

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Introduction
Severe infection is frequently accompanied by disturbances in the hemostatic balance. The most severe manifestation of these disturbances is known as the clinical syndrome of disseminated intravascular coagulation (DIC). DIC is characterized by extensive activation of the coagulation system, amplified by inhibition of anticoagulant pathways, such as the protein C-protein S-thrombomodulin system, and the fibrinolytic system. In addition, DIC can be associated with an inhibition of the fibrinolytic system. Cytokines are small proteins produced by many different cell types. They are important mediators of inflammatory processes at local tissue level. However, during overwhelming sepsis, abundant cytokine production can result in organ damage such as observed during the “systemic inflammatory response syndrome” induced by sepsis. In addition, cytokines have been found to influence both procoagulant and anticoagulant pathways. In this brief overview we will outline current knowledge on how cytokines may influence the coagulation system and the fibrinolytic system during DIC and sepsis.
Production of Cytokines
The cytokine network can be arbitrarily divided in three parts, i.e., pro-inflammatory cytokines, anti-inflammatory cytokines and soluble inhibitors of pro-inflammatory cytokines.1,2 Pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF), interleukin (IL)-1, IL-12 and interferon-γ (IFN-γ), facilitate inflammation. Anti-inflammatory cytokines, among which IL-4, IL-10 and IL-13, can inhibit the production of pro-inflammatory cytokines and, in addition, can exert a number of other inhibitory effects on inflammatory processes. The activity of pro-inflammatory cytokines can be further modulated by naturally occurring inhibitors, such as soluble TNF receptors, soluble IL-1 receptors, and IL-1 receptor antagonist (IL-1ra).
Many different investigations have documented elevated concentrations of cytokines in the circulation of patients with sepsis.3 Pro-inflammatory cytokines can usually only be detected in a subset of patients, while anti-inflammatory cytokines and soluble inhibitors can be found in the vast majority of patients with sepsis and even in healthy individuals. Pro-inflammatory cytokines detected in the circulation likely are derived primarily from the site of the infection. This notion is supported by findings in patients with unilateral community-acquired pneumonia, who had much higher concentrations of pro-inflammatory cytokines, including TNF and IL-1b, in bronchoalveolar lavage fluid obtained from the infected lung than in lavage fluid derived from the contralateral lung or in serum.4 Similar observations have been made in patients with peritonitis, in whom cytokine concentrations were higher in fluid obtained from the peritoneal cavity than in serum.5 In contrast to pro-inflammatory cytokines, anti-inflammatory cytokines and cytokine antagonists, especially IL-10, soluble TNF receptors and the type II soluble IL-1 receptor, can usually be detected in circulating blood. In fact, it has been hypothesized that during sepsis this anti-inflammatory part of the cytokine family may be produced at least in part from sites of the body that are distant from the site of infection.6 In comparison with other cytokines, IL-6 has been found in the circulation of septic patients most consistently, although the actual levels of IL-6 show considerable variation.3 IL-10, an important anti-inflammatory cytokine in sepsis, was detectable in 80–100 percent of sepsis patients, whereas IL-10 usually is not detectable in plasma from normal individuals.7,8 Furthermore, the circulating concentrations of soluble TNF receptors, soluble IL-1 receptor type II and IL-1ra are elevated in patients with sepsis, likely reflecting an attempt of the host to limit systemic TNF and IL-1 toxicity.8
The kinetics of cytokine release in vivo has been investigated thoroughly in healthy human volunteers injected with a low dose of lipopolysaccharide (LPS; the toxic moiety of the outer membrane of gram-negative bacteria) and in baboons infused with a lethal dose of live Escherichia coli. 2 The release of TNF follows a highly similar pattern in both experimental models. In both endotoxemic volunteers and bacteremic baboons, TNF is the first cytokine that becomes detectable in the circulation, reaching peak concentrations after 90 minutes.9–11 In the lethally infected baboons, circulating TNF levels are orders of magnitude higher, however, than in healthy humans treated with LPS. IL-6, IL-10, IL-1ra and soluble TNF receptors can be detected in the circulation shortly after the first release of TNF during experimental sepsis and endotoxemia, while the detection of IL-1β, IL-12 and IFN-γ, also secreted shortly after TNF, is confined to models of severe sepsis.9–16 It should be noted that the early TNF production plays a role in the subsequent induction of both pro- and anti-inflammatory mediators, as indicated by the finding that anti-TNF treatment attenuated the increase in plasma or serum concentrations of IL-1β, IL-1ra, IL-6, IL-10 and soluble TNF receptors after administration of endotoxin and/or live bacteria to humans or nonhuman primates.9,10,12,14,16
Role of Cytokines in Organ Toxicity in Systemic Infection and Inflammation
Ample evidence exists that the systemic action of pro-inflammatory cytokines can damage tissues and can diminish organ function. Recombinant TNF or IL-1 reproduced many of the clinical and metabolic features of sepsis when injected into laboratory animals.17,18 Concurrent injection of both cytokines was associated with a synergistic systemic toxic effect, substantiating the tight interactions between the various members of the cytokine network.18 Repeated administration of IL-12 can also cause significant toxicity, although not as acutely as TNF and IL-1.19 Although circulating IL-6 concentrations demonstrate a strong positive correlation with an adverse clinical outcome in patients with sepsis, endogenously produced IL-6 does not directly contribute to tissue injury and organ failure during severe sepsis. This conclusion is based on findings that intravenous infusion of recombinant IL-6 in humans only induced relatively mild clinical symptoms, such as chills and fever, and was by far not as toxic as intravenous TNF or IL-1,20,21 and that high doses of IL-6 were not able to induce hypotension or other hemodynamic effects in dogs.22
Several cytokines have been implicated in the development of fatal organ failure in sepsis. Elimination of TNF or IL-1 by administration of anti-TNF antibodies and recombinant IL-1ra respectively was highly protective against lethality when given before intravenous infusion of a LD100 dose of LPS or live bacteria.23–26 In healthy humans and chimpanzees, neutralization of endogenous TNF activity attenuated many inflammatory responses induced by low dose LPS, including cytokine release and activation of neutrophils.12,13 IL-6 is not essential for the development of tissue damage during severe endotoxemia, considering that IL-6 gene deficient mice displayed a sensitivity to endotoxin effects that was similar to that of normal wild type mice.27 By contrast, anti-IFN-γ or anti-IL-12 therapy markedly reduced lethality induced by LPS in mice.28,29 Similarly, mice that lack functional IFN-γ were relatively resistant to the toxic effects of LPS.30 The anti-inflammatory cytokine IL-10 has a protective role during endotoxemia, as documented by experiments in IL-10 gene deficient mice that showed an increased susceptibility to LPS-induced death,31 and studies in normal mice that could be protected against LPS-induced lethality by administration of recombinant IL-10.32 Also, recombinant IL-13 could protect mice from death induced by high doses of LPS.33
Effect of Cytokines on Tissue Factor and Coagulation Activation
DIC is associated with a massive activation of the coagulation system, ultimately leading to fibrin formation. Several mechanisms have been implicated in the pathogenesis of coagulation activation during DIC.34 Tissue factor is generally accepted as the pivotal mediator of initiation of coagulation activation in the setting of severe sepsis and endotoxemia. In DIC, activation of the coagulation system is accompanied by concurrent inhibition of anticoagulant mechanisms, including the availability of antithrombin, a reduced function of the protein C-protein S- thrombomodulin system and impairment of the fibrinolytic response. Cytokines can influence these pathways at multiple levels.
