 16.
- Rosenblatt DS, Fenton WA. Inherited disorders of folate and cobalamin transport and metabolism. In: Scriver CR, Beaudet AL, Sly WS et al, eds. The Metabolic & Molecular Bases of Inherited Disease.8th ed. New York: McGraw-Hill. 2001:3897–3933.
- 17.
- Goyette P, Frosst P, Rosenblatt DS. et al. Seven novel mutations in the methylenetetrahydrofolate reductase gene and genotype/phenotype correlations in severe MTHFR deficiency. Am J Hum Genet. 1995;56:1052–1059. [PMC free article: PMC1801446] [PubMed: 7726158]
- 18.
- Goyette P, Christensen B, Rosenblatt DS. et al. Severe and mild mutations in cis for the methylenetetrahydrofolate reductase (MTHFR) gene, and description of 5 novel mutations in MTHFR. Am J Hum Genet. 1996;59:1268–1275. [PMC free article: PMC1914869] [PubMed: 8940272]
- 19.
- Selzer RR, Rosenblatt DS, Laxova R. et al. Adverse effect of nitrous oxide in a child with 5,10-methylenetetrahydrofolate reductase deficiency. N Engl J Med. 2003;349:45–50. [PubMed: 12840091]
- 20.
- Sibani S, Leclerc D, Weisberg IS. et al. Characterization of mutations in severe methylenetetrahydrofolate reductase deficiency reveals an FAD-responsive mutation. Hum Mutation. 2003;21:509–520. [PubMed: 12673793]
- 21.
- Sibani S, Christensen B, O'Ferrall E. et al. Characterization of six novel mutations in the methylenetetrahydrofolate reductase (MTHFR) gene in patients with homocystinuria. Hum Mutation. 2000;15:280–287. [PubMed: 10679944]
- 22.
- Kluijtmans LA, Wendel U, Stevens EM. et al. Identification of four novel mutations in severe methylenetetrahydrofolate reductase deficiency. Eur J Hum Genet. 1998;6:257–65. [PubMed: 9781030]
- 23.
- Homberger A, Linnebank M, Sewell A. et al. Severe methylenetetrahydrofolate reductase deficiency: Two novel genotypes with different clinical course. J Inherit Metab Dis. 2001;24(Suppl 1):50.
- 24.
- Tonetti C, Amiel J, Munnich A. et al. Impact of new mutations in the methylenetetrahydrofolate reductase gene assessed on biochemical phenotypes: A familial study. J Inherit Metab Dis. 2001;24:833–42. [PubMed: 11916316]
- 25.
- Goyette P, Rozen R. The thermolabile variant 677C→T can further reduce activity when expressed in CIS with severe mutations for human methylenetetrahydrofolate reductase. Hum Mutation. 2000;16:132–38. [PubMed: 10923034]
- 26.
- Shan X, Wang L, Hoffmaster R. et al. Functional characterization of human methylenetetrahydrofolate reductase in Saccharomyces cerevisiae. J Biol Chem. 1999;274:32613–32618. [PubMed: 10551815]
- 27.
- Weisberg IS, Jacques PF, Selhub J. et al. The 1298A>C polymorphism in methylenetetrahydrofolate reductase (MTHFR): In vitro expression and association with homocysteine. Atherosclerosis. 2001;156:409–15. [PubMed: 11395038]
- 28.
- Guenther BD, Sheppard CA, Tran P. et al. The structure and properties of methylenetetrahydrofolate reductase from Escherichia coli suggest how folate ameliorates human hyperhomocysteinemia. Nat Struct Biol. 1999;6:359–365. [PubMed: 10201405]
- 29.
- Frischmeyer PA, Dietz HC. Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet. 1999;8:1893–1900. [PubMed: 10469842]
- 30.
- Christensen B, Frosst P, Lussier-Cacan S. et al. Correlation of a common mutation in the methylenetetrahydrofolate reductase gene with plasma homocysteine in patients with premature coronary artery disease. Arterioclerosis Thrombosis and Vascular Biology. 1997;17:569–573. [PubMed: 9102178]
- 31.
- Jacques PF, Bostom AG, Wiliams RR. et al. Relation between folate status, a common mutation in methylenetetrahydrofolate reductase, and plasma homocysteine concentrations. Circulation. 1996;93:7–9. [PubMed: 8616944]
- 32.
