

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

During the entire process of neural crest development from specification till final differentiation, delamination and migration are critical steps where nascent crest cells face multiple challenges: within a relatively short period of time that does not exceed several hours, they have to change drastically their cell- and substrate-adhesion properties, lose cell polarity and activate the locomotory machinery, while keeping proliferating, surviving and maintaining a pool of precursors in the neural epithelium. Then, as soon as they are released from the neural tube, neural crest cells have to adapt to a new, rapidly-changing environment and become able to interpret multiple cues which guide them to appropriate target sites and prevent them to distribute in aberrant locations. It appears from recent studies that, behind an apparent linearity and unicity, neural crest development is subdivided into several independent steps, each being governed by a multiplicity of rules and referees. Here resides probably one of the main reasons of the success of neural crest cells to accomplish their task.
Introduction
The early development of the neural crest in vertebrate embryos can be likened to the history of a number of European peoples during the last centuries. Briefly, it starts with a long and obscure time period when minorities are dominated by their potent neighbors, their original territories occupied and often divided into separate entities, and their traditions and identities severely repressed. As time goes by, the minorities express signs of identity, first timidly and cryptically, then progressively more markedly. This period terminates in a sudden and paroxystic step, typical of revolutions, with the revolts of minorities, their rejection of all external influences, and their declarations of independence. This is inevitably followed by an unfortunate cohort of conflicts, wars and population displacements and emigrations. However, as populations are completely separated, they can express freely their individual characteristics and conflicts become less acute. Quite often, this situation is favorable for establishment of new, more stable and balanced contacts between the previously-fighting populations, and it is accompanied by the mutual recognition of the identities as well as the revival of a common past. This state is supposed to persist unless a new, emerging empire strikes again, but obviously this has not occured yet in Europe and is pure science fiction.
Thus, the initial step of neural crest development would correspond to the domination period. The original territory of crest cells lies at the boundary between the ectoderm and the neural tube and is not well delimited. Prospective crest cells are most likely recruited from cells located on either side of the boundary but cannot be identified with certainty during early neurulation. Nascent neural crest cells become progressively specified and express a number of specific markers. However, they remain integrated at the dorsal aspect of the neural tube and are morphologically indistinguishable from the other neural epithelial cells. Then, the delamination step occurs. Undubiously, this event is the best sign of crest cells individualization from the rest of the neural tube and constitutes their declaration of independence. It is associated with profound changes in crest morphological and molecular features, with loss of previous properties and acquisition of new characters. At this step, neural crest cells are clearly distinct from neural tube cells and are easily recognizable both molecularly and morphologically. In most species, once cells are segregated from the neural tube, they immediately venture away from the tube in multiple directions. During migration, neural crest cells may face hostile environments that may repulse them or cause their death, but they may also occupy more accessible areas where they survive, grow and sometimes settle to undergo differentiation. Interestingly, as observed among European countries, neural crest cells that once have made secession with the neural tube often reestablish intimate contacts with it, such as at the sensory and motor nerve entry and exit points, and express a number of common molecular markers with the central nervous system. Thus, the analogy between neural crest cells and the origins, migrations and fates of European peoples appears to be very large, particularly during delamination and migration, the two critical steps when neural crest traits become manifest: how to become different while sharing a common history and how to survive, move and develop in a sometimes hostile environment.
Neural crest ontogeny has been extensively covered in the past by numerous reviews.1-6 Here, I will focus on the recent advances and trends in some specific aspects that have considerably modified our view of the delamination and migration stages and discuss the current questions that are being examined and those that remain unexplained.
Neural Crest Delamination: Their Declaration of Independence
Delamination (also refered as to emigration, individualization or segregation from the neural tube) encompasses the series of events that allow the physical separation of neural crest cells from the rest of the neural tube. Although the term delamination is now widely employed, it may not be entirely appropriate and may be misleading to some extent, because neural crest cells do not appear by a process involving splitting of apposed laminae. Delamination can be viewed as both the final step of the whole process of neural crest cell specification, allowing cells to become irreversibly segregated from the neural tube, and the transition toward migration. It must be, however, clearly distinguished from the specification and migration steps since, as discussed below, these events appear now to be driven by independent and discernable cellular and molecular mechanisms. Yet, analyzing delamination has long been a difficult and elusive task: indeed, this step lags during a rather limited time period (so far it has not been possible either to predict or even to catch the moment when an individual neural crest cell dissociates from the neural fold); until recently, there was virtually no good molecular markers for this step in contrast to specification and migration (the best sign for delamination is the presence of cells at the periphery of the neural epithelium, in between the basal surfaces of the neural tube and the ectoderm); and there are no clear separations or transitions between the specification, delamination and migration steps which instead overlap at the level of the population, as the whole process evolves continuously in most species (thus, when the pioneer crest cells are undergoing migration, others are still being specified or delaminating). However, because it constitutes the first tangible sign of neural crest formation as an individual cell population, delamination is a key step during neural crest ontogeny that has long attracted great interest. In addition, it provides a paradigm to analyze cell dissemination, a process often encountered under normal and pathological situations both during embryogenesis and adulthood, e.g., during tumor metastasis and tissue repair.
Because of the lack of adapted experimental designs, neural crest delamination has been at first exclusively the matter of descriptive studies. However, in the early 1990s, following the pioneer studies of Newgreen,7 a breakthrough has been made possible essentially using an ex vivo culture approach, based on the comparison of the molecular and cellular properties of cells migrating out of neural tube explants at different stages of neurulation.3,8 But it is only quite recently that the process of delamination has been amenable to in situ experimentation, in particular owing to the rapid development of transgenic approaches in mouse, zebrafish, frog, and chick. Finally, because it is highly suitable for both in vitro/ex vivo cultures and in ovo electroporation allowing real time analyses, the chick embryo has become the most popular model for studying neural crest delamination, but interesting information have been obtained with the Xenopus and mouse embryos. Curiously, although the zebrafish embryo allows a direct visualization of neural crest delamination and proved to be a valuable model for studying determination and migration, so far it has not been the matter of intense studies to address specifically the question of delamination.
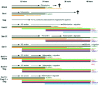
Morphological and Cellular Events Define Neural Crest Cell Delamination Primarily as an Epithelium-to-Mesenchyme Transition
Morphological and immunohistological studies in various species ranging from fish to mammals have permitted a detailed description of the main cellular events that accompany neural crest cell delamination.3 Although the topographies of the embryos in Vertebrates are notably different and influence timing of neural crest emigration relative to neural tube folding and closure (for example, cranial neural crest emigration in the mouse occurs when the neural tube is wide open while in the chick it appears coincident with neural fold fusion), a number of basic events can be recognized in the processes leading to the separation of the neural crest population from the neural tube (Fig. 1). These events are in many aspects similar to those occuring in any epithelium-to-mesenchyme transition (EMT). However, although neural crest cell delamination is often regarded as a model of EMT, it presents a number of specific features that make it somewhat different and atypical compared with other EMT such as somite disruption, tumor dissemination, wound healing, or dispersion of epithelial cell lines in vitro. In particular, in the trunk region, at the time nascent crest cells depart from the neural tube, the neural epithelium does not manifest signs of complete disorganization and disruption of tissular cohesion. Rather, it remains morphologically intact and crest cells are only gradually expelled from the epithelium in the extracellular matrix underneath, suggesting that the process of delamination is tightly controled both spatially and temporally and that there are mechanisms ensuring continuously the replacement of cells that have emigrated.

As they segregate from the neural tube, neural crest cells change shape progressively: from regular, elongated, and radially-oriented, they become at first more rounded and irregular in shape with increasing numbers of filopodia protruding out of the neural tube.9-11 Once cells are entirely separated from the neural tube, they flatten over the surface of the neural tube and extend tangentially to it. These morphological changes correlate at least in vitro with reorganizations of the actin cytoskeleton, from a dense fibrillar network at the cell periphery in association with junctions to a more diffuse and labile organization.12
Important alterations in cellular cohesion have also been reported. Interestingly, these appear considerably more complex than originally appreciated. Indeed, at onset of neurulation in the chick, all cells in the neuroectoderm exhibit junctional complexes typical of polarized epithelial cells with tight junctions and adherens junctions containing E-cadherin. However, not all epithelial features exist in neuroepithelial cells: there are no desmosomes and the intermediate filaments are not composed of cytokeratins.13 As the neural folds elevate to form the neural tube, i.e., long before any sign of crest cell delamination, tight junctions are lost gradually from the prospective neural epithelium along a ventro-dorsal gradient, but remain intact in the superficial ectoderm.14 In addition, although adherens junctions are retained, E-cadherin is replaced by N-cadherin and cadherin-6B until onset of migration.15-17 Whether N- and E-cadherins can coexist transiently in individual cells is not known precisely. Slightly before emigration, coincident with change in cell shape, adherens junctions become disrupted though expression of N-cadherin on the cells' surface is not repressed.17 N-cadherin and cadherin-6B are lost from neural crest cells, but only after their complete exclusion from the neural epithelium.17-19 Finally, neural crest cells undergoing migration start expressing cadherin-7 and/or cadherin-11,19,20 two types of cadherin expressed by fibroblastic cells. In the mouse, this cadherin sequence is different as neural crest cells start expressing cadherin-6, a close relative of chick cadherin-6B, prior to delamination but retain it on their surface during early migration, instead of shifting to cadherin-7.21 The overall significance of this complex series of changes in the repertoire of cadherins in the neural epithelium during neural crest cell emigration is unknown at present, but it is likely that it is a prerequisite for the step-by-step occurrence of defined cellular events leading to the correct segregation of cells from the neural tube. Indeed, qualitative and quantitatives differences in cadherin expression in neighboring cells have been found to intruct cell segregation and influence cell fate,22 and the fact that cells have never been seen delaminating from the superficial ectoderm may be related with the absence of cadherin shift in this tissue. In addition, perturbation experiments aimed at altering this sequence all result in severe deficiencies in neural crest delamination. Thus, mouse embryos mutated for SIP-1, a repressor of E-cadherin expression, show persistent E-cadherin labeling thoughout epidermis and neural tube, and this is associated with a complete lack of neural crest delamination in cranial regions.23 Likewise, forced expression of N-cadherin in prospective neural crest cells causes a deficit in their emigration and their accumulation into the lumen of the neural tube.19 Finally, precocious overexpression of cadherin-7 in neural crest cells also prevents migration instead of producing anticipated emigration.19
Beside changes in cell-cell adhesion, prospective neural crest cells undergo a number of modifications in their interactions with the extracellular matrix that are believed to favor their release from the neural tube. First, there is no basement membrane covering the dorsal aspect of neural tube where cells delaminate, but this absence does not correlate strictly with the onset of delamination, indicating that although it is necessary, it is not a key triggering event.24 Nevertheless, in vitro studies clearly indicate that neural crest cells respond differently to extracellular matrix material prior to and after delamination,8 but it is not clear yet whether these changes result chiefly from modifications in the repertoire of integrins as observed in the chick embryo25,26 or from activation of distinct downstream signaling pathways. In addition, in vivo and in vitro perturbation experiments suggest that neural crest cell delamination is fostered by matrix metalloprotease-2 (MMP-2), a type IV collagenase, even though it is only produced in the late phase of delamination, once cells are released in the extracellular environment.27 Thus, neural crest interactions with the extracellular matrix are clearly altered during delamination, but the means and kinetics of these modifications remain ill-defined.
An important issue concerns the establishement of the spatial, temporal, and functional hierarchies in the various events affecting cell shape, cell-cell adhesion and cell-matrix interactions. In other words, what is the exact sequence of cellular events that accompany crest delamination; which of them play a critical role; how are they coordinated; are some of them dispensable and which is the last event necessary to trigger complete delamination ? At the present time, there are no clear answers to these questions. However, several clues suggest that delamination is not a linear cascade of events in which each step relies directly on the occurrence of the previous ones. For example, the fact that N-cadherin down-regulation is a late event taking place just prior to crest cell dissociation from the neural tube would suggest that this is a prerequisite for triggering delamination, and the N-cadherin overexpression experiments mentionned above tend to support this view.19 Yet, recent in vivo studies reveal that complete delamination of neural epithelial cells followed by active migration can occur without down-regulation of N-cadherin from the cells' surface.28,29 Likewise, using an in vitro approach, Newgreen and colleagues showed that affecting N-cadherin-mediated cell adhesion, blocking atypical PKC or challenging integrin-dependent matrix adhesion result in the same outcome, clearly demonstrating that delamination can be achieved by various routes, thereby excluding any obvious hierarchies among the different cellular events occuring during EMT.12,30
Candidates for orchestrating cellular events during neural crest EMT involve Rho GTPases -Cdc42, Rac and Rho- known to control cell adhesion and motility through dynamic regulation of the actin cytoskeleton. So far, two members of the family, Rho-A and Rho-B, have been identified in the neural epithelium at the time of neural crest cell migration.31 Nonsurprisingly, Rho-A, the most common and best characterized member of the family implicated in actin bundling and focal contact formation, is ubiquitous in the neural tube. In contrast, Rho-B, a more divergent member whose function and cellular targets remain largely unknown, exhibits a very dynamic expression pattern in prospective crest cells prior to and during early migration, thereby suggesting that it plays a specific role during delamination. Indeed, blockade of Rho activity using the C3 exotoxin inhibits neural crest delamination in vitro,31 but it should be stressed that as the C3 exotoxin shows no selectivity for any particular Rho, this experiment did not allow to ascribe a delamination-promoting activity exclusively to Rho-B. Nevertheless, forced expression of a dominant-active form of Rho-B in the neural tube has been found to cause massive cell delamination, resulting in a severe distortion of the neural tube morphology. 29 The cellular events regulated by Rho-B in neural crest cells remain to be identified. Studies in cultured cell lines have shown that it is poorly involved in cytoskeletal organization, and its close association with endocytic vesicles argues instead for a role in intracellular transport of cell-surface receptors.32 Interestingly, cells delaminating from neural tubes transfected with an activated form of Rho-B show a marked exclusion of N-cadherin staining from adherens junctions.29 It is therefore plausible that Rho-B might promote delamination of neural crest cells by affecting N-cadherin trafficking in the cells. Alternatively, it cannot be excluded that it may also function in the dynamics of crest cell locomotion as primary mouse embryo fibroblasts derived from Rho-B-/- strains display a marked defect in cell motility.33 Although Rho-B presents a number of additional features pertinent to a prominent role in the control of neural crest delamination (unlike most GTPases which are relatively stable, it is turned over quickly and its synthesis is tightly regulated by growth factors), it is likely that it operates in concert with other Rho GTPases, notably Rac and Cdc42. Indeed, in other examples of EMT, changes in cell cohesion has been found to correlate with subtle modifications in the balance between the different Rho GTPases. In particular, during condensation of the lateral plate mesoderm into the somite, Cdc42 activity levels appear critical for the binary decision that defines the epithelial and mesenchymal somitic compartments whereas proper levels of Rac-1 are necessary for somitic epithelialization.34
A Combinatorial Transcriptional Code for Controling EMT during Neural Crest Delamination
The search for early molecular determinants of neural crest cell specification led to the identification of a large variety of transcription factors that appeared to be expressed in prospective crest cells at least until early migration (Fig. 1). This suggested that specification of dorsal neural epithelial cells into neural crest progenitors is necessary and sufficient to initiate a linear signaling cascade characterized by a precise sequence of expression of transcription factors and ultimately leading to their delamination and migration, provided cells are confronted with the appropriate environment. However, in the absence of studies at the single cell level, there is no direct proof for a strict correlation between expression of early neural crest markers and the cellular capacity to undergo delamination. Rather, several observations made essentially in chick and Xenopus suggest that neural crest specification, delamination, and migration are causally independent events.