Tissue factor is a glycoprotein expressed on a number of cell types upon stimulation with endotoxin (and other bacterial products), including monocytes and vascular endothelial cells. Tissue factor binds and activates factor VII, leading to the formation of tissue factor-VIIa complexes, which can activate clotting factors X and IX. The activity of the factor VIIa-tissue factor complex can be inhibited by tissue factor pathway inhibitor (TFPI). TFPI can bind factor Xa, and thereafter inhibit factor VIIa-tissue factor activity by forming a quaternary Xa-TFPI-VIIa complex. Several lines of evidence point to a crucial role of tissue factor in DIC. Enhanced expression of tissue factor at the surface of circulating monocytes has been reported in patients and nonhuman primates with severe sepsis,35,36 and inhibition of the activation of the tissue factor-VIIa pathway by either antibodies directed against tissue factor or factor VII/VIIa, active site inhibited factor VIIa (Dansyl-Glu-Gly-Arg chloromethylketone or DEGR-VIIa) or TFPI, prevented the coagulation activation in endotoxemic chimpanzees and septic baboons.37–43
A number of cytokines have been found to influence tissue factor expression on endothelial cells and/or monocytes/macrophages in vitro. Cytokines that increase tissue factor expression are TNF, IL-1α, IL-1β, IL-6, IL-8, leukemia inhibitory factor, IFN-γ and monocyte chemoattractant protein 1 (MCP-1).44–61 The anti-inflammatory cytokines transforming growth factor (TGF)-β, IL-4, IL-10 and IL-13 diminished tissue factor expression induced by various stimuli including LPS, TNF, IL-1, MCP-1 and C-reactive protein.48–51,55–57,62,63 In cocultures of endothelial cells and monocytes, LPS strongly upregulated endothelial cell tissue factor activity, which was predominantly mediated by endogenous IL-1β.64 Indeed, an anti-IL-1β antibody almost completely inhibited the enhanced endothelial cell expression of tissue factor activity while anti-IL-1α or anti-TNF antibodies were without a significant effect.64 In another study, activation of endothelial cells by either LPS, TNF or IL-1 lead to adhesion of monocytes and subsequent induction of monocyte tissue factor.65 Interestingly, lymphocytes also may alter tissue factor expression on monocytes, as indicated by experiments in which mitogen stimulation enabled both unfractionated T cells and their CD4 and CD8 subsets to promote tissue factor activity on monocytes.55 After stimulation, T cell clones with a T helper 1 (Th1) cytokine profile induced monocytic tissue factor production, while Th2 clones were unable to do so. Optimal tissue factor synthesis by monocytes was found to require both cell-cell contact with activated Th1 cells and Th1 cytokines, in particular IFN-γ, while Th2 cytokines (i.e., IL-4, IL-10 and IL-13) were inhibitory.55 Together, these studies point to cross-talk between monocytes, endothelial cells and lymphocytes, regulating tissue factor expression by mechanisms that at least in part are mediated by direct cell contact and an interplay between different cytokines.
The role of cytokines in coagulation activation in vivo has been the subject of a number of investigations. These studies have identified TNF, IL-1, IL-6, IL-12 and IL-2 as cytokines that can induce thrombin generation in humans and nonhuman primates. A single intravenous bolus injection of recombinant TNF into normal humans induced an early activation of factor X, peaking after 45 minutes, followed by a more gradual and sustained activation of prothrombin, as reflected by elevated plasma levels of the prothrombin activation peptide F1+2 for 6–12 hours (Fig. 1).66 Activation of the intrinsic route could not be detected using a series of sensitive and specific assays, suggesting that tissue factor was important for the procoagulant response to TNF.66 Likewise, cancer patients infused with relatively high doses of recombinant TNF demonstrated coagulation activation, as indicated by rises in the plasma concentrations of F1+2 and fibrinopeptide A.67 TNF can bind to two distinct transmembrane receptors, the p55 (or type I) and p75 (or type II) TNF receptors. In baboons, only a TNF mutant protein with exclusive affinity for the p55 TNF receptor and wild type TNF, but not a p75 activating TNF mutant, elicited coagulation activation.68 In vitro, both TNF receptor subtypes may contribute to the induction of tissue factor on endothelial cells, i.e., antagonistic antibodies against either the p55 or p75 receptor reduced TNF-induced tissue factor expression on human endothelial cells, although blockade of the p55 receptor had the most potent inhibiting effect.53,54 Further, agonistic antibodies against either the p55 or p75 receptor were able to enhance tissue factor expression.54 In contrast, in a human ex vivo native blood flow system, tissue factor mediated fibrin depositions on endothelial cells could only be induced by a TNF mutant with exclusive affinity for the p55 TNF receptor, not by a p75 binding TNF mutant.52 Together, it seems reasonable to conclude that the p55 TNF receptor is the predominant receptor mediating TNF-induced tissue factor expression by endothelial cells in vitro, and that this TNF receptor type also is responsible for the procoagulant activity of this cytokine in vivo.