- Yamada K, Chen Z, Rozen R. et al. Effects of common polymorphisms on the properties of recombinant human methylenetetrahydrofolate reductase. Proc Natl Acad Sci USA. 2001;98:14853–14858. [PMC free article: PMC64948] [PubMed: 11742092]
- 33.
- Kluijtmans LA, van den Heuve lLP, Boers GH. et al. Molecular genetic analysis in mild hyperhomocysteinemia: A common mutation in the methylenetetrahydrofolate reductase gene is a genetic risk factor for cardiovascular disease. Am J Hum Genet. 1996;58:35–41. [PMC free article: PMC1914961] [PubMed: 8554066]
- 34.
- Gallagher PM, Meleady R, Shields DC. et al. Homocysteine and risk of premature coronary heart disease. Evidence for a common gene mutation. Circulation. 1996;94:2154–2158. [PubMed: 8901666]
- 35.
- Mills JL, McPartlin JM, Kirke PN. et al. Homocysteine metabolism in pregnancies complicated by neural-tube defects. Lancet. 1995;345:149–151. [PubMed: 7741859]
- 36.
- Steegers-Theunissen RP, Boers GH, Blom HJ. et al. Neural tube defects and elevated homocysteine levels in amniotic fluid. Am J Obstet Gynecol. 1995;172:1436–1441. [PubMed: 7755050]
- 37.
- van der Put NMJ, Steegers-Theunissen RPM, Frosst P. et al. Mutated methylenetetrahydrofolate reductase as a risk factor for spina bifida. Lancet. 1995;364:1070–1072. [PubMed: 7564788]
- 38.
- Bellamy MF, McDowell IF. Putative mechanisms for vascular damage by homocysteine. J Inherit Metab Dis. 1997;20:307–15. [PubMed: 9211203]
- 39.
- Rosenquist TH, Ratashak SA, Selhub J. Homocysteine induces congenital defects of the heart and neural tube: Effect of folic acid. Proc Natl Acad Sci USA. 1996;93:15227–1532. [PMC free article: PMC26385] [PubMed: 8986792]
- 40.
- Friso S, Choi S-W, Girelli D. et al. A common mutation in the 5,10-methylenetetrahydrofolate reductase gene affects genomic DNA methylation through an interaction with folate status. Proc Natl Acad Sci. 2002;99:5606–5611. [PMC free article: PMC122817] [PubMed: 11929966]
- 41.
- Bagley PJ, Selhub J. A common mutation in the methylenetetrahydrofolate reductase gene is associated with an accumulation of formylated tetrahydrofolates in red blood cells. Proc Natl Acad Sci. 1998;95:13217–13220. [PMC free article: PMC23763] [PubMed: 9789068]
- 42.
- Botto LD, Yang Q. 5-methylenetetrah ydrofolate reductase gene variants and congenital anomalies: A HuGE review. Am J Epidemiol. 2000;151:862–877. [PubMed: 10791559]
- 43.
- Ogino S, Wilson RB. Genotype and haplotype distributions of MTHFR677C>T and 1298A>C single nucleotide polymorphisms: A meta-analysis. J Hum Genet. 2003;48:1–7. [PubMed: 12560871]
- 44.
- Guillen M, Corella D, Portoles O. et al. Prevalence of the methylenetetrahydrofolate reductase 677>C mutation in the Mediterranean Spanish population. Association with cardiovascular risk factors. Eur J Epidemiol. 2001;17:255–261. [PubMed: 11680544]
- 45.
- Rosenberg N, Murata M, Ikeda Y. et al. The frequent 5,10-methylenetetrahydrofolate reductase C677T polymorphism is associated with a common haplotype in Whites, Japanese, and Africans. Am J Hum Genet. 2002;70:758–762. [PMC free article: PMC384952] [PubMed: 11781870]
- 46.
- Linnebank M, Homberger A, Nowak-Goettl U. et al. Linkage disequilibrium of the common mutation 677C→T and 1298A→C of the human methylenetetrahydrofolate reductase gene as proven by the novel polymorphisms 129C→T, 1068C→T. Eur J Ped. 2000;159:472–473. [PubMed: 10867857]
- 47.
- Viel A, Dall'Agnese L, Simone F. et al. Loss of heterozygosity at the 5,10-methylenetetrahydrofolate reductase locus in human ovarian carcinomas. Br J Cancer. 1997;75:1105–1110. [PMC free article: PMC2222800] [PubMed: 9099956]
- 48.