The Snail Family of Zn-Finger Transcription Factors
Slug and Snail were the first transcription factors to be identified in the neural crest, about a decade ago.35 They belong to the Snail family of Zn-finger transcription factors and are most commonly used as neural crest markers.36 In the chick, neural crest cells express Slug but not Snail,37 whereas in the mouse and zebrafish,37,38 it is the reverse situation, crest cells express Snail instead of Slug, and, lastly, in Xenopus, both factors coexist in crest cells but are induced separately.39 Slug and Snail have been shown to play similar roles and be interchangeable in some experimental systems,40 although they may also perform distinct functions in cells.39 There are numerous indications that Snail transcription factors are involved in EMT.36 Beside prospective neural crest cells, they are expressed in multiple embryonic regions known to undergo EMT such as in the primitive streak and mesoderm during gastrulation, during sclerotome dispersal, and during formation of the heart cushions. Snail mutant mice die at gastrulation, most likely due to defective ingression of the mesoderm in the primitive streak.41 In tumors, Snail triggers EMT through direct repression of E-cadherin, and its expression correlates with the invasive phenotype in cell lines as well as in vivo, in chemically-induced skin tumors.42,43 Likewise, overexpression of Slug in cultured epithelial cells causes desmosome dissociation followed by cell dispersion, upregulation of vimentin, and fibronectin redistribution.44 With regard to the neural crest, loss-of-function experiments in chick and Xenopus based on antisense oligonucleotides to Slug or Snail result in a strong deficit in migrating neural crest cells.35,39,45 For all these reasons, Slug has been presented as a major player necessary for neural crest delamination. However, other studies tend to contradict this view and suggest that Slug may be neither sufficient nor necessary for delamination at least at some axial levels. First, with the possible exception of the cranial levels in the mouse,42 Slug expression is often delayed with respect to the exclusion of E-cadherin from the neural tube particularly at truncal levels. It can be argued that Slug might also repress N-cadherin expression, but this has never been described so far. More significantly, in Xenopus, although Slug or Snail overexpression in whole embryos leads to the expansion of prospective neural crest territory and a greater number of melanocytes, their effect is limited to areas contiguous with endogenous neural-crest-forming regions.39,45 In addition, Slug is unable to induce by itself crest formation in ectodermal explants, suggesting that its sole expression is insufficient to direct a program of neural crest ontogeny.45 This is further supported by cell-tracing experiments which revealed that not all Slug/Snail-expressing cells are fated to become migrating neural crest cells.46 In chick, overexpression of Slug in the neural tube using in ovo electroporation increased the number of neural crest cells migrating out of the dorsal side of the neural tube associated with an increase in Rho-B expression, but this occured only at cranial levels.40 In addition, as observed in Xenopus, only cells situated in the most dorsal side of the embryo, i.e., in the neural crest prospective region, were able to emigrate, whereas cells situated immediately more ventrally exhibited no signs of delamination and expressed no neural crest markers.29,40 Slug overexpression in the trunk region caused only a slight expansion of the prospective crest cell region, but this was not accompanied by greater numbers of migrating cells. Finally, neural crest delamination can be severely affected in trunk of chick embryos, without detectable repression of Slug expression.47 Thus, paradoxically, despite convincing data on Slug/Snail function in numerous examples of EMT, their precise role in neural crest delamination remains elusive: they may promote delamination in a specific cellular context in the dorsal part of the neural tube but may be insufficient to drive by themselves EMT of neural tube cells situated more ventrally.
The Winged Helix-Forkhead Transcription Factor Foxd-3
Foxd-3 is a transcription factor of the winged helix-forkhead family whose temporal expression also closely matches neural crest induction, delamination, and migration. In Xenopus, chick, and mouse, its expression starts early during neural crest induction approximately coincident with or slightly before that of Slug, but in contrast to the latter, it remains expressed in most neural crest cells throughout migration, except for melanocyte precursors.48-51 As for Slug/Snail, targeted inactivation of Foxd-3 in the mouse is embryonic lethal at very early stages of development, before implantation52 and, thus, informations about its possible contribution to neural crest specification and delamination come essentially from gain- and loss-of-function analyses in frog and chick. In the chick, forced expression of Foxd-3 in the trunk neural tube was found to suppress interneuron differentiation and induce precocious and robust expression of HNK-1, a marker for migrating neural crest cells, in the whole neural tube by 24 hours post-transfection. Foxd-3 is also able to provoke ectopic delamination of cells but not until 24 hours post-transfection. Delamination is evident only after 36-48 hours and is accompanied by down-regulation of N-cadherin, up-regulation of integrins and cadherin-7, and disruption of the basement membrane lining the neural tube.28,29,49 In Xenopus embryos, when ectopically overexpressed, Foxd-3 is a potent inducer of neural crest markers, including itself and Slug, but it also promotes expression of neural markers.50 Interestingly, in contrast to Slug, Foxd-3 can induce expression of neural crest markers in distant locations from the neural crest region. Conversely, attenuation of Foxd-3 activity by overexpression of a dominant-negative form of the molecule inhibits neural crest differentiation.50 Thus, although these studies did not directly address Foxd-3 function in delamination, they clearly highlight its critical role in neural crest formation and support observations made in the chick. In conclusion, unlike Slug, Foxd-3 seems to be a potent inducer of crest cell specification and delamination, but the delay for its effect indicates that it does so only indirectly, possibly by activating intermediate genes, and in an uncontroled manner as it induces markers of migrating neural crest cells prior to delamination and at the expense of other cell populations of the dorsal neural tube.
The Sox-E Subgroup of HMG-Box Transcription Factors
Recently, the Sox-E subgroup of HMG box-containing transcription factors attracted much attention, because in all species and at all axial levels examined, they are specifically expressed in a temporal order in neural crest cells from early determination to late migration.28,29,53 Sox-9 is the first to appear in prospective crest cells and it is closely followed by Sox-10 and, later, by Sox-8, just before neural crest cells exit the neural tube. At the onset of migration, while Sox-10 is retained by migrating cells, Sox-8 and Sox-9 are rapidly down-regulated. Later during differentiation, Sox genes are reexpressed in distinct neural crest subpopulations and have been shown alternatively to contribute the maintenance of neural crest multipotency or to participate to specific differentiation programs. Thus, Sox-E genes, and particularly Sox-10, compose at present the most universal neural crest cell markers. As observed for Foxd-3, production of neural crest cells is strongly agcted in Xenopus embryos upon knockdown or overexpression of Sox-9 and Sox-10. However, while both factors are critical for early crest determination and melanocyte differentiation, they are apparently relatively dispensable for delamination and subsequent migration.54-56 Likewise, in mouse embryos lacking Sox-9, neural crest cells are specified and able to start migration but they rapidly undergo programmed cell death shortly after.29 Very recently, implication of Sox-E transcription factors in delamination has been further investigated in detail in chick by two different laboratories, but their results differ in some significant aspects.28,29,53 Both studies found that forced expression in the trunk neural tube of either Sox-8, Sox-9 or Sox-10, but not of Sox-2, a Sox gene from a different subgroup, convert within less than 24 hours neural tube cells into neural crest-like cells expressing the HNK-1 marker. In addition, both studies showed that Sox-E genes induces cadherin-7 expression but no downregulation of N-cadherin and that they rapidly turn off Rho-B expression. However, while Cheung and coworkers did not document any marked ectopic delamination of neural tube in the electroporated side of the neural tube and concluded that Sox-E genes by themselves are not capable of triggering cell delamination, McKeown and colleagues observed at variance extensive migration. This was seen at all levels along the dorsoventral axis, including in the floor plate, about 36 hours after electroporation, i.e., rather late after induction of a neural crest phenotype and, 48 hours after electroporation, the transfected side of the neural tube was almost entirely disrupted and almost all cells were released into the neighboring sclerotome.28 At present, there are no obvious explanations for these discrepancies, but whatever the exact role of Sox-E genes in neural crest development, it appears that, like Foxd-3, they can elicit cell delamination only secondarily, after induction of HNK-1 and cadherin-7, two markers normally expressed after delamination. In addition, delamination induced by Sox-E genes as well as by Foxd-3 is massive, leaving the neural tube as an empty bag or a flat tire from which the whole content has been poured out, a situation which is never observed normally, therefore suggesting that these transcription factors cause delamination by an aberrant and uncontroled sequence of events at the expense of the other neural cell types.
Recently, beside Sox-E genes, another Sox transcription factor of the Sox-D subgroup, Sox-5, has been characterized at cranial levels in the chick.57 It is expressed in premigratory crest cells, slightly later than Slug and is maintained in most neural crest cells during migration as well as in glial cells of cranial ganglia. Misexpression of Sox-5 in the cephalic neural tube leads to an exquisite phenotype contrasting with the massive effects obtained with Sox-E genes. In the dorsal neural tube, it augments both spatially and temporally the production of crest cells, associated with up-regulation of Foxd-3, Slug, Pax-7, Sox-10 and Rho-B, whereas in more ventral regions of the neural tube, it induces Rho-B expression, but not Foxd-3 or Sox-10 and its capacity to induce delamination is only marginal. Thus, like Slug, Sox-5 effect might be dependent on the cellular context within the neural tube.
Other Families of Transcription Factors
Neural crest cells have been found to express several additional transcription factors at the time of delamination, among which Pax-3, AP-2, Myc and members of the Zic family are the most remarkable.58 The role of these factors in neural crest delamination has not been addressed directly and their possible implication in this process cannot then be formally excluded. However, it is clear that because they are not restricted to prospective neural crest cells, they cannot pretend to play a major role by themselves. Pax-3, for example, has been shown to be genetically upstream of Foxd-349 and mouse Splotch embryos in which its gene is mutated exhibit strong defects in neural crest cell generation and migration, possibly as a result from decreased cell-cell adhesion due to oversialylation of N-CAM molecules.59,60 Yet, the precise role of Pax-3 in the control of cell adhesion remains unclear, as it has been also observed in vitro that its forced expression in mesenchymal cells may induce their aggregation of into multi-layered condensed cell clusters with epithelial characteristics.61 The protooncogene Myc has been implicated in Xenopus in crest cell determination independently of its proliferation role62 and, in the chick, it has been shown to stimulate massive crest cell migration followed by their differentiation into neurons.63 Finally, mice deficient in the AP-2 gene show severe defects causing embryonic lethality and affecting primarily development of the neural crest: failure of neural tube closure, craniofacial anomalies and absence of cranial ganglia.64
It is striking that, among the different transcription factors characterized in neural crest cells at the time of their segregation from the neural tube, none of them exhibit expression patterns matching precisely with delamination, suggesting that this step is essentially dependent on transcriptional events occuring during the previous specification step. However, recent studies allowed to pin down factors that mark precisely crest cell delamination more reliably than Slug or Rho-B for example. Ets-1, a member of the Ets family of winged helix-turn-helix transcription factors has been found to be dynamically expressed in delaminating crest cells at cranial levels (ref. 65 and E. Théveneau, M. Altabef, and J.-L. Duband, unpublished results). At the midbrain level, for example, its expression starts in prospective crest cells just after apposition of neural folds, at the 5-6 somite stage, i.e., about 4-6 hours before onset of migration, and it persists in the dorsal neural tube until cell delamination ceases, i.e., at the 11-somite stage. In addition, migrating neural crest cells almost immediately turn Ets-1 expression off as soon as they become fully segregated from the neural tube and leave its vicinity. Ets-1 has been previously implicated in various EMTs and migratory events during embryonic development and, in contrast to most other transcription factors expressed by crest cells, a detailed list of its potential target genes has been established: these include key molecules for cell locomotion such as integrins, cadherins and MMPs.66-69 Ectopic expression of Ets-1 in the chick neural tube by in ovo electroporation results in delamination of neural tube cells, at both cranial and truncal levels, although Ets-1 is not prominent in trunk crest cells. Interestingly, Ets-1-induced cell delamination presents unique characteristics that are not observed with forced expression of Foxd-3 or Sox-E genes (E. Théveneau, M. Altabef, and J.-L. Duband, unpublished results). It is rapid, cells being seen delaminating within 12 hours posttransfection; it occurs primarily at the basal side of the neural tube, but also less frequently at its apical (luminal) side; unlike Foxd-3, Sox-9 or Sox-10, it is not massive but rather progressive, leaving the neural tube intact in a very similar manner to the normal delamination of neural crest cells; cells exiting from the neural tube upon Ets-1 overexpression do not express neural crest cell markers such as HNK-1 or Slug, but show local disruption of the basement membrane, indicating that Ets-1 most likely triggers delamination by activating expression of MMPs; finally, delamination is not followed by migration, cells remaining for a while at the close vicinity of the neural tube, before undergoing apoptosis. Thus, at least at cranial levels, Ets-1 might regulate late cellular events accompanying neural crest cell delamination independently of a neural crest phenotype, thereby illustrating that specification, delamination, and migration are separable events.