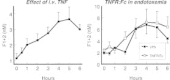
Although TNF can activate coagulation in vivo, and can induce tissue factor in vitro, it is unlikely that this cytokine plays a significant role in activation of the coagulation system during endotoxemia or gram-negative sepsis. Complete elimination of endogenous TNF activity in healthy humans or chimpanzees injected with LPS, by infusion of a recombinant TNF receptor IgG fusion protein or a neutralizing TNF antibody, did not influence coagulation activation (Fig. 1).12,13,69 In accordance, baboons infused with an otherwise lethal dose of Escherichia coli were protected from lethality by treatment with an anti-TNF antibody in spite of the fact that the antibody had little or no effect on the occurrence of DIC.24Together these data suggest that (1) although administration of exogenous TNF is able to elicit coagulation activation, endogenously produced TNF is not required for a procoagulant response during endotoxemia and sepsis, and (2) that mortality and activation of coagulation are not necessarily linked phenomena.
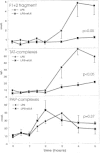
IL-1α administration to baboons, at a dose that caused a transient shock-like state, also resulted in activation of the common pathway of the coagulation system within the first hours after the infusion.70 In addition, infusion of recombinant human IL-1ra inhibited coagulation activation in baboons with lethal bacteremia, suggesting that endogenous IL-1 is important for the procoagulant response in this model.70Furthermore, experimental data indicate that IL-6 may also play a role in the initiation of the coagulation system in vivo. In patients with renal cell carcinoma, intravenous administration of recombinant IL-6 was associated with rises in the plasma concentrations of F1+2 and thrombin-antithrombin III (TAT) complexes, reaching maximal values at the end of the 4-hour infusion.71 In chimpanzees exposed to low dose LPS, treatment with an anti-IL-6 antibody prevented coagulation activation (Fig. 2).72 A single injection of recombinant human IL-12 caused sustained activation of the coagulation system in chimpanzees that reached a plateau between 8 and 48 hours after the injection.73 IL-2 infusion into cancer patients resulted in a more rapid procoagulant response, as reflected by a transient increase in the plasma concentrations of TAT complexes, peaking after 4 hours.74 The anticoagulant potential of IL-10 in vitro, by virtue of its inhibitory effect on tissue factor expression (see above), has been confirmed in humans injected with LPS.75 Indeed, recombinant IL-10 (25 μg/kg) inhibited the procoagulant response induced by low dose Escherichia coli LPS (4 ng/kg) in healthy subjects.75However, infusion of recombinant IL-10 at a much higher dose (500 μg/kg) did not influence activation of the coagulation system in baboons challenged with high dose Salmonella LPS (500 μg/kg), in spite of a strong inhibition of proinflammatory cytokine release.76 Hence, further studies are required to define the potency of exogenous IL-10 and the role of endogenous IL-10 in coagulation activation during sepsis.
In summary, tissue factor plays a central role in the initiation of coagulation activation in in vivo models of endotoxemia and bacteremia. Many cytokines can influence tissue factor expression on monocytes and endothelial cells in vitro, and direct cell-cell interactions, mediated by cytokines, may be important for the regulation of tissue factor expression at sites of inflammation. TNF, IL-1, IL-6, IL-12 and IL-2 are cytokines that have been found capable of activating the coagulation system in vivo, whereas IL-10 can act as an anticoagulant cytokine in vivo.
Effect of Cytokines on the Protein C-Protein S-Thrombomodulin System
Besides activation of coagulation mediated by tissue factor, impairment of anticoagulant mechanisms seems to play a role in the amplification of coagulation in sepsis. One such anticoagulant pathway is the protein C-protein S-thrombomodulin system.77 Thrombin mediates the activation of protein C after binding to thrombomodulin, a receptor at the surface of endothelial cells. Protein S serves as a cofactor for activated protein C (APC), which proteolytically inactivates the coagulation factors Va and VIIIa. Protein C activation by the thrombin-thrombomodulin complex on the cell surface is augmented by protein C binding to the endothelial cell protein C receptor (EPCR).78The impairment of the protein C system during sepsis is the result of increased consumption of protein S and protein C, and decreased activation of protein C by downregulation of thrombomodulin on endothelial cells. Furthermore, protein S can be bound by the acute phase response protein C4b-binding protein, thereby reducing the biological availability of this important cofactor for protein C. EPCR can be shed from vascular endothelium, and soluble EPCR can block the APC anticoagulant activity.79
The availability of APC is important for the prevention of excessive fibrin formation. Infusion of APC into septic baboons prevented hypercoagulability and death. Conversely, inhibition of activation of endogenous protein C by a monoclonal antibody exacerbated the response to a lethal Escherichia coli infusion, and converted a sublethal model produced by a LD10 dose of Escherichia coli into a severe shock response associated with DIC and death.80Similarly, infusion of an anti-EPCR monoclonal antibody, reducing the efficiency by which protein C can be activated by the thrombin-thrombomodulin complex, also was associated with an exacerbation of a sublethal Escherichia coli infection to lethal sepsis with DIC in baboons,81 and reduction of the bioavailability of protein S by administration of C4b binding protein resulted in similar changes.82
Cytokines can influence the protein C-protein S-thrombomodulin pathway at various levels, overall impairing the anticoagulant function of this physiological system. TNF and IL-1 diminished thrombomodulin activity and gene expression in endothelial cell cultures.44,46,83–85 TNF did not affect thrombomodulin expression by THP-1 monocytic cells84 or keratinocytes.8TNF has been reported to downregulate EPCR in endothelial cells,86 whereas IL-1β stimulated the release of EPCR from cultured endothelial cells in vitro, reducing EPCR expression sufficiently to decrease the rate of protein C activation.87 Little is known about the effects of cytokines on the protein C-protein S-thrombomodulin system in vivo. TNF may be involved in the release of soluble thrombomodulin into the circulation of baboons with severe Escherichia coli bacteremia, since this response could be attenuated by a neutralizing anti-TNF antibody.88 Mouse studies indicate that TNF and IL-1 may enhance the procoagulant response to sepsis by downregulation of protein C synthesis at tissue level, i.e., injection of recombinant TNF into mice caused decreased protein C mRNA expression in liver, kidney and testis; IL-1 administration was assocociated with reduced protein C mRNA expression in liver and testis, but not in kidney.89
Importantly, APC has been demonstrated to exert a number of anti-inflammatory activities that are not directly linked to its anticoagulant potential. These effects are discussed below.