- Weisberg I, Tran P, Christensen B. et al. A second genetic polymorphism in methylenetrahydrofolate reductase (MTHFR) associated with decreased enzyme activity. Mol Genet Metabol. 1998;64:169–172. [PubMed: 9719624]
- 49.
- Rady PL, Szucs S, Grady J. et al. Genetic polymorphisms of methylenetetrahydrofolate reductase (MTHFR) and methionine synthase reductase (MTRR) in ethnic populations in Texas: A report of a novel MTHFR polymorphic site, G1793A. Am J Med Genet. 2002;107:162–168. [PubMed: 11807892]
- 50.
- Robien K, Ulrich CM. 5 10-Methylenetetrahydrofolatereductase polymorphisms and leukemia risk: A HuGE minireview. Am J Epidemiol. 2003;157:571–582. [PubMed: 12672676]
- 51.
- van der Put NMJ, Eskes TKAB, Blom HJ. Is the common 677C→T mutation in the methylenetetrahydrofolate reductase gene a risk factor for neural tube defects ? A meta-analysis. Q J Med. 1997;90:111–115. [PubMed: 9068801]
- 52.
- Zetterberg H, Rymo L, Coppola A. et al. Reply to “MTHFR C677T and A1298C polymorphisms and mutated sequences occuring in cis” Eur J Human Genet. 2002;10:579–582.
- 53.
- Trembath D, Sherbondy AL, Vandyke DC. et al. Analysis of select folate pathway genes, PAX3 and human T in a midwest neural tube defect population. Teratorogy. 1999;59:331–341. [PubMed: 10332959]
- 54.
- Antonarakis S, Ashburner M, Auerbach AD. et al. For the nomenclature working group. Recommendations for a nomenclature system for human gene mutations. Hum Mutation. 1998;11:1–3. [PubMed: 9450896]
- 55.
- Chen Z, Karaplis AC, Ackerman SL. et al. Mice deficient in methylenetetrahydrofolate reductase exhibit hyperhomocysteinemia and decreased methylation capacity, with neuropathology and aortic lipid deposition. Human Mol Genet. 2001;10:433–443. [PubMed: 11181567]
- 56.
- Chen Z, Ge B, Hudson TJ. et al. Microarray analysis of brain RNA in mice with methylenetetrahydrofolate reductase deficiency and hyperhomocysteinemia. Gene Expression Patterns. 2002;1:89–93. [PubMed: 15018804]
- 57.
- Ho PI, Ortiz D, Rogers E. et al. Multiple aspects of homocysteine neurotoxicity: Glutamate excitotoxicity, kinase hyperactivation and DNA damage. J Neuroscience Res. 2002;70:694–702. [PubMed: 12424737]
- 58.
- Schwahn BC, Chen Z, Laryea MD. et al. Homocysteine-betaine interactions in a murine model of 5,10-methylenetetrahydrofolate reductase deficiency. FASEB J. 2003;17:512–514. [PubMed: 12551843]
- Introduction
- DNA and Genomic Structure of MTHFR
- MTHFR/Mthfr Transcripts
- MTHFR Pseudogene
- Mutations in Severe MTHFR Deficiency
- Polymorphisms in MTHFR
- Biochemical and Molecular Studies
- Clinical Impact
- Recommendations for Nomenclature of MTHFR Mutations and Numbering of Bases
- Animal Model of MTHFR Deficiency
- Conclusion
- References
- Molecular Biology of Methylenetetrahydrofolate Reductase (MTHFR) and Overview of...Molecular Biology of Methylenetetrahydrofolate Reductase (MTHFR) and Overview of Mutations/Polymorphisms - Madame Curie Bioscience Database
- Structural Components and Architectures of RNA Exosomes - Madame Curie Bioscienc...Structural Components and Architectures of RNA Exosomes - Madame Curie Bioscience Database
- Comparison of Muscle Development in Drosophila and Vertebrates - Madame Curie Bi...Comparison of Muscle Development in Drosophila and Vertebrates - Madame Curie Bioscience Database
- Cancer Invasion and Metastasis: Molecular and Cellular Perspective - Madame Curi...Cancer Invasion and Metastasis: Molecular and Cellular Perspective - Madame Curie Bioscience Database
- Lipoxins as an Immune-Escape Mechanism - Madame Curie Bioscience DatabaseLipoxins as an Immune-Escape Mechanism - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...