Cooperative Activity of Transcription Factors during Neural Crest Cell EMT
The above studies reveal that delamination elicited by transcription factors ectopically expressed in the intermediate and ventral neural tube is either partial or disordered, and that none of them is able to induce a complete neural crest phenotype (Fig. 2). Thus, they do not allow to draw a coherent sketch of the transcriptional network controling neural crest delamination. A possible clue is to identify the epistatic relationships between these transcription factors in prospective neural crest cells.58 In Xenopus, gain- and loss-of-functions approaches revealed highly complex crossregulation of Snail, Slug, Sox-9, Sox-10 and Foxd-3 genes which can influence each other via direct transcriptional activation of repression or through secondary factors, thereby excluding any obvious linear hierarchy among these factors.39,50,55 In chick, the situation contrasts with that observed in Xenopus in that Slug, Foxd-3, and Sox-9 signals are apparently independent and display distinct sets of targets.28,29,40,49 In addition, Foxd-3 and Sox-9 lie upstream the Sox-10 and Sox-8 genes, consistent with their precocious expression in prospective crest cells. In the mouse, lastly, while the Foxd-3 and Snail mutants were not informative because of premature lethality, the Sox9 mutant provided interesting informations about the functional interactions between Foxd-3, Snail, Sox-10 and Sox-9. It appears that Snail, but not Foxd-3 and Sox-10, is dramatically downregulated in premigratory crest cells in mutant embryos compared with their wildtype littermates, indicating that in this species, Snail is downstream of Sox-9.29 Recently, using the chick system, the Briscoe's laboratory further investigated possible cooperative activities between Slug, Foxd-3 and Sox-9 in the control of truncal crest delamination by comparing the effects of these factors individually or in various combinations.29 While forced expression of Slug showed no obvious effect 24 hours after electroporation, Sox-9 induced HNK-1 expression within 12 hours in the intermediate and ventral neural tube but, after 24 hours, it was not capable of triggering significant cell delamination with no disruption of the basement membrane and of N-cadherin junctions and no integrin upregulation. In contrast, Sox-9 and Slug in conjunction induced robust HNK-1 expression, disorganization of the neural epithelium with degradation of the basement membrane, delocalization of N-cadherin out of adherens junctions, but no increase in integrins. Thus, confirming previous observations, Slug is effective in inducing cell delamination only if cells are specified as neural crest cells. Foxd-3 by itself was sufficient to induce first Sox-10 after 12 hours followed by HNK-1 expression after 24 hours associated with a decrease in N-cadherin expression. Ultimately, after 36-48 hours, it provoked an increase in integrins, a breakdown of the basement membrane allowing delamination and migration. Finally, combination of all three factors in neural tube cells caused cells to express neural crest markers, to delaminate entirely from the neural tube and to move actively in the surrounding tissues: this was associated with the complete breakdown of the basement membrane, the disappearance of N-cadherin from the cell surface, and the up-regulation of integrins. Thus, Sox-9, Foxd-3 and Slug ectopically expressed in the neural tube can recapitulate most of the events observed during neural crest delamination except that, unlike for endogenous neural crest cells, delamination is massive and uncontroled and leaves the neural tube totally disorganized (Fig. 2).
From these studies, some of the basic traits of the interplay between transcription factors during the transition from neural crest determination to early migration are now taking shape. First, delamination as well as specification and migration require the cooperating activities of at least three members of distinct families of transcription factors, Snail/Slug, Foxd-3 and Sox-9. Second, there is no simple linear hierarchies among these factors, rather a complex network of mutual interactions. Third, deployment of delamination is complete and efficient only if its basic cellular events are properly ordered in connection with crest cell specification and migration. For example, although neural crest specification is not sufficient to induce complete delamination (as suggested by experiments of Sox-9 overexpression) and that, conversely, delamination can be induced independently of specification (as suggested by overexpression of Rho-B or Ets-1), delamination is followed by active migration only if cells are specified into neural crest cells.
Regulation of Neural Crest EMT by a Balance between BMP-4, Noggin and Sonic Hedgehog
Long before transcription factors were identified in prospective neural crest cells as a response to inducing signals, it has been established that neural crest delamination is under the control of extrinsic factors released in the environment by the neighboring tissues, i.e., ectoderm, neural tube and paraxial mesoderm.3 Because of their implication in the regulation of cell-substrate adhesion, members of the transforming growth factor-β (TGF-β) family have been suspected to play a critical role in neural crest delamination. Thus, our laboratory has been able to show that TGF-β1 and TGF-β2 induces a precocious emigration of neural crest cells from avian neural tube explants possibly by increasing adhesion of cells to their substrate.8 The kinetics of TGF-β effect suggests that it functions primarily through integrin activation. More recently, the Kalcheim's laboratory confirmed and further extended this observation also in the avian system.47 In particular, it was found that BMP-4 is expressed in the dorsal neural tube, and that addition of BMP-4 alone to neural tube explants stimulates production of neural crest cells. Consistent with this, delaminating and early migrating neural crest cells express the BMP receptor IA.70 However, BMP-4 expression is not restricted to the time window when neural crest cells are released, and instead it is expressed uniformly throughout a long portion of the neural axis, in a pattern consistent with a role not only in delamination but also in specification and early migration. Interestingly, Noggin, a BMP-4-specific antagonist, shows a dynamic expression in the dorsal neural tube along a caudorostral gradient that coincides precisely with onset of neural crest emigration, thereby suggesting that BMP-4 activity may be regulated spatially and temporally by Noggin in relation with delamination. In agreement with this assumption, addition of Noggin to neural tube explants or grafting Noggin-producing cells in embryos in the vicinity of the neural tube at the time of neural crest migration prevent neural crest cell migration. Later studies by the same laboratory demonstrated that Noggin expression is under the control of the paraxial mesoderm.71 More specifically, the dorsomedial region of the dissociating somite was found to be the source of an inhibitory factor of an as-yet unknown nature that downregulates Noggin expression in the dorsal neural tube. Hence, neural crest delamination would be triggered by a signaling cascade elicited by BMP-4 interacting with its receptor BMPR-IA. The timing of crest emigration would be dictated by factors extrinsic to the neural epithelium, such as the dorsomedial portion of somite that controls Noggin expression in the dorsal neural tube. Additional cross-talks between the somite and neural tube cannot be excluded in order to further coordinate neural crest delamination and neural tube patterning with the maturation and subdivision of the paraxial mesoderm. However, this appealing model may not apply to all truncal levels but only for a very limited portion as it has been known for long that neural crest departure is not strictly synchronized with somitogenesis.2 Additional regulatory mechanisms may then be required for controling timing of emigration. Furthermore, it remains to be determined whether this model can be transposed to cranial levels where the paraxial mesoderm is not partitioned into somites like in the trunk. Nonetheless, cranial neural crest formation and migration in the mouse has also been found to be under the influence of BMPs, as BMP-2 mutants show marked defect in neural crest development.72
The BMP-4-signaling cascade controls neural crest cell delamination primarily through regulation of adhesion events associated with EMT. As mentionned above, when added to chick neural tube explants, both TGF-β and BMP-4 promote substrate-adhesion of neural crest cells through activation of β1-integrins (ref. 8 and A. Jarov, C. Fournier-Thibault and J.-L. Duband, unpublished). In addition, BMP-4 can induce in a temporal sequence expression of, first, Slug and cadherin-6B, then Rho-B and finally cadherin-7.31 Conversely, inhibition of BMP-4 by Noggin in chick embryo causes a severe repression of Rho-B, cadherin-6B but, surprisingly, not of Slug.47 A likely explanation is that Slug is regulated by several independent processes that might compensate for the lack of BMP signals. In support of this, functional Lef-binding sequences have been isolated in the Xenopus Slug-gene promoter, suggesting that it might also be controled by Wnt signals.73 However, the timing of appearance of these factors in prospective neural crest is not compatible with the expression pattern of BMP-4 and Noggin. Cadherin-6B and Slug, for example, are expressed in the dorsal neural tube long before the downregulation of Noggin and therefore prior to the time when BMP-4 signals are activated. This suggests that BMP-4 is not the endogenous inducer of these genes but that it is merely involved either in their maintenance or in their upregulation. Alternatively, these genes may be induced at thresholds of BMP-4 doses much lower than those necessary to cause delamination. As to the other key players in neural crest EMT are concerned, i.e., Foxd-3 and Sox-E genes, the possible influence of BMPs on their expression has surprisingly not been explored and documented yet. Of interest, members of the forkhead family to which Foxd-3 belongs have been found to be part of TGF-β-signaling pathways74 whereas, in other systems such as limb cartilage differentiation, Sox9 has been proposed to function independently of, but in concert, with BMP.75
Although neural crest cells are the only cell type within the neural tube that is endowed with delaminating and migratory properties, it has become clear over the last decade that other neural epithelial cells also possess at least transiently some migratory capacity (Fig. 3a-e). When challenged with BMPs, ventral portions of neural tubes explanted in vitro can generate cells that display some of the migratory characteristics of neural crest cells.76,77 Likewise, as extensively described above, ectopic expression of Rho-B, Foxd-3, Sox-9 or Ets-1 in the intermediate or ventral neural tube can induce cell delamination sometimes followed by migration. Furthermore, our laboratory has observed that all neural epithelial cells can naturally disperse in vitro on fibronectin or laminin substrates even in the complete absence of exogenous factors, provided they were derived from early, immature neural plates in the most caudal region of the embryo.78 Interestingly, the migratory potential of neural epithelial cells is only transient and declines gradually along a ventrodorsal gradient with the progressive maturation of the neural tube. Thus, in contrast to cells from early neural plates which are all able to disperse, only cells from the dorsal half retain this ability in more mature neural tube adjacent to the anterior, unsegmented mesoderm. At axial levels corresponding to the epithelial somites, only neural crest cells situated at the apex of the neural tube are able to migrate. Later, after the last neural crest has emigrated, the neural tube remains compact when explanted in vitro, and virtually no cell is seen delaminating from it. This progressive restriction in the migration potential of the neuroepithelium along a ventrodorsal gradient is suggestive of an inhibitory action of diffusible factors originating from the ventral neural tube. This inhibitory activity is most likely attributable to Sonic hedgehog (Shh), a morphogen produced by the notochord and the floor plate that plays a citical role in the patterning of the neural tube. Indeed, in addition to its well-characterized function in driving differentiation of ventral neural tube cells and promoting their survival and proliferation, Shh has been found to control the substrate-adhesive properties of both dorsal and ventral neuroepithelial cells.78,79 When Shh is presented under an immobilized form onto their substrate or produced by neuroepithelial cells themselves after transfection, neural tube explants or neural crest cells fail to disperse and instead form compact structures. Shh effect on cell adhesion is immediate, reversible and can be accounted for by inactivation of surface β1-integrins combined with an increase in N-cadherin mediated cell cohesion. In agreement with these in vitro data, forced expression of Shh in the dorsal neural epithelium after in ovo electroporation results in the cellular detachment of neural tube cells from the basement membrane followed by their collapse into the lumen, most likely due to inhibition of integrin function (C. Fournier-Thibault and J.-L. Duband, unpublished).
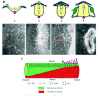
All together, these observations suggest that the adhesive properties of neural epithelial cells, both cell-cell and cell-substratum, are essentially regulated by the antagonistic activities of BMPs and Shh although they do not exclude the possible implication of other signaling molecules such as FGF, Wnt or retinoic acid. Dorsally, BMP-4 is produced by the ectoderm and the roof plate and its migration-promoting activity is restricted, spatially to the margin of the neural tube, because of its limited diffusion properties80 and, temporally due to the antagonistic activity of Noggin. Conversely, Shh produced ventrally in the floor plate gradually diffuses toward the dorsal aspect of the neural tube and progressively restricts the capacity of neuroepithelial cells to disperse by lowering the activity of integrins and reinforcing cadherin contacts. The activity of Shh in the dorsal neural tube would be limited at least transiently by the production of GAS-1, a specific Shh antagonist.81,82 The outcome of this exquisite regulation of cell adhesion by the interplay between morphogens and their respective antagonists would be that neural crest EMT is restricted spatially and temporally to the dorsal side of the neural tube (Fig. 3f).
Several Possible Scenarios for Neural Crest Delamination
Segregation of neural crest cells has so far been essentially regarded as an EMT. However, most the experiments aimed at manipulating EMT in the neural tube ended with the same striking outcome: the complete disorganization of the neural tube and the absence of replacement of the emigrating cells. Therefore, additional mechanisms are to be required to operate in parallel to or in combination with EMT to account for all the aspects of crest cell delamination, including for the correct spatio-temporal coordination and regulation of the molecular cascades and cellular events elicited by BMPs, Slug, Foxd-3 and Sox-9. Insight into these mechanisms can be gained if specification and delamination of neural crest cells are viewed as a question of generation of cellular diversity among neuroepithelial cells. Generating cellular diversity is usually achieved through different means, such as EMT coupled or not with cell migration, proliferation of precursor or stem cells, asymmetric cell division and lateral inhibition, and all contribute to the establishment of frontiers between neighboring cell populations or to the segregation of individuals or groups of cells that subsequently follow distinct fates and acquire specific functions (Fig. 4).