Effect of Cytokines on the Fibrinolytic System
Plasmin is the active end-enzyme of the fibrinolytic system, which is generated from plasminogen by an action of tissue type and urokinase type plasminogen activators (tPA and uPA). The main inhibitor of both tPA and uPA is plasminogen activator inhibitor type I (PAI-1), whereas plasmin is inhibited by α2-antiplasmin leading to the formation of plasmin-α2-antiplasmin (PAP) complexes.
In models of endotoxemia and bacteremia, both the coagulation system and the fibrinolytic system become activated. Originally, it was believed that the fibrinolytic response represented a mere reaction to the formation of thrombin and fibrin. This supposition has now been proven to be incorrect. Several lines of evidence support this conclusion: (1) administration of LPS or E. coli to humans and nonhuman primates was associated with an early and transient activation of the fibrinolytic system, reflected by a brisk rise in the plasma concentrations of tPA, which preceeded the activation of the coagulation system,12,13,38,39,43,69,72,75,76,90 and (2) prevention of coagulation activation by several anti-tissue factor/VIIa strategies did not affect activation of fibrinolysis during human and primate endotoxemia.38,39,43 Hence, the fibrinolytic response should be viewed as an anticoagulant pathway that at least in part is regulated by mediators that are not directly linked to the clotting cascade.
Cytokines play an important role in the regulation of fibrinolysis. In vitro, both TNF and IL-1 exerted anti-fibrinolytic effects on vascular endothelium, primarily by stimulating the production and release of PAI-1. Furthermore, TNF and IL-1 may diminish the release of tPA by endothelial cells.50,91–96 Lymphotoxin, IL-2 and TGF-β also enhanced endothelial cell PAI-1 release in vitro.92,93,96 TNF, lymphotoxin, IL-1, IL-2, IL-4 increased uPA release by endothelial cells,93,95–98 whereas IFN-γ inhibited TNF-induced uPA release.95,97 Interestingly, IFNg also inhibited PAI-1 release stimulated by LPS or thrombin, but not PAI-1 secretion triggered by TNF or IL-1.94 Cytokines can also modulate the production and/or activity of fibrinolytic mediators by cell types other than endothelial cells, including hepatocytes, monocytes, fibroblasts and keratinocytes.99–102
Administration of LPS or E. coli results in a fibrinolytic response that is characterized by an early release of tPA antigen and activity, followed by a rise in PAI-1 levels, which is associated with an abrupt decline in plasminogen activator activity in plasma.12,13,38,39,43,69,72,75,76,90,103 TNF appears to be of pivotal importance for fibrinolytic activation in vivo. Infusion of recombinant TNF in human volunteers induced initial activation and subsequent inhibition of plasmin generation similar to that observed during bacteremia or endotoxemia.104 The time-difference between LPS-induced changes in coagulation as compared to the TNF-induced kinetics coincided with the 90-minutes delay in the appearance of TNF after the injection of LPS. In patients with cancer, TNF was also capable of eliciting the release of tPA, uPA and PAI-1 into the circulation.105,106 Importantly, neutralization of endogenous TNF activity in healthy humans or chimpanzees injected with LPS resulted in a complete prevention of the fibrinolytic response, affecting both tPA and PAI-1 release.12,13,69 Anti-TNF treatment attenuated the LPS-induced increase in plasma PAI-1 levels in mice, concurrently reducing PAI-1 mRNA expression in the liver.107
IL-1α infusion in baboons was associated with an early activation of the fibrinolytic system, with the characteristic subsequent rises in the plasma concentrations of tPA and PAI-1 also registered after administration of LPS, E. coli or TNF.70 The role of endogenous IL-1 in the fibrinolytic response to sepsis has been examined by administration of recombinant IL-1ra, blocking both IL-1α and IL-1β effects, in baboons with lethal bacteremia. In these animals, both tPA and PAI-1 release was diminished by IL-1ra treatment; the plasma levels of PAP complexes were not influenced by IL-1ra, presumably because PAP levels were already maximal (after 1–1.5 hours) before IL-1 appears in the circulation in this model.70 IL-1β gene deficient mice had increases in plasma PAI-1 and liver PAI-1 mRNA levels upon LPS injection that were indistinguishable from the rises measured in normal wild type mice, suggesting that IL-1β does not play an important role in this LPS effect.108 However, PAI-1 production triggered by administration of turpentine, giving rise to a more prolonged sterile inflammation, was reduced in IL-1β gene deficient mice.108 IL-2 stimulated tPA and PAI-1 release in cancer patients.74 IL-12 injection into chimpanzees induced a delayed fibrinolytic response, characterized by rises in tPA and PAP complexes in plasma from 24 hours postinjection and onward.73 Remarkably, PAI-1 levels did not increase after IL-12 administration.73 IL-6 did not influence fibrinolysis when infused into patients with renal cell carcinoma,71 and anti-IL-6 treatment did not alter the fibrinolytic response to LPS in chimpanzees (Fig. 2).72 Finally, in healthy humans exposed to LPS, IL-10 attenuated the systemic release of tPA, PAI-1, PAP complexes and D-dimer, indicating that this anti-inflammatory cytokine not only negatively controls the procoagulant response to intravenous LPS, but also the activation of the fibrinolytic system.75
In summary, although a number of cytokines can influence fibrinolysis in vitro and in vivo, the role of individual cytokines in the fibrinolytic response to sepsis and endotoxemia remains to be established. It is beyond doubt that TNF plays a pivotal role in the activation of the fibrinolytic system during endotoxemia. Conclusive evidence is available that, at least in experimental endotoxemia, fibrinolysis is not triggered by the formation of thrombin or fibrin, but proceeds as an inflammatory reaction not linked to activation of coagulation.
Interplay Between Cytokines and Mediators of Hemostasis
Mediators traditionally implicated in the regulation of coagulation and fibrinolysis can influence other inflammatory pathways, including the cytokine network. End products of the coagulation system can trigger inflammatory responses in vitro. Coagulation of whole blood in vitro resulted in expression of IL-1β mRNA in blood cells.109 In accordance, thrombin significantly enhanced LPS-induced IL-1 activity produced by guinea pig macrophages.110 Prothrombin and factor Xa also increased IL-1 release although not as potent as thrombin.110 Similarly, coagulating blood produced IL-8 in vitro.111,112 Addition of LPS to coagulating blood resulted in a synergistic increase in IL-8 release, which could be inhibited by the thrombin inhibitor hirudin or TFPI. Monocytes were responsible for IL-8 production by coagulating blood, and these cell types could also be stimulated directly with a-thrombin.111,112 Thrombin also was able to induce IL-8 expression in the monocytic cell line U937, an effect that could be mimicked by thrombin receptor-activating peptide, indicating that this response was signaled via the specific thrombin receptor.113 Factor Xa, thrombin and fibrin can also activate endothelial cells, eliciting the synthesis of IL-6 and/or IL-8.114–117 Furthermore, thrombin increased mRNA levels of IL-8, MCP-1, E-selectin, and PAI-1 in cultured endothelial cells, and potentiated TNF-induced E-selectin expression.118 Together, these data indicate that end products of the clotting cascade can interfere with cytokine production (and other inflammatory reactions) by different cell types in vitro.
Mediators of anticoagulant pathways can also influence inflammation in vitro. APC may exert anti-inflammatory activities on monocytes/macrophages. APC inhibited TNF, IL-1b, IL-6 and IL-8 release by LPS-stimulated monocytes/macrophages.119–122 One study reported an inhibitory effect of protein S on LPS-induced cytokine production by monocytes, and a synergistic inhibition by concurrent addition of APC and protein S.119 Of interest, APC may upregulate IL-6 and IL-8 production by endothelial cells, an effect that was enhanced by protein S.123 Moreover, mediators of fibrinolysis can influence the synthesis of cytokines. PAI-1 inhibited LPS-induced TNF production by mononuclear cells in vitro.124,125 An anti-uPA monoclonal antibody attenuated TNF release by monocytic THP-1 cells stimulated with LPS, whereas exogenous uPA enhanced LPS-induced TNF secretion through an effect independent of plasmin activity.126 uPA may also regulate cytokine activity, as indicated by the finding that U937 monocytic cells can produce cell-associated uPA that is able to inactivate IFN-γ by proteolysis.126
Cross-talk between coagulation and inflammation has been documented further by in vivo studies in baboons. Prevention of DIC by either inhibition of the factor VIIa-tissue factor pathway or enhancement of the protein C-protein S-thrombomodulin system in models of severe bacteremia was also protective against lethality.40–42,80–82 The link between DIC and death is not self-explanatory, however. Indeed, although anti-TNF completely prevented lethality in baboons with E. coli sepsis, it did not influence DIC.24 In addition, administration of factor Xa blocked in its active center (DEGR-Xa), which inhibits the coagulation cascade downstream from tissue factor, failed to influence lethality of bacteremic baboons, while completely inhibiting the development of DIC.127 Together these findings suggest that elimination of the VIIa-tissue factor pathway or enhancement of the function of the protein C-protein S-thrombomodulin system protect against death by an effect that is not directly linked to their effect on the coagulation system. It is therefore likely that the VIIa-tissue factor pathway and the protein C-protein S-thrombomodulin system have effects on inflammatory responses different from the hemostatic mechanism. Several experimental findings support this hypothesis. TFPI or DEGR-VIIa treatment not only prevented DIC in bacteremic baboons, but also reduced IL-6 and IL-8 levels.40,42 Further, APC not only is able to reduce monocyte cytokine production in vitro (see above), but also diminished LPS-induced TNF release in rats in vivo,119,121,122 while DEGR-Xa was without effect.121 Treatment with an anti-APC antibody was associated with increased TNF secretion upon exposure of rats to LPS.122 In accordance, treatment with an anti-EPCR antibody resulted in enhanced IL-6 and IL-8 release in baboons with sublethal Escherichia coli bacteremia,81 while infusion of C4b binding protein resulted in a detectable TNF response in this model.82 Other evidence for the anti-inflammatory properties of the protein C-protein S-thrombomodulin system is derived from studies in which APC and recombinant thrombomodulin protected rats against LPS-induced lung injury by inhibition of leukocyte activation, not by an effect on the coagulation system,121,128 and by an investigation in which APC reduced the severity of compression-induced spinal cord injury in rats by inhibiting the activation of leukocytes, in association with a reduction in brain TNF concentrations.129 The bidirectional interplay between coagulation and inflammation may vary depending upon the severity of the bacterial challenge, as suggested by recent findings in healthy humans challenged with low dose LPS. In these subjects TFPI treatment did not influence cytokine release or other inflammatory responses in spite of a complete prevention of coagulation activation, which contrasts with observations in baboons with severe sepsis (see above).43 Presumably, in lethal sepsis other cell types contribute to the systemic inflammatory response, which is also more sustained and severe than in normal humans subjected to transient and mild endotoxemia.