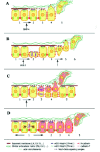
Proliferation of Precursor Cells
There are now convincing, both direct and indirect evidence that formation of the neural crest involves proliferation of precursor cells situated in the dorsal neural tube. On the basis of genetic analyses in the mouse, Foxd-3 has been proposed to play a critical role in the establishement or maintenance of proliferating and self-renewing progenitor cell populations.52 Although it has not been formally demonstrated in the case of neural crest cells, this role most likely also applies to them since Foxd-3 is restricted to few embryonic cell types, all exhibiting properties of multipotent progenitor cells, i.e., the blastocyst, epiblast, neural crest, neuroblasts and ES cells. Moreover, recent elegant experiments in the chick and Xenopus embryos have shown that segregation of neural crest cells from the neural tube is intimately coupled with cell division. In Xenopus, Kee and Bronner-Fraser found that depletion of Id3, a member of the Id family of helix-loop-helix transcription regulators expressed in nascent and migrating neural crest cells, results in the absence of neural crest progenitors.83 This appears to be mediated by cell cycle inhibition followed by the death of the pool of neural crest precursors, rather than a cell fate switch. Conversely, overexpression of Id3 increases cell proliferation and results in a greater number of migrating neural crest cells. These observations therefore highlight a critical role for cell proliferation in the generation of neural crest cells and ascribe to Id3 a unique regulatory role in mediating the decision of neural crest precursors to proliferate or to die, independent of cell fate determination. In the chick, Burstyn-Cohen and Kalcheim established that truncal neural crest cells synchronously emigrate from the neural tube in the S phase of the cell cycle.84 Inhibition of the G1/S transition in vivo or in explants specifically blocks delamination, without affecting expression of Slug, cadherin-6B, Rho-B or Pax-3. In contrast, arrest at the S or G2 phases has no immediate effect on delamination. It has been known for long that in neuroepithelial cells, the nucleus shuttles from the luminal side at mitosis to the basal side in the S phase. Neural crest cells would then delaminate at the favor of the most proximal position of their nuclei to their site of release. This appealing model accounts for the very progressive release of neural crest cells at truncal levels where delamination has been estimated to last during at least 24-30 hours, and it is compatible with the constant replacement of cells that have exited. Interestingly, at cranial levels where delamination is more sudden and massive (it lasts during less than 12 hours), fewer cells are in the S phase once they are released out of the neural tube, suggesting that in this region, delamination may be driven chiefly by EMT (E. Théveneau, M. Altabef and J.-L. Duband).
However, it remains to determine whether cell division in the dorsal neural tube is sufficient to account for the total number of cells that delaminate from the neural tube. This will require a precise estimate of the number of divisions that occur in the neural tube as well as the number of neural crest cells that are produced at each axial level. Moreover, it will be necessary to determine why cells are released out of the neural tube only in its dorsal aspect since this nuclear migration event occurs in all neuroepithelial cells. The lack of an organized, continuous basal lamina along the dorsal neural tube may be one of the cues to allow the release of cells once they become located basally. Alternatively, cell cycle would be coupled with other cellular events involved in the detachment of the cells from the neural tube. In this regard, further studies in the avian embryo reported recently by the Kalcheim's laboratory propose that G1/S transition in neural crest cells accompanying cell delamination is linked to BMP/Noggin signaling through the canonical Wnt signaling.85 They found that Noggin overexpression inhibits G1/S transition, while blocking G1/S transition abrogates BMP-induced EMT. Moreover, Wnt-1 expression is stimulated by BMP and interfering with Wnt signaling by blocking β-catenin and Lef-Tcf inhibits G1/S transition, cell delamination and transcription of several BMP-dependent genes. However, several previous observations are at variance with this study, and the precise function of Wnt signals in neural crest emigration remains obscure. First, although Wnt-1 presents a localized expression in the dorsal neural tube at the time of crest delamination, onset of its expression matches closer with somitogenesis than with neural crest delamination and, unlike BMP4, it persists in the dorsal neural tube long after cessation of emigration. Such an expression pattern is therefore not compatible with a direct role in the triggering of emigration. Second, targeted inactivation of Wnt-1 and Wnt-3a as well as genetic analyses in the mouse aimed at altering the canonical Wnt signaling pathway in neural crest cells mostly implicated Wnt signals in lineage specification rather than in delamination.86-88 Third, neural crest cells produce Wnt antagonists at the time of their emigration, thereby raising the intriguing possibility that they may not be responsive to Wnt signals at this step.89-91 Finally, overstimulation of Wnt-β-catenin signals in neural crest cells in vitro has been found to provoke a severe inhibition of delamination and migration, as a result of a decrease in substrate adhesion and reduction of proliferation.92
Asymmetric Cell Division
During asymmetric cell division, cells become polarized in response to extrinsic or intrisic signals before mitosis. As a result, cell-fate determinants present in the cytoplasm become localized to one pole of the cell in alignment with the mitotic spindle and, upon cytokinesis, they are unequally partitioned to the two daughter cells.93 At present, there are no definitive proof for the contribution of asymmetric cell division to neural crest cell delamination. In particular, although both horizontal (i.e., symmetrical) and vertical (i.e., asymmetrical) cleavage plans can be detected in the dorsal neural epithelium at the time of neural crest delamination, the cell-fate determinant Numb does not show a polarized distribution in mitotic neural epithelial cells prior to neurogenesis.94 Several indirect observations would in contrast argue in favor of the implication of asymmetrical division in neural crest cell delamination. Cell tracing experiments using lipophilic dyes such as DiI revealed the existence of a common precursor for neural crest and dorsal neural tube cells.95,96 In Drosophila, neuroepithelial cells are polarized along the apical-basal axis and divide symmetrically; upon deterioration of adherens junctions, cell divisions are converted from symmetric to asymmetric,97 suggesting that orientation of the mitotic spindle is under the control of adherens junctions. Although this mechanism has not been specifically addressed in neural crest cells, disruptions of adherens junctions have also been detected among dorsal neuroepithelial cells, coincidently with delamination.17 In Drosophila again, Snail proteins play an essential role in the generation of neuroblasts by controling expression of cell fate determinants during asymmetric cell division.98
Lateral Inhibition
Lateral inhibition is the process by which a cell both adopts a distinct fate from its neighbors, through signal echanges mediated by Notch and its receptor Delta, and prevents them to follow the same differentiation program. It allows to explain how a regularly-spaced array of structure can develop from a uniform field such as the development of the nervous system in Drosophila.99 It is not clear yet whether Notch signals directly affect neural crest cell delamination or whether they are merely involved in the establishement of frontiers between the ectoderm, neural crest and neural tube domains, but it is worth-mentionning that Notch has been found to promote EMT via Snail during cardiac cushion tissue formation in Zebrafish,100 a process presenting numerous similarities with crest cell delamination. In addition, it has been shown in the avian embryo that Notch has a dual function during neural crest formation, first, in maintaining expression of BMP-4 in the ectoderm and, second, in inhibiting Slug expression also in the ectoderm, possibly to prevent aberrant delamination of cells from this tissue.101 Conversely, in Drosophila, during differentiation of the mesoderm, Snail shows a dual activity on the Notch signaling pathway: it stimulates Notch signaling in some cells while repressing Notch target genes in others, thereby contributing to create precise boundaries among the tissue.102 Finally, at the time of neural crest delamination, lunatic fringe, a positive modulator of Notch signaling, is expressed throughout the neural tube with the notable exception of the prospective neural crest area, thereby delineating a border between the neural crest and neural tube domains. Overexpression of lunatic fringe in the cranial neural tube by retrovirally-mediated gene transfer causes a significant increase in the number of migrating neural crest cells as a result of activated cell proliferation.103
Other Possible Means
Other morphogenetic events may influence emigration of neural crest cells from the neural tube. One possibility is the elevation of the neural folds and the closure of the neural tube. In most species, delamination of neural crest cells is coupled to neural tube closure, but untill recently there was no easy and appropriate mean to manipulate the mechanical events accompanying neurulation to investigate their possible impact of neural crest delamination. Recent studies identifying new genes involved in the dynamics of neurulation may however provide new insights into this process.104 The other mechanism that can be put forward is repulsion from the neural tube. Although this possibility has not been seriously explored yet, it cannot be excluded since expression of molecules with repulsive activities has been reported in the dorsal neural tube at the time of neural crest delamination. For example, both Slit-1 and Slit-2, two proteins known to be potent chemorepellents for a variety of axons in Drosophila and in Vertebrates show a conspicuous expression in the roof plate. The available informations about the temporal expression of slits in relation with neural crest emigration are not sufficient to speculate on their implication in this process, but it is worthmentionning that trunk neural crest cells show a dual response to Slit-2 in vitro: they avoid cells expressing slit-2 and they migrate farther when exposed to soluble slit.105 BMP-7 is also a candidate molecule for repulsing neural crest cells out of the neural tube by analogy with commisural neurons. These neurons, located near the dorsal midline, send axons ventrally and across the floor plate but not dorsally through the roof plate. The latter has been found to express a diffusible factor that repels commisural axons and orients their growth within the dorsal spinal cord, and this chemorepellent activity is mediated by BMP-7 produced by roof plate cells.106
If it is confirmed that these mechanisms actually participate to the control of neural crest cell delamination, it will be of importance to determine at the single cell level whether they are purely independent or whether they may reflect a genuine requirement for distinct signaling processes. Moreover, such a multiplicity of cellular events is likely to represent a mechanism to establish precocious heterogeneity within the neural crest, even prior to delamination.107 It is then conceivable that different subsets of neural crest progenitors are specified independently and sequentially by distinct signaling cascades and are released at the periphery of the neural tube by different cellular processes (Fig. 4).
Cessation of Delamination
Important efforts have been put on the signaling cascades that induce neural crest cell delamination and migration and we are now relatively close to a fair appreciation of the basic mechanisms that govern these events. In comparison, very few is known about the mechanisms controling cessation of delamination, although it is likely to be precisely regulated in connection with the further development of the spinal cord. Several mechanisms may account for cessation of emigration. If neural crest cells are generated by a limited pool of precursors, cessation of emigration naturally occurs with the last precursor cell to segregate from the neural tube. This hypothesis supposes that the neural crest precursor, yet to be identified, lacks the capacity to give rise to cells other than neural crest cells and, reciprocally, that roof plate cells cannot differentiate into neural crest-like cells after cessation of emigration. However, although the existence of a unique neural crest precursor cannot be ruled out, cell lineages studies have clearly shown that neural crest or neural tube cells can be generated at the expense of one another, therefore arguing in favor of a great flexibility in the differentiation potential of neural epithelial cells at the time of neural crest emigration. Moreover, even if neural crest stem cells have been identified, they are very rare and their proportion cannot account for the production of all emigrating neural crest cells. Consistent with this, as mentionned above, strong heterogeneity has been observed among neural crest cells even before delamination. A more likely alternative then is that neural crest cells are generated from multipotent cells in the dorsal neural tube and that cessation of migration results as an impoverishment of this potential due to an intrinsic developmental program defined by a sort of internal clock and/or in response to external influences. If it is so, it would be then conceivable to induce experimentally a prolonged production of neural crest cells without major alteration in the morphology of the neural tube. So far, the only documented example concerns the secreted glycoprotein Noelin-1. In chick, Noelin-1 messages are expressed in a graded pattern in the closing neural tube and later they are restricted to the dorsal neural folds and migrating crest. Overexpression of Noelin-1 causes an excess of neural crest emigration and extends the time that the neural tube is competent to generate neural crest cells.108 How Noelin-1 mediates its effect at the molecular level has not been investigated yet partly because it has been found to exhibit divergent expression patterns in frogs, mouse and chick and thus may not perform the same functions.109
Neural Crest Migration: How to Survive, Move and Develop in a Hostile Environment
Much data has accumulated over the years on the process of neural crest cell migration, notably the road maps and the driving code. Interestingly, new concepts are progressively emerging from recent studies using different model systems and new insights are to be expected with the development of powerful real-time imaging techniques.
Transition between Delamination and Migration
With the exception of the Axolotl in which neural crest cells stand for a while on top of the neural tube prior to undergoing migration (hence the term neural crest), migration immediately follows the delamination step; yet, as discussed above, there are experimental evidence that both events are driven by independent and discernible mechanisms. However, the nature of the signals triggering onset of migration is not known at the present time. A candidate molecule for triggering neural crest motility is the hepatocyte growth factor (HGF) also known as scatter factor, but so far its expression has not been reported in neural crest cells, although they can respond to exogenous HGF in in vitro cultures (M. Delannet and J.-L. Duband, unpublished). Efficient neural crest migration may be achieved in vitro in the complete absence of external growth factors added to the culture medium or originating from the neural tube, indicating that induction of migration may be cell autonomous and would depend directly on cellular events occuring during delamination. Disruption of cadherin-mediated cell contacts and activation of integrins may be one of such events. We have shown previously that, in migrating neural crest cells, surface distribution and activity of N-cadherin are precisely regulated by intracellular signals elicited by integrins, thereby revealing that direct coupling between adhesion receptors provides the necessary interplay between cell-cell and cell-substrate adhesion during migration.110 Indeed, in vitro, neural crest cells express intact N-cadherin molecules on their surface but, contrasting with nonmotile cells, the bulk of these molecules is maintained excluded from the regions of cell-cell contacts, thus causing their instability. Stable contacts can be restored upon addition to the cells of specific inhibitors of integrin function and signaling activity. A possible target of this signaling pathway is β-catenin, known to play a critical role in both intercellular adhesion and cell signaling.111-113 Interestingly, it has been found in other cellular systems that cadherin binding can cause a massive recruitment of β-catenin to the cell membrane, thereby sequestering it and preventing its nuclear localization.114 This ability of cadherins to regulate the pool of β-catenin available for signaling therefore raises the intriguing possibility that β-catenin function in neural crest cells would be possibly driven by modulations in cellular cohesion during migration. Thus, in migrating neural crest cells, N-cadherin activity would be repressed by signals emanating from integrins, thereby resulting in an increase in the cytoplasmic pool of β-catenin that would be in turn allocated to the nucleus. Conversely, upon cessation of cell migration, e.g., after inhibition of integrin function, N-cadherin-mediated cell-cell contacts would be restored and β-catenin would be mostly recruited to them and would no longer be available for signaling. Consistent with this model, we reported recently that β-catenin is essentially associated with N-cadherin at the cell surface of actively migrating neural crest cells and that it is detected in their nuclei in association with Lef-1 only at the time of their segregation from the neural tube. However, manipulating N-cadherin-mediated cell contacts in migrating neural crest cells had no obvious impact on the nuclear localization of β-catenin, indicating that the membrane and nuclear pools of β-catenin are not directly connected at least during migration.92 Thus, the putative role of β-catenin as an inducer of neural crest cell migration awaits further developments and the identity of the molecular switch between delamination and migration remains elusive.
Maintenance of Survival during Neural Crest Migration
The problem of neural crest cell survival after delamination and during migration has long been underestimated. It was generally believed that, once released at the periphery of the neural tube, neural crest cells are naturally endowed with the ability to move, proliferate, and invade tissues and are marginally concerned with survival problems except in regions that they are not supposed to occupy. However, during the 1990s, it became obvious that in all animal organisms, only with rare exceptions, each cell type is programed to survive in a very peculiar tissular environment. Consequently, when a cell escapes from its tissue, it is immediately confronted with a new environment that may present very different features and does not support their survival: such cells unavoidably undergo apoptosis, thus eliminating any risk of aberrant cellular interactions. This “environmentalist” view has for example revolutionized our conception of cancer, which can be defined as the series of intracellular alterations allowing a cell to proliferate in an uncontroled manner, to be freed from its environmental constraints and to become able to occupy new territories. The same view also applies to migrating neural crest cells.