Conclusion
Severe sepsis almost invariably is accompanied by pathological alterations in the hemostatic balance. Activation of coagulation is stimulated by enhanced expression of tissue factor at the surface of endothelial cells and monocytes. Concurrently, anticoagulant pathways are inhibited, in particular the protein C-protein S-thrombomodulin system and the fibrinolytic system. Cytokines can influence procoagulant and anticoagulant mechanisms at multiple levels. Proinflammatory cytokines such as TNF, IL-1, IL-6 and IL-12 can induce coagulation activation in vivo; TNF and IL-1 can also trigger subsequent release of tPA and PAI-1, indicative of activation and rapid inactivation of fibrinolysis. IL-10 is an anti-inflammatory cytokine that can inhibit both LPS-induced activation of coagulation and fibrinolysis. Studies in which the endogenous activity of individual cytokines was blocked have identified TNF as a crucial mediator of the fibrinolytic response to LPS, whereas IL-6 and IL-1 likely contribute to activation of the coagulation system. There is ample evidence that the tissue factor-factor VIIa pathway, which is crucial for the initiation of coagulation activation during experimental endotoxemia and sepsis, also influences inflammatory responses not directly related to the clotting cascade. In addition, the protein C-protein S-thrombomodulin system not only functions as an important anticoagulant pathway, but also exerts anti-inflammatory effects. There is extensive cross-talk between the cytokine network and the hemostatic mechanism only part of which has been elucidated at the present time.
References
- 1.
- Hack CE, Aarden LA, Thijs LG. Role of cytokines in sepsis. Adv Immunol. 1997;66:101–195. [PubMed: 9328641]
- 2.
- Van der Poll T, van Deventer S J H. Cytokines and anti-cytokines in the pathogenesis of sepsis. Infectious Disease Clinics of North America. 1999;13:413–426. [PubMed: 10340175]
- 3.
- Lowry SF, Calvano SE, van der Poll T. Measurement of inflammatory mediators in clinical sepsis In: Sibbald WJ, Vincent JL, eds. Clinical Trials for the Treatment of Sepsis New York: Springer-Verlag, 199586–105.
- 4.
- Dehoux MS, Boutten A, Ostinelli J, Seta N, Dombret MC, Crestani B. et al. Compartimentalized cytokine production within the human lung in unilateral pneumonia. Am J Respir Crit Care Med. 1994;150:710–716. [PubMed: 8087341]
- 5.
- Fröhlich D, Eiber RM, Jochum M, Billing A. Perioperative pattern of peritoneal interleukin 8, tumour necrosis factor-α, and granulocyte elastase release in human secondary peritonitis. Cytokine. 1997;9:288–292. [PubMed: 9112338]
- 6.
- Olszyna DP, Prins JM, Buis B, van Deventer S J H, Speelman P, van der Poll T. Levels of inhibitors of tumor necrosis factor-α and interleukin 1 in urine and sera of patients with urosepsis. Infect Immun. 1998;66:3527–3534. [PMC free article: PMC108383] [PubMed: 9673230]
- 7.
- Marchant A, Deviere J, Byl B, de Groote D, Vincent JL, Goldman M. Interleukin-10 production during septicaemia. Lancet. 1994;343:707–710. [PubMed: 7907683]
- 8.
- Van der Poll T, de Waal Malefyt R, Coyle SM, Lowry SF. Antiinflammatory cytokine responses during clinical sepsis and experimental endotoxemia: sequential measurements of plasma soluble interleukin (IL)-1 receptor type II, IL-10 and IL-13 concentrations. J Infect Dis. 1997;175:118–122. [PubMed: 8985204]
- 9.
- Van Zee KJ, Kohno T, Fischer E, Rock SC, Moldawer LL, Lowry SF. Tumor necrosis factor soluble receptors circulate during experimental and clinical inflammation and can protect against excessive tumor necrosis factor α in vitro and in vivo. Proc Natl Acad Sci USA. 1992;89:4845–4849. [PMC free article: PMC49184] [PubMed: 1317575]
- 10.
- Fong Y, Tracey KJ, Moldawer LL, Hesse DG, Manogue KR, Kenney JS. et al. Antibodies to cachectin/tumor necrosis factor reduce interleukin 1β and interleukin 6 appearance during lethal bacteremia. J Exp Med. 1989;170:1627–1633. [PMC free article: PMC2189514] [PubMed: 2809510]
- 11.
- Van der Poll T, van Deventer S J H. Endotoxemia in healthy subjects as a human model of inflammation In: Cohen J, Marshall J, eds. The Immune Response in the Critically Ill New York: Springer-Verlag, 1999335–357.
- 12.
- Van der Poll T, Levi M, van Deventer S J H, ten Cate H, Haagmans BL, Biemond BJ. et al. Differential effects of anti-tumor necrosis factor monoclonal antibodies on systemic inflammatory responses in experimental endotoxemia in chimpanzees. Blood. 1994;83:446–451. [PubMed: 8286742]
- 13.
- Van der Poll T, Coyle SM, Levi M, Jansen PM, Dentener M, Barbosa K. et al. Effect of a recombinant dimeric tumor necrosis factor receptor on inflammatory responses to intravenous endotoxin in normal humans. Blood. 1997;89:3727–3734. [PubMed: 9160678]
- 14.