The importance of maintaining cell survival during neural crest ontogeny emerged in recent studies investigating the function of molecular players originally thought to play a role in cell specification and delamination. Neural epithelial cells forced to undergo EMT by expressing Rho-B or Ets-1 fail to migrate and to survive after their release at the periphery of the neural tube, in contrast to cells forced to express Foxd-3 or Sox-9 that do not show any sign of apoptosis. Phenotypic characterization of these cells reveal that Rho-B- or Ets-1-transfected cells show a striking difference with the Foxd-3- or Sox-9-transfected cells in that they express none of neural crest cell markers, such as HNK-1, Slug and cadherin-7 (refs. 28, 29, 53 and E. Théveneau, M. Altabef, and J.-L. Duband, unpublished data), suggesting that acquisition of a neural crest phenotype may protect cells from apoptosis once they are segregated from the neural epithelium. Accordingly, neural crest cell death can be rescued in Rho-B-transfected neural tubes by cotransfecting Sox-9.29 Among the numerous genes expressed in premigratory neural crest cells, Slug (or Snail in mouse) is likely to play a major role in maintaining neural crest survival. In Sox-9 mutant embryos, neural crest cells are specified and are able to undergo EMT, yet they die soon after the onset of migration. Interestingly, almost all the essential neural crest genes, including Foxd-3, are expressed in neural crest cells from these embryos, with the notable exception of Snail.29 In C. elegans, several lines of evidence point to a direct role for Snail superfamily members in the control of cell death.36 Finally, further experiments using cultured cell lines and mouse and chick embryos convincingly demonstrated that Slug and Snail confer resistance to cell death induced by the withdrawal of survival factors or by pro-apoptotic signals.115 Thus, Snail proteins may not play an essential role in triggering EMT among prospective neural crest cells, but they may directly connect cell survival with EMT and possibly with other cellular events such as cell division and Notch signaling, thereby providing to crest cells a selective advantage to migrate and populate distant territories. It is not much their ability to migrate actively that make neural crest cells a peculiar cell population among the neural epithelium, but it is mostly their capacity to survive once they become irreversibly separated from the neural tube.
Though it may be important for cell survival, Slug may not be the sole factor involved in protection from cell death. In particular, ectopic expression of Foxd-3 in the neural tube induces cell delamination but no apoptosis despite the absence of Slug induction.29,49 It cannot be excluded that Foxd-3 itself may drive a survival program in neural crest cells directly or indirectly by alternative means. Indeed, ectopic expression of Foxd-3 has been found to upregulate adhesion molecules of the cadherin and integrin families, which have been found to prevent apoptosis of epithelial cells, a process called anoikis.116 Moreover, Slug is only transiently expressed by migrating neural crest cells, particularly at truncal levels, therefore raising the problem of how this system is relayed to ensure maintenance of cell survival. A possibility is that neural crest cells produce their own survival factors or become responsive to growth factors secreted by tissues neighboring their migration routes. In this respect, it has been demonstrated that the neural tube produces factors that selectively support survival and proliferation of neural crest sub-populations and that this activity can be mimicked by FGF-2 or BDNF.117,118
Integrin Function Is Not Limited to the Control of Cell Adhesion and Locomotion
The organization and molecular composition of the migration routes of neural crest cells have been amply documented. Neural crest cells follow defined, restricted pathways that contain a fibrillar network of extracellular matrix material and are lined by the basal laminae of epithelia,2,119 although it is plausible that under certain conditions, they may also use the surface of cells encountered during migration as a support for locomotion. Numerous functional studies have provided convincing evidence that the matrix encountered by crest cells serves as a scaffold onto which they migrate and that integrin receptors for matrix molecules play a prominent role in this process. In vitro, neural crest cells are able to spread and migrate in an integrin-dependent manner onto a variety of matrix components, including fibronectin, laminin-1, vitronectin and collagens120-123 and, in vivo, antibodies to matrix molecules or integrins, competitor peptides, or antisense oligonucleotides to integrins or matrix constituents all perturb crest cell migration.124-127 Thus, integrin play a critical role in the mechanics of neural crest cell migration.
However, it is now increasingly clear that integrin functions during neural crest development are not limited stricly to substrate anchorage and cell motion. Integrins are heterodimers of a and β subunits that are present and conserved in all metazoan animals and are primarily involved in physical aspects of cell adhesion to the substrate, in cell traction and matrix assembly, by providing a bridge between the matrix and the cytoskeleton.128,129 Central to their function is their unique ability to promote actin assembly to generate tension locally via the recuitment of a wide array of molecules that directly activate the actin polymerization machinery or physically link it to adhesion sites.130,131 In addition to their structural and mechanosensory roles, integrins are able to activate upon engagement with their ligands a large variety of tyrosine kinases and GTPases to induce multiple downstream signaling pathways.132,133 Furthermore, integrin signals have been found to cooperate with and modulate signaling events initiated by growth factor receptors.134 Thus, because of this dual physical and chemical signaling activity, integrins influence numerous aspects of cell behavior, including migration, proliferation, survival, and differentiation.
It has been found that crest cells migrating in vitro express a multiplicity of integrins (at least three vitronectin receptors, three laminin-1 receptors and up to seven fibronectin receptors), and that not all of them are implicated in adhesion and migration.120,121,135 Such a diversity of matrix receptors certainly reflects the very changing nature of the environment to which crest cells are confronted during migration, but is also presumably related with additional roles for integrins not directly related to matrix adhesion. For example, as discussed earlier, integrins have been found to control cell-cell interactions during migration by repressing the surface distribution and activity of N-cadherin, thus ensuring rapid and flexible coordination between adhesion systems.110 Integrins are also involved in the maintenance of cell survival as revealed by functional studies in avian embryos136 and by genetic analyses in mouse and zebrafish.137-139 In fact, the primary defect observed in neural crest cells of embryos depleted in individual integrin α subunits is anoikis during migration, thereby revealing that anchorage-dependent survival signals elicited by integrins are of paramount importance for cells confronted with a continuously-changing environment in which supplies in growth factors are limited. Finally, both changes in the integrin repertoire at the time of neuronal differentiation in peripheral ganglia and the numerous alterations observed in conditional β1-integrin gene deletion argue for a possible implication of integrins in late neural crest development, such as lineage segregation, cell differentiation, and final maturation of the peripheral nervous system.25,140,141 All together, these observations suggest that integrins may control multiple cellular events during neural crest development. Yet, the signaling cascades that are responsible for their coordination over time and space remain to be established.
Novel Guidance Mechanisms for Neural Crest Cells
One of the main goals of migration is to segregate neural crest lineages from a uniform population and to drive them to specific locations at the right time so that they can undergo differentiation and establish appropriate contacts with their neighboring tissues. Hence a complex driving code which guides cells to their targets and prevents them from invade wrong pathways and occupying aberrant tissues. In most cases, this code is essentially a repressing code, with a complex combination of repulsive molecules (Fig. 5). This is particularly well-documented in the case of the somite which imposes a partition of the truncal neural crest population into several streams, each at the origin of the sensory, sympathetic, Schwann cell and melanocyte lineages. At least 10 different molecules have been implicated in the restriction of neural crest cell migration into the rostral half of the somite.142 These molecules are primarily recruited among extracellular matrix components or surface molecules released into the matrix, e.g., tenascin, F-spondin and semaphorins, as well as among surface receptors such as the ephrins and slits. The same mechanism is likely to apply also to the colonization of the branchial arches by hindbrain neural crest cells.143,144 Recently, two studies provided interesting information about the mechanisms by which the various neural crest subpopulations can interpret this code and distribute differently along separate pathways. In the first one, De Bellard and coworkers found that slit-1, slit-2, and slit-3 are expressed in the mesenchyme adjacent to the ventral aorta and the gut, which is selectively invaded by vagal, but not truncal, neural crest cells, suggesting that they may prevent ventral migration of trunk crest cells.105 Accordingly, truncal, but not vagal, neural crest cells express Robo-1 and Robo-2, two receptors for slits, and avoid Slit-expressing cells in vivo and in vitro, clearly indicating that Slit may contribute to the differential ability of neural crest population to populate and innervate the gut.105 In the second study, Santiago and Erickson investigated how neural crest cells fated to sensory and sympathetic lineages only migrate ventrally and are prevented from migrating laterally into the skin, whereas melanoblast precursors are directed only toward the lateral pathway.145 They found that ephrin-B ligands are expressed in the lateral pathway at the stage when neural crest cells migrate ventrally, consistent with a putative repulsive activity. Non surprisingly, inhibition of ephrin receptor function by addition of soluble ephrin-B ligand relieves the blockade of migration, thus allowing precocious invasion of the lateral pathway by neural crest cells normally migrating only ventrally. However, ephrin-B ligands unexpectedly continue to be expressed at later stages during melanoblast migration. Furthermore, when signaling of the Eph receptors for ephrins is disrupted in vivo, melanoblasts fail to migrate laterally, suggesting that ephrin-B ligands not only favor but are required for melanoblast migration. Thus, ephrins act as bifunctional guidance cues: they first repel sensory and sympathetic neural crest precursors from the lateral pathways, and later stimulate migration of melanoblast precursors into this pathway. The mechanisms by which ephrins regulate repulsion or attraction in crest cells are unknown, but most likely reside in the downstream signaling cascades elicited by the Eph receptor.
Chemotactism has long been proposed to account for the precision by which neural crest cells reach their target sites, particularly for those subpopulations that colonize sites situated at long distances from the source. But numerous arguments have been opposed against chemotactism in the case of neural crest cells. First, neural crest migration is essentially centrifugal, from a single source, the neural tube, to multiple target sites and this is not considered as compatible with chemotaxis which instead is highly efficient for centripetal migrations towards a unique final destination, as e.g., germ cells invading the gonads. Second, neural crest cells undergo migration sometimes well before their target tissue are elaborated in the embryo and this is difficult to reconcile with chemotactism. Finally, manipulations of the neural tube in ovo such as rotation along the dorsoventral axis revealed that neural crest cells can move backward along their migratory paths. However, although the existence of a unique chemotactic mechanism is unlikely to account for all neural crest directions, it is plausible that this process may regulate locally migration of distinct neural crest subpopulations such as melanoblast precursors and enteric neural crest cells. Thus, the dermis has been proposed to attract melanoblast precursors to the lateral path by a chemotactic mechanism, based on the observations that melanoblasts cannot enter the lateral path until emergence of the dermis and that grafts of dermis explants distally in the lateral pathway elicits precocious migration of neural crest cell into this pathway.146 Likewise, GDNF, a growth factor critical for the survival of enteric neural crest cells, has been found to promote oriented migration of neural crest cells throughout the gastrointestinal tract and to prevent them from dispersing out of the gut.147,148
Cell Communications during Migration
During the course of their migration, neural crest cells continuously establish contact with the neighboring tissues as well as with the other neural crest cells. Analyses of static images of embryonic sections stained with neural crest markers, notably HNK-1, and videomicroscopic studies of cells cultured in vitro clearly established that neural crest cells migrate as a cohort of cells remaining in close contact and that only few pioneer cells migrated as individuals (e.g., see ref. 149). Partly based on these observations, Thomas and Yamada proposed in 1992 the new, intriguing concept that migration could be stimulated by cell-cell contacts instead of being inhibited as originally described for isolated fibroblasts by Abercrombie.150,151 Unfortunately, the molecular cascade involved has not been deciphered and this hypothesis has been poorly visited since then. It is only recently that the importance of cell communication during migration has been reconsidered seriously owing to numerous technical developments, including embryo or explants cultures, high resolution confocal microscopy and in ovo imaging methods combined with the great improvement of cell labeling and transient transfection techniques. Young and coworkers used mice in which the expression of GFP is under the control of the ret promoter to visualize enteric neural crest cell migration in the embryonic gut in organ culture.152 In a similar approach, Teddy and Kulesa and Kasemeler-Kulesa and coworkers used chick embryos in which the neural tube is electroporated with GFP to explore hindbrain and truncal neural crest migration in whole embryo explants maintained in vitro.153,154 These studies revealed that neural crest cell migrate in chain-like formations that displayed complicated patterns of migration with sudden and frequent changes in migratory speed and trajection very much like in in vitro cultures. Pioneer cells formed a scaffold onto which following cells migrated. Morover, cells maintained nearly constant contacts with other migrating neural crest cells both at short and long distances up to 100 μm. Interestingly, cell-cell contacts often stimulated a cell to change direction, thereby revealing intense communication between cells and their possible implication in directional guidance.
The identity of the mediators of cell communication between neural crest cells is currently unknown but plausible candidates include cadherins and connexins. For example, cadherin-7 which is expressed in neural crest cells as soon as they segregate from the neural tube may be involved in cell recognition among navigating neural crest cells although this possibility has not yet been addressed directly. This putative function is compatible with the fact that cadherin-7 does not mediate strong cell-cell interactions in comparison with N-cadherin or E-cadherin and that its expression does not interfere with cell locomotion.155 Additional studies on mouse neural crest cells revealed that, in N-cadherin-deficient embryos, neural crest cell migration is affected. Videomicroscopic analyses further indicated that neural crest cells lacking N-cadherin exhibit an elevated speed of locomotion but that cell directionality was reduced, therefore pointing to the implication of N-cadherin in cell-cell communication during migration.156 Likewise, transgenic mice exhibiting inhibition of connexin-43 in cardiac neural crest cells showed a deficit in neural crest cells in the outflow tract due to a reduced cell migration. Conversely, an elevation of connexin-43 expression caused outflow tract obstruction and conotruncal heart malformation as a result of an enhanced rate of neural crest migration and an increase in the abundance of neural crest-derived cells in the outflow tract.157
Cessation of Migration
As for delamination, cessation of migration has been poorly explored and numerous questions remain open. Supposedly, it involves the converse sequence of events of delamination and onset of migration, i.e., inactivation of integrins consecutive to the loss of substrate adhesion, increase in cell cohesion, reorganisation of the cytoskeleton and more in-depth modifications of cells, such as engagement into a differentiation program, thereby rendering the process of cessation of migration irreversible. Curiously, although neural crest cells navigate through a large variety of territories and interact with numerous cell types, once they stop migration, they regroup into homogenous clusters composed essentially of neural crest cells and they do not mingle with other cells. This is particularly true for the major neural crest derivatives such as peripheral ganglia (spinal, sympathetic, ciliary, cranial and enteric) and for the skull. There are however a few notable exceptions to this rule, such as the melanocytes, some cranial ganglia and connective tissues of the face and neck, where neural crest cells mingle with the other cell populations in the invaded tissues.