- Van der Poll T, Jansen J, Levi M, ten Cate H, ten Cate JW, van Deventer S J H. Regulation of interleukin 10 release by tumor necrosis factor in humans and chimpanzees. J Exp Med. 1994;180:1985–1988. [PMC free article: PMC2191735] [PubMed: 7964475]
- 15.
- Jansen PM, van der Pouw Kraan T C T M, de Jong IW, Van Mierlo G, Wijdenes J, Chang AA. et al. Release of interleukin-12 in experimental Escherichia coli septic shock in baboons: Relation to plasma levels of interleukin-10 and interferon-γ Blood. 1996;87:5144–5151. [PubMed: 8652827]
- 16.
- Jansen J, van der Poll T, Levi M, ten Cate H, Gallati H, ten Cate JW. et al. Inhibition of the release of soluble tumour necrosis factor receptors in experimental endotoxemia by an anti-tumor necrosis factor-α antibody. J Clin Immunol. 1995;15:45–50. [PubMed: 7759600]
- 17.
- Tracey KJ, Beutler B, Lowry SF, Merryweather J, Wolpe S, Milsark IW. et al. Shock and tissue injury induced by recombinant human cachectin. Science. 1986;234:470–474. [PubMed: 3764421]
- 18.
- Okusawa S, Gelfland JA, Ikejima T, Connolly RJ, Dinarello CA. Interleukin 1 induces a shock-like state in rabbits. \ill\Synergism with tumor necrosis factor and the effect of cyclooxygenase inhibition. J Clin Invest. 1988;81:1162–1172. [PMC free article: PMC329645] [PubMed: 3258319]
- 19.
- Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, Atkins MB. et al. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-g production. Blood. 1997;90:2541–2548. [PubMed: 9326219]
- 20.
- Van Gameren MM, Willemse P H B, Mulder NH, Limburg PC, Groen H J M, Vellenga E. et al. Effects of recombinant human interleukin-6 in cancer patients: A phase I/II study. Blood. 1994;84:1434–1441. [PubMed: 8068939]
- 21.
- Stouthard J M L, Romijn JA, van der Poll T, Endert E, Klein S, Bakker P J M. et al. Endocrine and metabolic effects of interleukin-6 in humans. Am J Physiol. 1995;268:E813–E819. [PubMed: 7762632]
- 22.
- Preiser JC, Schmartz D, van der Linden P, Content J, Vanden Bussche P, Buurman W. et al. Interleukin-6 administration has no acute hemodynamic or hematologic effect in the dog. Cytokine. 1991;3:1–4. [PubMed: 1883951]
- 23.
- Tracey KJ, Fong Y, Hesse DG, Manogue KR, Lee AT, Kuo GC. et al. Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature. 1987;330:662–664. [PubMed: 3317066]
- 24.
- Hinshaw LB, TekampOlson P, Chang A C K, Lee PA, Taylor F B Jr., Murray CK. et al. Survival of primates in LD100 septic shock following therapy with antibody to tumor necrosis factor (TNF). Circ Shock. 1990;30:279–292. [PubMed: 2178801]
- 25.
- Ohlsson K, Björk P, Bergenfeldt M, Hageman R, Thompson RC. Interleukin 1 receptor antagonist reduces mortality from endotoxin shock. Nature. 1990;348:550–552. [PubMed: 2147233]
- 26.
- Fischer E, Marano MA, Van Zee KJ, Rock CS, Hawes AS, Thompson WA. et al. Interleukin-1 receptor blockade improves survival and hemodynamic performance in Escherichia coli septic shock, but fails to alter host responses to sublethal endotoxemia. J Clin Invest. 1992;89:1551–1557. [PMC free article: PMC443028] [PubMed: 1533231]
- 27.
- Fattori E, Cappelletti M, Costa P, Sellitto C, Cantoni L, Carelli M. et al. Defective inflammatory response in interleukin-6-deficient mice. 1994;180:1243–1250. [PMC free article: PMC2191674] [PubMed: 7931061]
- 28.
- Silva AT, Cohen J. Role of interferon-g in experimental gram-negative sepsis. J Infect Dis. 1992;166:331–335. [PubMed: 1634804]
- 29.
- Wysocka M, Kubin M, Vieira LQ, Ozmen L, Garotta G, Scott P. et al. Interleukin-12 is required for interferon-γ production and lethality in lipopolysaccharide-induced shock in mice. Eur J Immunol. 1995;25:672–676. [PubMed: 7705395]
- 30.
- Car BD, Eng VM, Schnyder B, Ozmen L, Huang S, Gallay P. et al. Interferon-γ receptor deficient mice are resistant to endotoxic shock. J Exp Med. 1994;179:1437–1444. [PMC free article: PMC2191498] [PubMed: 8163930]
- 31.
- Berg DJ, Kühn R, Rajewsky K, Müller W, Menon S, Davidson N. et al. Interleukin-10 is a central regulator of the response to LPS in murine models of endotoxic shock and the Shwartzman reaction but not endotoxin tolerance. J Clin Invest. 1995;96:2339–2347. [PMC free article: PMC185885] [PubMed: 7593621]
- 32.
- Marchant A, Bruyns C, Vandenabeele P, Ducarme M, Gérard C, Delvaux A. et al. IL-10 controls IFN-γ and TNF production during experimental endotoxemia. Eur J Immunol. 1994;24:1167–1171. [PubMed: 8181527]
- 33.
- Muchamuel T, Menon S, Pisacane P, Howard MC, Cockayne DA. IL-13 protects mice from lipopolysaccharide-induced lethal endotoxemia: Correlation with down-modulation of TNF-α, IFN-γ, and IL-12 production. J Immunol. 1997;158:2898–2903. [PubMed: 9058827]
- 34.