Several processes have been put forward to explain in causal terms the cessation of migration of neural crest cells, but they mostly apply to the first category of neural crest derivatives. Spatial restriction of migration involving coincidently the lack of available space, the absence of a suitable extracellular matrix and/or the presence of physical barriers (such as dense connective tissues or epithelia) certainly contributes at least to a transient blockade of migration, but so far it has not been possible to design appropriate experimental devices to test this hypothesis directly. In contrast, factors implicated in the directional guidance of neural crest cells have been clearly demonstrated to induce arrest of migration. For example, mouse embryos deficient in the semaphorin Sema-3A or its neuropilin receptor exhibit marked alterations in the formation of the sympathetic nervous system. In these animals, sympathetic precursors are not accumulated at their target sites around the dorsal aorta but dispersed widely. Consistent with this, when confronted with Sema-3A-secreting cells in vitro, sympathetic neurons lose their locomotory activity, coalesce into compact cell masses and emit thick bundles of neurites. These data therefore indicate that Sema-3A functions both as a stop signal to prohibit migration of the neural crest cells of sympathetic neuron lineage into inappropriate regions of embryos and as a signal to promote aggregation of sympathetic neurons into tightly packed cell masses at defined target sites to produce the stereotyped sympathetic nerve pattern (ref. 158 and Fig. 5).
Recently, morphogens have also been shown to control arrest of migration. In particular, while BMP have been implicated in the initiation of migration by activating integrins and downregulating cadherins, Shh appeared to play an opposite function during cessation of migration. Shh can directly modulate substrate adhesion of neural crest cells in vitro by shifting integrins from an active to an inactive state.79 Moreover, migrating neural crest cells cannot penetrate embryonic regions where Shh is produced, notably the perinotochordal area,159 and tend to accumulate and aggregate at the periphery of these regions: for example the spinal ganglia form in a region along the ventral neural tube which harbors no obvious clues for spatial restriction of migration or repulsive cues, but which is situated at the vicinity of the notochord. Thus, like Sema-3A, Shh activity may have on neural crest cells a broader impact that simple repulsion: it may inhibit migration, induce compaction and promote differentiation, as already observed for neuroepithelial cells.78 Accordingly, grafts of notochord or of Shh-producing cells along the dorsal mesencephalon in the chick induce formation of ectopic, trigeminal-like sensory ganglia while mice deficient in Shh show poorly-condensed trigeminal and spinal ganglia distributed in aberrant sites.160 Similarly, Zebrafish mutations in the Shh signaling pathway result in the absence of spinal ganglia and in the loss of expression of neurogenin-1, a gene required for determination of DRG precursors, albeit early neural crest migration occurs almost normally (ref. 161 and Fig. 5).
Certain neural crest populations, such as melanoblasts and enteric neuron precursors, display the striking ability to leave cells behind them during migration so that they finally distribute evenly along their migratory pathway while others, e.g., sensory and sympathetic ganglia, migrate en masse and accumulate in a unique site. This raises the puzzling question of the molecular and cellular mechanisms that selectively promote migration of cells at the front of the population or induce their arrest at the rear. Although this complex problem is far from being elucidated, time-lapse imaging studies and analyses of mouse mutants revealed an intricate interplay between morphogens and growth factors in the coordination of enteric neural crest migration, proliferation and differentiation that might contribute to the typical pattern of zenteric ganglia along the entire gut.152,162 Thus, GDNF promotes proliferation, differentiation and oriented migration of enteric neural crest cells; Shh in contrast inhibits differentiation, but it promotes proliferation, and restricts GDNF-induced migration; finally, endothelin-3 inhibits cell differentiation and shows the same effect on migration that Shh.
Conclusion
An overview of the molecular mechanisms underlying the neural crest development reveals apparent paradoxes that have greatly influenced appreciation of this process and the ways problems were tackled: a great diversity of independent events combined into an apparent linear and unique process. Intriguingly, the same paradox applies to Europe: a great diversity of peoples with distinct cultures and languages sharing a long History and many values. Indeed, albeit ontogeny of the neural crest evolves as a continuous process apparently obeying to a preestablished genetic program, each step is independent and can be at least experimentally separated from the others. This is in accord with the situation observed in a number of pathological situations, where neural crest derivatives can be generated in aberrant positions despite the fact that cells failed to delaminate or migrate properly. Rather than a linear cascade, neural crest cell delamination and migration must then be considered as the result of a conjunction of a great variety of cellular events that ultimately control cell and matrix interactions, cell proliferation, cell fate and cell survival. On the other hand, neural crest cells are most likely generated by multiple processes rather than a single one and they follow numerous migration paths, each governed by specific rules rather than a unique, common one. Such a multiplicity of processes and rules is likely to contribute to establish precocious diversity among the cell population.
Acknowledgments
Many thanks for Jean-Françs Colas and Claire Fournier-Thibault for stimulating discussions. Work form the author's laboratory is supported by the CNRS and the Université Pierre et Marie Curie and by a grant from ARC (No. 4260).
References
- 1.
- Le Douarin NM, Kalcheim C. The Neural Crest. 2nd ed. New York: Cambridge University Press. 1999
- 2.
- Erickson CA, Perris R. The role of cell-cell and cell-matrix interactions in the morphogenesis of the neural crest. Dev Biol. 1993;159:60–74. [PubMed: 8365575]
- 3.
- Duband J-L, Monier F, Delannet M. et al. Epithelium-mesenchyme transition during neural crest development. Acta Anat. 1995;154:63–78. [PubMed: 8714290]
- 4.
- LaBonne C, Bronner-Fraser M. Molecular mechanisms of neural crest formation. Annu Rev Cell Dev Biol. 1999;15:81–112. [PubMed: 10611958]
- 5.
- Nieto MA. The early steps of neural crest development. Mech Dev. 2001;105:27–35. [PubMed: 11429279]
- 6.
- Kalcheim C. Mechanism of early neural crest development: From cell specification to migration. Int Rev Cytol. 2000;200:143–196. [PubMed: 10965468]
- 7.
- Newgreen DF, Gibbins IL. Factors controlling the time of onset of the migration of neural crest cells in the fowl embryo. Cell Tiss Res. 1982;224:145–160. [PubMed: 7094004]
- 8.
- Delannet M, Duband J-L. Transforming growth factor-b control of cell-substratum adhesion during avian neural crest cell emigration in vitro. Development. 1992;116:275–287. [PubMed: 1483393]
- 9.
- Tosney KW. The early migration of neural crest cells in the trunk region of the avian embryo: An electron microscopic study. Dev Biol. 1978;62:317–333. [PubMed: 627310]
- 10.
- Spieth J, Keller RE. Neural crest cell behavior in white and dark larvae of Ambystoma mexicanum: Differences in cell morphology, arrangement and extracellular matrix as related to migration. J Exp Zool. 1984;229:91–107. [PubMed: 6699590]
- 11.
- Raible DW, Wood A, Hodson W. et al. Segregation and early dispersal of neural crest cells in the embryonic zebrafish. Dev Dyn. 1996;195:29–42. [PubMed: 1292751]
- 12.
- Newgreen DF, Minichiello J. Control of epitheliomesenchymal transformation. II. Cross-modulation of cell adhesion and cytoskeletal systems in embryonic neural cells. Dev Biol. 1996;176:300–312. [PubMed: 8666126]
- 13.
- Erickson CA, Tucker RP, Edwards BF. Changes in the distribution of intermediate-filament types in Japanese quail embryos during morphogenesis. Differentiation. 1987;34:88–97. [PubMed: 2442055]
- 14.
- Aaku-Saraste E, Hellwig A, Huttner WB. Loss of occludin and functional tight junctions, but not ZO-1, during neural tube closureremodeling of the neuroepithelium prior to neurogenesis. Dev Biol. 1996;180:664–679. [PubMed: 8954735]
- 15.
- Thiery JP, Delouvée A, Gallin W. et al. Ontogenetic expression of cell adhesion molecules: L-CAM is found in epithelia derived from the three primary germ layers. Dev Biol. 1984;102:61–78. [PubMed: 6365655]
- 16.
- Hatta K, Takagi S, Fujisawa H. et al. Spatial and temporal expression pattern of N-cadherin cell adhesion molecules correlated with morphogenetic processes of chicken embryos. Dev Biol. 1987;120:215–227. [PubMed: 3817290]
- 17.
- Duband J-L, Volberg T, Sabanay I. et al. Spatial and temporal distribution of the adherens-junctionassociated adhesion molecule A-CAM during avian embryogenesis. Development. 1988;103:325–344. [PubMed: 3224557]
- 18.
- Nakagawa S, Takeichi M. Neural crest cell-cell adhesion controlled by sequential and subpopulation-specific expression of novel cadherins. Development. 1995;121:1321–1332. [PubMed: 7540531]
- 19.
- Nakagawa S, Takeichi M. Neural crest emigration from the neural tube depends on regulated cadherin expression. Development. 1998;125:2963–2971. [PubMed: 9655818]
- 20.
- Vallin J, Girault J-M, Thiery JP. et al. Xenopus cadherin-11 is expressed in different populations of migrating neural crest cells. Mech Dev. 1998;75:171–174. [PubMed: 9739138]
- 21.
- Pla P, Moore R, Morali OG. et al. Cadherins in neural crest cell development and transformation. J Cell Physiol. 2001;189:121–132. [PubMed: 11598897]
- 22.
- Duguay D, Foty RA, Steinberg MA. Cadherin-mediated cell adhesion and tissue segregation: Qualitative and quantitative determinants. Dev Biol. 2003;253:309–323. [PubMed: 12645933]
- 23.
- Van de Putte T, Maruhashi M, Francis A. et al. Mice lacking Zfhx1b, the gene that codes for Smad-interacting protein-1, reveal a role for multiple neural crest cell defects in the ethiology of Hirschsprung disease-mental retardation syndrome. Am J Hum Genet. 2003;72:465–470. [PMC free article: PMC379238] [PubMed: 12522767]
- 24.
- Martins-Green M, Erickson CA. Basal lamina is not a barrier to neural crest cell emigration: Documentation by TEM and by immunofluorescent and immunogold labelling. Development. 1987;101:517–533. [PubMed: 3332260]
- 25.
- Bronner-Fraser M, Artinger M, Muschler J. et al. Developmentally regulated expression of α6 integrin in avian embryos. Development. 1992;115:197–211. [PubMed: 1638980]
- 26.
- Kil SH, Krull CE, Cann G. et al. The a4 subunit of integrin is important for neural crest cell migration. Dev Biol. 1998;202:29–42. [PubMed: 9758701]
- 27.
- Duong TD, Erickson CA. MMP-2 plays an essential role in producing epithelial-mesenchymal transformations in the avian embryo. Dev Dyn. 2004;229:42–53. [PubMed: 14699576]
- 28.
- McKeown SJ, Lee VM, Bronner-Fraser M. et al. Sox10 overexpression induces neural crest-like cells from all dorsoventral levels of the neural tube but inhibits differentiation. Dev Dyn. 2005;233:430–444. [PubMed: 15768395]
- 29.
- Cheung M, Chaboissier M-C, Mynett A. et al. The transcriptional control of trunk neural crest induction, survival, and delamination. Dev Cell. 2005;8:179–192. [PubMed: 15691760]
- 30.
- Newgreen DF, Minichiello J. Control of epitheliomesenchymal transformation. I. Events in the onset of neural crest cell migration are separable and inducible by protein kinase inhibitors. Dev Biol. 1995;170:91–101. [PubMed: 7541378]
- 31.
- Liu J-P, Jessel TM. A role for rhoB in the delamination of neural crest cells from the dorsal neural tube. Development. 1998;125:5055–5067. [PubMed: 9811589]
- 32.
- Gampel A, Parker PJ, Mellor H. Regulation of epîrmal growth factor receptor traffic by the small GTPase rhoB. Curr Biol. 1999;9:955–958. [PubMed: 10508588]
- 33.
- Liu AX, Rane N, Liu JP. et al. RhoB is dispensable for mouse development, but it modifies susceptibility to tumor formation as well as cell adhesion and growth factor signaling in transformed cells. Mol Cell Biol. 2001;21:6906–6912. [PMC free article: PMC99867] [PubMed: 11564874]
- 34.
- Nakaya Y, Kuroda S, Katagiri YT. et al. Mesenchymal-epithelial transition during somitic segmentation is regulated by differential roles of Cdc42 and Rac1. Dev Cell. 2004;7:425–438. [PubMed: 15363416]
- 35.
- Nieto MA, Sargent MG, Wilkinson DG. et al. Control of cell behavior during vertebrate development by Slug, a zinc finger gene. Science. 1994;264:835–839. [PubMed: 7513443]
- 36.
- Nieto M. The Snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol. 2002;3:155–166. [PubMed: 11994736]
- 37.
- Sefton M, Sanchez S, Nieto MA. Conserved and divergent roles for members of the Snail family of transcription factors in the chick and mouse embryo. Development. 1998;125:3111–3121. [PubMed: 9671584]
- 38.
- Thisse C, Thisse B, Schilling TF. et al. Structure of the zebrafish snail-1 gene and its expression in wild-type, spadetail and no-tail mutant embryos. Development. 1993;119:1203–1215. [PubMed: 8306883]
- 39.
- Aybar MJ, Nieto MA, Mayor R. Snail precedes slug in the genetic cascade required for the specification and migration of the Xenopus neural crest. Development. 2003;130:483–494. [PubMed: 12490555]
- 40.
- del BarrioMG, Nieto MA. Overexpression of Snail family members highlights their ability to promote chick neural crest formation. Development. 2002;129:1583–1593. [PubMed: 11923196]
- 41.
- Carver EA, Jiang R, Lan Y. et al. The mouse snail gene encodes a key regulator of the epithelial-mesenchymal transition. Mol Cell Biol. 2001;21:8184–8188. [PMC free article: PMC99982] [PubMed: 11689706]
- 42.
- Cano A, Pérez-Moreno MA, Rodrigo I. et al. The transcriptional factor Snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83. [PubMed: 10655586]
- 43.
- Batlle E, Sancho E, Franci C. et al. The transcription factor Snail is a repressor of E-cadherin gene expression in epithelial tumor cells. Nat Cell Biol. 2000;2:84–89. [PubMed: 10655587]
- 44.
- Savagner P, Yamada KM, Thiery JP. The Zinc-finger protein Slug causes desmosome dissociation, an initial and necessary step for growth factor-induced epithelial-mesenchymal transition. J Cell Biol. 1997;137:1403–1419. [PMC free article: PMC2132541] [PubMed: 9182671]
- 45.
- LaBonne C, Bronner-Fraser M. Snail-related transcriptional repressors are required in Xenopus for both the induction of the neural crest and its subsequent migration. Dev Biol. 2000;221:195–205. [PubMed: 10772801]
- 46.
- Linker C, Bronner-Fraser M, Mayor R. Relationship between gene expression domains of Xsnail, Xslug, and Xtwist and cell movement in the prospective neural crest of Xenopus. Dev Biol. 2000;224(2):215–225. [PubMed: 10926761]
- 47.
- Sela-Donenfeld D, Kalcheim C. Regulation of the onset of neural crest migration by coordinated activity of BMP4 and Noggin in the dorsal neural tube. Development. 1999;126:4749–4762. [PubMed: 10518492]
- 48.
- Kos R, Reedy MV, Jonhson RL. et al. The winged-helix transcription factor FoxD3 is important for establishing the neural crest lineage and repressing melanogenesis in avian embryos. Development. 2001;128:1467–1479. [PubMed: 11262245]
- 49.
- Dottori M, Gross MK, Labosky P. et al. The winged-helix transcription factor Foxd3 suppresses interneuron differentiation and promotes neural crest cell fate. Development. 2001;128:4127–4138. [PubMed: 11684651]
- 50.
- Sasai N, Mizuseki K, Sasai Y. Requirement of FoxD3-class signaling for neural crest determination in Xenopus. Development. 2001;128:2525–2536. [PubMed: 11493569]
- 51.
- Labosky PA, Kaestner KH. The winged helix transcription factor Hfh2 is expressed in neural crest and spinal cord during mouse development. Mech Dev. 1998;76(1-2):185–190. [PubMed: 9767163]
- 52.
- Hanna LA, Foreman RK, Tarasenko IA. et al. Requirement for Foxd3 in maintaining pluripotent cells of the early mouse embryo. Gen Dev. 2002;16:2650–2661. [PMC free article: PMC187464] [PubMed: 12381664]
- 53.
- Cheung M, Briscoe J. Neural crest development is regulated by the transcription factor Sox9. Development. 2003;130:5681–5693. [PubMed: 14522876]
- 54.
- Lee YH, Aoki Y, Hong CS. et al. Early requirement of the transcriptional activator Sox9 for neural crest specification in Xenopus. Dev Biol. 2004;275:93–103. [PubMed: 15464575]
- 55.
- Aoki Y, Saint-Germain N, Gyda M. et al. Sox10 regulates the development of neural crest-derived melanocytes in Xenopus. Dev Biol. 2003;259:19–33. [PubMed: 12812785]
- 56.
- Honore SM, Aybar MJ, Mayor R. Sox-10 is required for the early development of prospective neural crest in Xenopus embryos. Dev Biol. 2003;260:79–96. [PubMed: 12885557]
- 57.
- Perez-Alcala S, Nieto MA, Barbas JA. LSox5 regulates RhoB expression in the neural tube and promotes generation of the neural crest. Development. 2004;131:4455–4465. [PubMed: 15306568]
- 58.
- Meulemans D, Bronner-Fraser M. Gene-regulatory interactions in neural crest evolution and development. Dev Cell. 2004;7:291–299. [PubMed: 15363405]
- 59.
- Moase CE, Trasler DG. Delayed neural crest cell emigration from Sp and Spd mouse neural tube explants. Teratology. 1990;42:171–182. [PubMed: 2218944]
- 60.
- Moase CE, Trasler DG. N-CAM alterations in splotch neural tube defect mouse embryos. Development. 1991;113:1049–1058. [PubMed: 1821845]
- 61.
- Wiggan O, Fadel MP, Hamel PA. Pax3 induces cell aggregation and regulates phenotypicmesenchymalepithelial interconversion. J Cell Sci. 2001;115:517–529. [PubMed: 11861759]
- 62.
- Bellmeyer A, Krase J, Lindgren J. et al. The protooncogene c-Myc is an essential regulator of neural crest formation in Xenopus. Dev Cell. 2003;4:827–839. [PubMed: 12791268]
- 63.
- Wakamatsu Y, Watanabe Y, Nakamura H. et al. Regulation of the neural crest cell fate by N-myc: Promotion of ventral migration and neuronal differentiation. Development. 1997;124:1953–1962. [PubMed: 9169842]
- 64.
- Schorle H, Meier P, Buchert M. et al. Transcription factor AP-2 essential for cranial closure and craniofacial development. Nature. 1996;381:235–238. [PubMed: 8622765]
- 65.
- Tahtakran SA, Selleck MA. Ets-1 expression is associated with cranial neural crest migration and vasculogenesis in the chick embryo. Gene Expr Patterns. 2003;3:455–458. [PubMed: 12915311]
- 66.
- Fafeur V, Tulasne D, Quéva C. et al. The ets1 transcription factor is expressed during epithelial-mesenchymal transitions in the chick and is activated in scatter factor-stimulated MDCK epithelial cells. Cell Growth Differ. 1997;8:655–665. [PubMed: 9185999]
- 67.
- Maroulakou IG, Papas TS, Green JE. Differential expression of ets-1 and ets-2 proto-oncogenes during murine embryogenesis. Oncogene. 1994;9:1551–1565. [PubMed: 8183549]
- 68.
- Rosen GD, Barks JL, Iademarco MF. et al. An intricate arrangement of binding sites for the Ets family of transcription factors regulates activity of the a4 integrin gene promoter. J Biol Chem. 1994;269:15652–15660. [PubMed: 8195215]
- 69.
- Wasylyk B, Hagman J, Gutierrez-Hartmann A. Ets transcription factors: Nuclear effectors of the Ras-MAP-kinase signaling pathway. Trends Biochem Sci. 1998;23:213–216. [PubMed: 9644975]
- 70.
- Sela-Donenfeld D, Kalcheim C. Localized BMP-4-noggin interactions generate the dynamic patterning of noggin expression in somites. Dev Biol. 2002;246:311–328. [PubMed: 12051818]
- 71.
- Sela-Donenfeld D, Kalcheim C. Inhibition of Noggin expression in the dorsal neural tube by somitogenesis: A mechanism for coordinating the timing of neural crest emigration. Development. 2000;127:4845–4854. [PubMed: 11044399]
- 72.
- Kanzler B, Foreman RK, Labosky PA. et al. BMP signaling is essential for development of skeletogenic and neurogenic cranial neural crest. Development. 2000;127:1095–1104. [PubMed: 10662648]
- 73.
- Vallin J, Thuret R, Giacomello E. et al. Cloning and characterization of three Xenopus slug promoters reveal direct regulation by Lef/β-catenin signaling. J Biol Chem. 2001;276(32):30350–30358. [PubMed: 11402039]
- 74.
- Chen X, Rubock MJ, Whitman M. A transcriptional partner for MAD proteins in TGF-β signalling. Nature. 1996;383(6602):691–696. [PubMed: 8878477]
- 75.
- Chimal-Monroy J, Rodriguez-Leon J, Montero JA. et al. Analysis of the molecular cascade responsible for mesodermal limb chondrogenesis: Sox genes and BMP signaling. Dev Biol. 2003;257(2):292–301. [PubMed: 12729559]
- 76.
- Liem KF, Tremml G, Roelink H. et al. Dorsal differentiation of neural plate cells induced by BMP-mediated signals from epidermal ectoderm. Cell. 1995;82:969–979. [PubMed: 7553857]
- 77.
- Basler K, Edlund T, Jessell TM. et al. Control of cell pattern in the neural tube: Regulation of cell differentiation by dorsalin-1, a novel TGFb family member. Cell. 1993;73:687–702. [PubMed: 7916656]
- 78.
- Jarov A, Williams KP, LE L. et al. A dual role for Sonic hedgehog in regulating adhesion and differentiation of neuroepithelial cells. Dev Biol. 2003;261:520–536. [PubMed: 14499657]
- 79.
- Testaz S, Jarov A, Williams KP. et al. Sonic hedgehog restricts adhesion and migration of neural crest cells independently of the Patched-Smoothened-Gli signaling pathway. Proc Natl Acad Sci USA. 2001;98:12521–12526. [PMC free article: PMC60086] [PubMed: 11592978]
- 80.
- Hu Q, Ueno N, Behringer RR. Restriction of BMP4 activity domains in the developing neural tube of the mouse embryo. EMBO Rep. 2004;5(7):734–739. [PMC free article: PMC1299095] [PubMed: 15218525]
- 81.
- Lee CS, Fan CM. Embryonic expression patterns of the mouse and chick Gas1 genes. Mech Dev. 2001;101(1-2):293–297. [PubMed: 11231094]
- 82.
- Lee CS, Buttitta L, Fan CM. Evidence that the WNT-inducible growth arrest-specific gene 1 encodes an antagonist of sonic hedgehog signaling in the somite. Proc Natl Acad Sci USA. 2001;98(20):11347–11352. [PMC free article: PMC58732] [PubMed: 11572986]
- 83.
- Kee Y, Bronner-Fraser M. To proliferate or to die: Role of Id3 in cell cycle progression and survival of neural crest progenitors. Genes Dev. 2005;19(6):744–755. [PMC free article: PMC1065727] [PubMed: 15769946]
- 84.
- Burstyn-Cohen T, Kalcheim C. Association between the cell cycle and neural crest delamination through specific regulation of G1/S transition. Dev Cell. 2002;3:383–395. [PubMed: 12361601]
- 85.
- Burstyn-Cohen T, Stanleigh J, Sela-Donenfeld D. et al. Canonical Wnt activity regulates trunk neural crest delamination linking BMP/noggin signaling with G1/S transition. Development. 2004;131(21):5327–5339. [PubMed: 15456730]
- 86.
- Hari L, Brault V, Kléber M. et al. Lineage-specific requirements of β-catenin in neural crest development. J Cell Biol. 2002;159:867–880. [PMC free article: PMC2173383] [PubMed: 12473692]
- 87.
- Brault V, Moore R, Kutsch S. et al. Inactivation of the β-catenin gene by Wnt-1-Cremediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128:1253–1264. [PubMed: 11262227]
- 88.
- Ikeya M, Lee SMK, Johnson JE. et al. Wnt signalling required for expansion of neural crest and CNS progenitors. Nature. 1997;389:966–970. [PubMed: 9353119]
- 89.
- Baranski M, Berdougo E, Sandler JS. et al. The dynamic expression pattern of frzb-1 suggests multiple roles in chick development. Dev Biol. 2000;217:25–41. [PubMed: 10625533]
- 90.
- Ladher RK, Church VL, Allen S. et al. Cloning and expression of the Wnt antagonists Sfrp-2 and Frzb during chick development. Dev Biol. 2000;218:183–198. [PubMed: 10656762]
- 91.
- Jin E-J, Erickson CA, Takada S. et al. Wnt and BMP signaling govern lineage segregation of melanocytes in the avian embryo. Dev Biol. 2001;233:22–37. [PubMed: 11319855]
- 92.
- de MelkerAA, Desban N, Duband JL. Cellular localization and signaling activity of β-catenin in migrating neural crest cells. Dev Dyn. 2004;230(4):708–726. [PubMed: 15254905]
- 93.
- Knoblich JA. Asymmetric cell division during animal development. Nat Rev Mol Cell Biol. 2001;2:11–20. [PubMed: 11413461]
- 94.
- Wakamatsu Y, Maynard TM, Jones SU. et al. NUMB localizes in the basal cortex of mitotic avian neuroepithelial cells and modulates neuronal differentiation by binding to Notch-1. Neuron. 1999;23:71–81. [PubMed: 10402194]
- 95.
- Serbedzija GN, Bronner-Fraser M, Fraser SE. A vital dye analysis of the timing and pathways of avian trunk neural crest cell migration. Development. 1989;106:809–816. [PubMed: 2562671]
- 96.
- Bronner-Fraser M, Fraser S. Developmental potential of avian trunk neural crest cells in situ. Neuron. 1989;3:755–766. [PubMed: 2484346]
- 97.
- Lu B, Roegiers F, Jan LY. et al. Adherens junctions inhibit asymmetric division in the drosophila epithelium. Nature. 2001;409:522–525. [PubMed: 11206549]
- 98.
- Ashraf SI, Ip YT. The Snail protein family regulates neurablast expression of inscuteable and string, genes involved in asymmetry and cell division in Drosophila. Development. 2001;128:4757–4767. [PubMed: 11731456]
- 99.
- Baron M. An overview of the Notch signalling pathway. Semin Cell Develop Biol. 2003;14:113–119. [PubMed: 12651094]
- 100.
- Timmerman LA, Grego-Bessa J, Raya A. et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Gen Dev. 2004;18:99–115. [PMC free article: PMC314285] [PubMed: 14701881]
- 101.
- Endo Y, Osumi N, Wakamatsu Y. Bimodal functions of Notch-mediated signaling are involved in neural crest formation during avian ectoderm development. Development. 2003;129:863–873. [PubMed: 11861470]
- 102.
- Cowden J, Levine M. The Snail repressor positions Notch signaling in the Drosophila embryo. Development. 2002;129(7):1785–1793. [PubMed: 11923213]
- 103.
- Nellemann C, de BellardME, Barembaum M. et al. Excess lunatic fringe causes cranial neural crest over-proliferation. Dev Biol. 2001;235:121–130. [PubMed: 11412032]
- 104.
- Colas J-F, Schoenwolf GC. Assessing the contributions of gene products to the form-shaping events of neurulation: A transgenic approach in chick. Genesis. 2003;37:64–75. [PubMed: 14595842]
- 105.
- De BellardME, Rao Y, Bronner-Fraser M. Dual function of slit2 in repulsion and enhanced migration of trunk, but not vagal, neural crest cells. J Cell Biol. 2003;162:269–279. [PMC free article: PMC2172792] [PubMed: 12876276]
- 106.
- Augsburger A, Schuchardt A, Hoskins S. et al. BMPs as mediators of roof plate repulsion of commissural neurons. Neuron. 1999;24(1):127–141. [PubMed: 10677032]
- 107.
- Henion PD, Weston JA. Timing and pattern of cell fate restrictions in the neural crest lineage. Development. 1997;1997:4351–4359. [PubMed: 9334283]
- 108.
- Barembaum M, Moreno T, LaBonne C. et al. Noelin-1 is a secreted glycoprotein involved in generation of the neural crest. Nat Cell Biol. 2000;2:219–2225. [PubMed: 10783240]
- 109.
- Moreno TA, Bronner-Fraser M. Neural expression of mouse Noelin1/2 and comparion with other vertebrates. Mech Dev. 2002;119:121–125. [PubMed: 12385760]
- 110.
- Monier-Gavelle F, Duband J-L. Cross-talk between adhesion molecules: Control of N-cadherin activity by intracellular signals elicited by β1 and β3 integrins in migrating neural crest cells. J Cell Biol. 1997;137:1663–1681. [PMC free article: PMC2137812] [PubMed: 9199179]
- 111.
- Barth AIM, Näke IS, Nelson WJ. Cadherins, catenins and APC protein: Interplay between cytoskeletal complexes and signaling pathways. Curr Opin Cell Biol. 1997;9:683–690. [PubMed: 9330872]
- 112.
- Huber O, Bierkamp C, Kemler R. Cadherins and catenins in development. Curr Opin Cell Biol. 1996;8:685–691. [PubMed: 8939663]
- 113.
- Miller JR, Moon RT. Signal transduction through β-catenin and specification of cell fate during embryogenesis. Genes Develop. 1996;10:2527–2539. [PubMed: 8895655]
- 114.
- Orsulic S, Huber O, Aberle H. et al. E-cadherin binding prevents beta-catenin nuclear localization and β-catenin/Lef-1-mediated transactivation. J Cell Sci. 1999;112:1237–1245. [PubMed: 10085258]
- 115.
- Vega S, Morales AV, Ocana OH. et al. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. 2004;18(10):1131–1143. [PMC free article: PMC415638] [PubMed: 15155580]
- 116.
- Frisch SM, Screaton RA. Anoikis mechanisms. Curr Opin Cell Biol. 2001;13(5):555–562. [PubMed: 11544023]
- 117.
- Kalcheim C. Basic fibroblast growth factor stimulates survival of nonneuronal cells developing from trunk neural crest. Dev Biol. 1989;134:1–10. [PubMed: 2659407]
- 118.
- Kalcheim C, Barde Y-A, Thoenen H. et al. In vivo effect of brain-derived neurotrophic factor on the survival of developing dorsal root ganglion cells. EMBO J. 1987;6:2871–2873. [PMC free article: PMC553720] [PubMed: 3691474]
- 119.
- Perris R, Perissinotto D. Role of the extracellular matrix during neural crest cell migration. Mech Develop. 2000;95:3–21. [PubMed: 10906446]
- 120.
- Delannet M, Martin F, Bossy B. et al. Specific roles of the αVβ1, αVβ3 and αVβ5 integrins in avian neural crest cell adhesion and migration on vitronectin. Development. 1994;120:2687–2702. [PMC free article: PMC2710119] [PubMed: 7525179]
- 121.
- Desban N, Duband J-L. Avian neural crest cell migration on laminin: Interaction of the a1β1 integrin with distinct laminin-1 domains mediates different adhesive responses. J Cell Sci. 1997;110:2729–2744. [PubMed: 9427390]
- 122.
- Newgreen DF, Gibbins IL, Sauter J. et al. Ultrastructural and tissue-culture studies on the role of fibronectin, collagen and glycosaminoglycans in the migration of neural crest cells in the fowl embryo. Cell Tiss Res. 1982;221:521–549. [PubMed: 7034954]
- 123.
- Perris R, Paulsson M, Bronner-Fraser M. Molecular mechanisms of avian neural crest cell migration on fibronectin and laminin. Dev Biol. 1989;136:222–239. [PubMed: 2509262]
- 124.
- Bronner-Fraser M. An antibody to a receptor for fibronectin and laminin perturbs cranial neural crest development in vivo. Dev Biol. 1986;117:528–536. [PubMed: 2944780]
- 125.
- Boucaut J-C, Darribè T, Poole TJ. et al. Biological active synthetic peptides as probes of embryonic development: A competitive peptide inhibitor of fibronectin function inhibits gastrulation in amphibian embryos and neural crest cell migration in avian embryos. J Cell Biol. 1984;99:1822–1830. [PMC free article: PMC2113361] [PubMed: 6490722]
- 126.
- Kil SH, Lallier T, Bronner-Fraser M. Inhibition of cranial neural crest adhesion in vitro and migration in vivo using integrin antisens oligonucleotides. Dev Biol. 1996;179:91–101. [PubMed: 8873756]
- 127.
- Tucker RP. Abnormal neural crest cell migration after the in vivo knockdown of tenacin-C expression with morpholino antisense oligonucleotides. Dev Dyn. 2001;222:115–119. [PubMed: 11507773]
- 128.
- Hynes RO. Integrins: Versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. [PubMed: 1555235]
- 129.
- Hynes R, Zhao Q. The evolution of cell adhesion. J Cell Biol. 2000;150:F89–F95. [PubMed: 10908592]
- 130.
- Geiger B, Bershadsky A, Pankov R. et al. Transmembrane extracellular matrix-cytoskeleton crosstalk. Nat Rev Mol Cell Biol. 2001;2:793–805. [PubMed: 11715046]
- 131.
- DeMali KA, Wennerberg K, Burridge K. Integrin signaling to the actin cytoskeleton. Curr Opin Cell Biol. 2003;15:572–582. [PubMed: 14519392]
- 132.
- Giancotti FG, Tarone G. Positional control of cell fate through joint integrin/receptor protein kinase signaling. Annu Rev Cell Dev Biol. 2003;19:173–206. [PubMed: 14570568]
- 133.
- Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285:1028–1032. [PubMed: 10446041]
- 134.
- ffrench-Constant C, Colognato H. Integrins: Versatile integrators of extracellular signals. Trends Cell Biol. 2004;14:678–686. [PubMed: 15564044]
- 135.
- Testaz S, Delannet M, Duband J-L. Adhesion and migration of avian neural crest cells on fibronectin require the cooperating activites of multiple integrins of the b1 and b3 families. J Cell Sci. 1999;112:4715–4728. [PubMed: 10574719]
- 136.
- Testaz S, Duband J-L. Central role of the α4β1 integrin in the coordination of avian truncal neural crest adhesion, migration, and survival. Dev Dyn. 2001;222:127–140. [PubMed: 11668592]
- 137.
- Yang JT, Rayburn H, Hynes RO. Embryonic mesodermal defects in α5 integrin-deficient mice. Development. 1993;119:1093–1105. [PubMed: 7508365]
- 138.
- Haack H, Hynes RO. Integrin receptors are required for cell survival and proliferation during development of the peripheral glial lineage. Dev Biol. 2001;233:38–55. [PubMed: 11319856]
- 139.
- Crump JG, Swartz ME, Kimmel CB. An integrin-dependent role of pouch endoderm in hyoid cartilage development. PloS Biol. 2004;2:1432–1445. [PMC free article: PMC479042] [PubMed: 15269787]
- 140.
- Duband J-L, Belkin AM, Syfrig J. et al. Expression of a1 integrin, a laminin-collagen receptor, during myogenesis and neurogenesis in the avian embryo. Development. 1992;116:585–600. [PubMed: 1337741]
- 141.
- Pietri T, Eder O, Breau MA. et al. Conditional β1-integrin gene deletion in neural crest cells causes severe developmental alterations of the peripheral nervous system. Development. 2004;131:3871–3883. [PubMed: 15253938]
- 142.
- Krull CE. Segmental organization of neural crest migration. Mech Dev. 2001;105:37–45. [PubMed: 11429280]
- 143.
- Trainor PA, Krumlauf R. Hox genes, neural crest cells and branchial arch patterning. Curr Opin Cell Biol. 2001;13(6):698–705. [PubMed: 11698185]
- 144.
- Trainor PA, Sobieszczuk D, Wilkinson D. et al. Signalling between the hindbrain and paraxial tissues dictates neural crest migration pathways. Development. 2002;129(2):433–442. [PubMed: 11807035]
- 145.
- Santiago A, Erickson CA. Ephrin-B ligands play a dual role in the control of neural crest cell migration. Development. 2002;129(15):3621–3632. [PubMed: 12117812]
- 146.
- Tosney KW. Long-distance cue from emerging dermis stimulates neural crest melanoblast migration. Dev Dyn. 2004;229:99–108. [PubMed: 14699581]
- 147.
- Young HM, Hearn CJ, Farlie PG. et al. GDNF is a chemoattractant for enteric neural cells. Dev Biol. 2001;229(2):503–516. [PubMed: 11150245]
- 148.
- Yan H, Bergner AJ, Enomoto H. et al. Neural cells in the esophagus respond to glial cell line-derived neurotrophic factor and neurturin, and are RET-dependent. Dev Biol. 2004;272(1):118–133. [PubMed: 15242795]
- 149.
- Rovasio RA, Delouvée A, Yamada KM. et al. Neural crest cell migration: Requirements for exogenous fibronectin and high cell density. J Cell Biol. 1983;96:462–473. [PMC free article: PMC2112280] [PubMed: 6833366]
- 150.
- Thomas LA, Yamada KM. Contact stimulation of cell migration. J Cell Sci. 1992;103:1211–1214. [PubMed: 1487497]
- 151.
- Abercrombie M, Heaysman JEM. Observations on the social behaviour of cells in tissue culture. I. Speed of movement of chick heart fibroblasts in relation to their mutual contacts. Exp Cell Res. 1953;5:111–131. [PubMed: 13083622]
- 152.
- Young HM, Bergner AJ, Anderson RB. et al. Dynamics of neural crest-derived cell migration in the embryonic mouse gut. Dev Biol. 2004;270(2):455–473. [PubMed: 15183726]
- 153.
- Teddy JM, Kulesa PM. In vivo evidence for short- and long-range cell communication in cranial neural crest cells. Development. 2004;131(24):6141–6151. [PubMed: 15548586]
- 154.
- Kasemeier-Kulesa JC, Kulesa PM, Lefcort F. Imaging neural crest cell dynamics during formation of dorsal root ganglia and sympathetic ganglia. Development. 2005;132(2):235–245. [PubMed: 15590743]
- 155.
- Dufour S, Beauvais-Jouneau A, Delouvée A. et al. Differential function of N-cadherin and cadherin-7 in the control of embryonic cell motility. J Cell Biol. 1999;146:501–516. [PMC free article: PMC3206574] [PubMed: 10427101]
- 156.
- Xu X, Li WEI, Huang GY. et al. Modulation of mouse neural crest motility by N-cadherin and connexin 43 gap junction. J Cell Biol. 2001;154:217–229. [PMC free article: PMC2196865] [PubMed: 11449002]
- 157.
- Huang GY, Cooper ES, Waldo K. et al. Gap junction mediated cell-cell communication modulates mouse neural crest migration. J Cell Biol. 1998;143:1725–1734. [PMC free article: PMC2132985] [PubMed: 9852163]
- 158.
- Kawasaki T, Bekku Y, Suto F. et al. Requirement of neuropilin 1-mediated Sema3A signals in patterning of the sympathetic nervous system. Development. 2002;129(3):671–680. [PubMed: 11830568]
- 159.
- Pettway Z, Guillory G, Bronner-Fraser M. Absence of neural crest cells from the region surrounding implanted notochords in situ. Dev Biol. 1990;142:335–345. [PubMed: 2175277]
- 160.
- Fedtsova N, Perris R, Turner EE. Sonic hedgehog regulates the position of the trigeminal ganglia. Dev Biol. 2003;261(2):456–469. [PubMed: 14499653]
- 161.
- Ungos JM, Karlstrom RO, Raible DW. Hedgehog signaling is directly required for the development of zebrafish dorsal root ganglia neurons. Development. 2003;130(22):5351–5362. [PubMed: 13129844]
- 162.
- Fu M, Lui VC, Sham MH. et al. Sonic hedgehog regulates the proliferation, differentiation, and migration of enteric neural crest cells in gut. J Cell Biol. 2004;166(5):673–684. [PMC free article: PMC2172437] [PubMed: 15337776]
- Introduction
- Neural Crest Delamination: Their Declaration of Independence
- The Snail Family of Zn-Finger Transcription Factors
- The Winged Helix-Forkhead Transcription Factor Foxd-3
- The Sox-E Subgroup of HMG-Box Transcription Factors
- Other Families of Transcription Factors
- Cooperative Activity of Transcription Factors during Neural Crest Cell EMT
- Proliferation of Precursor Cells
- Asymmetric Cell Division
- Lateral Inhibition
- Other Possible Means
- Neural Crest Migration: How to Survive, Move and Develop in a Hostile Environment
- Conclusion
- Acknowledgments
- References
- Neural Crest Delamination and Migration: Integrating Regulations of Cell Interac...Neural Crest Delamination and Migration: Integrating Regulations of Cell Interactions, Locomotion, Survival and Fate - Madame Curie Bioscience Database
- Dynamic Connections of Nuclear Envelope Proteins to Chromatin and the Nuclear Ma...Dynamic Connections of Nuclear Envelope Proteins to Chromatin and the Nuclear Matrix - Madame Curie Bioscience Database
- Protein Kinase A - Madame Curie Bioscience DatabaseProtein Kinase A - Madame Curie Bioscience Database
- Opioid Receptors on Peripheral Sensory Neurons - Madame Curie Bioscience Databas...Opioid Receptors on Peripheral Sensory Neurons - Madame Curie Bioscience Database
- Nucleotide Exchange Factors for Hsp70 Molecular Chaperones - Madame Curie Biosci...Nucleotide Exchange Factors for Hsp70 Molecular Chaperones - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...