- Levi M, ten Cate H. Disseminated intravascular coagulation. N Engl J Med. 1999;341:586–592. [PubMed: 10451465]
- 35.
- Osterud B, Flaegstad T. Increased tissue thromboplastin activity in monocytes of patients with meningococcal infection: related to unfavourable prognosis. Thromb Haemost. 1983;49:5–7. [PubMed: 6845273]
- 36.
- Li A, Chang AC, Peer GT, Hinshaw LB, Taylor F B Jr. Comparison of the capacity of rhTNF-alpha and Escherichia coli to induce procoagulant activity by baoon mononuclear cells in vivo and in vitro. Shock. 1996;5:274–279. [PubMed: 8721387]
- 37.
- Taylor F B Jr., Chang A, Ruf W, Morrissey JH, Hinshaw L, Catlett R. et al. Lethal E. coli septic shock is prevented by blocking tissue factor with monoclonal antibody. Circ Shock. 1991;33:127–134. [PubMed: 2044206]
- 38.
- Levi M, ten Cate H, Bauer KA, Van der Poll T, Edgington TS, Büller HR. et al. Inhibition of endotoxin-induced activation of coagulation and fibrinolysis by pentoxifylline or by a monoclonal anti-tissue factor antibody in chimpanzees. J Clin Invest. 1994;93:114–120. [PMC free article: PMC293743] [PubMed: 8282778]
- 39.
- Biemond BJ, Levi M, ten Cate H, Soule HR, Morris LD, Foster DL. et al. Complete inhibition of endotoxin-induced coagulation activation in chimpanzees with a monoclonal Fab fragment against factor VII/VIIa. Thromb Haemost. 1995;73:223–230. [PubMed: 7792734]
- 40.
- Taylor F B Jr., Chang A C K, Peer G, Ezban M, Hedner U. Active site inhibited factor VIIa (DEGR VIIa) attenuates the coagulant and interleukin-6 and -8, but not tumor necrosis factor, responses of the baboon to LD100 Escherichia coli. Blood. 1998;91:1609–1615. [PubMed: 9473226]
- 41.
- Creasey AA, Chang A C K, Feigen L, Wün TC, Taylor F B Jr., Hinshaw LB. Tissue factor pathway inhibitor reduces mortality from Escherichia coli septic shock. J Clin Invest. 1993;91:2850–2860. [PMC free article: PMC443354] [PubMed: 8514893]
- 42.
- Carr C, Bild GS, Chang A C K, Peer GT, Palmier MO, Frazier RB. et al. Recombinant E.coli-derived tissue factor pathway inhibitor reduces coagulopathic and lethal effects in the baboon gram-negative model of septic shock. Circ Shock. 1995;44:126–137. [PubMed: 7600636]
- 43.
- De Jonge E, Dekkers P E P, Creasey AA, Hack CE, Paulson SK, Karim A. et al. Tissue factor pathway inhibitor (TFPI) dose-dependently inhibits coagulation activation without influencing the fibrinolytic and the cytokine response during human endotoxemia. Blood. 2000;95:1124–1129. [PubMed: 10666180]
- 44.
- Nawroth PP, Stern DM. Modulation of endothelial cell hemostatic properties by tumor necrosis factor. J Exp Med. 1986;163:740–745. [PMC free article: PMC2188058] [PubMed: 3753996]
- 45.
- Nawroth PP, Handley DA, Esmon CT, Stern DM. Interleukin 1 induces endothelial cell procoagulant while suppressing cell-surface anticoagulant activity. Proc Natl Acad Sci USA. 1986;83:3460–3464. [PMC free article: PMC323535] [PubMed: 3486418]
- 46.
- Scarpati EM, Sadler JE. Regulation of endothelial cell coagulant properties. Modulation of tissue factor, plasminogen activator inhibitors and thrombomodulin by phorbol 12-myristate 13-acetate and tumor necrosis factor. J Biol Chem. 1989;264:20705–20713. [PubMed: 2555368]
- 47.
- Conckling PR, Greenberg CS, Weinberg JB. Tumor necrosis factor induces tissue factor-like activity in human leukemia cell line U937 and peripheral blood monocytes. Blood. 1988;72:128–133. [PubMed: 3134064]
- 48.
- Herbert JM, Savi P, Laplace MC, Lale A. IL-4 inhibits LPS-, IL-1β- and TNFα-induced expression of tissue factor in endothelial cells and monocytes. FEBS Letters. 1992;310:31–33. [PubMed: 1526281]
- 49.
- Herbert JM, Savi P, Laplace MC, Lalé A, Dol F, Dumas A. et al. IL-4 and IL-13 exhibit comparable abilities to reduce pyrogen-induced expression of procoagulant activity in endothelial cells and monocytes. FEBS Letters. 1993;328:268–270. [PubMed: 8102337]
- 50.
- Martin NB, Jamieson A, Tuffin DP. The effect of interleukin-4 on tumour necrosis factor-alpha induced expression of tissue factor and plasminogen activator inhibitor-1 in human umbilical vein endothelial cells. Thromb Haemost. 1993;70:1037–1042. [PubMed: 8165597]
- 51.
- Schwager I, Jungi TW. Effect of human recombinant cytokines on the induction of macrophage procoagulant activity. Blood. 1994;83:152–160. [PubMed: 8274733]
- 52.
- Kirchhofer D, Tschopp TB, Hadvary P, Baumgartner HR. Endothelial cells stimulated with tumor necrosis factor-alpha express varying amounts of tissue factor resulting in inhomogenous fibrin deposition in a native blood flow system.Effects of thrombin inhibitors. J Clin Invest. 1994;93:2073–2083. [PMC free article: PMC294327] [PubMed: 8182139]
- 53.
- Paleolog EM, Delasalle S A J, Buurman WA, Feldmann M. Functional activities of receptors for tumor necrosis factor-α on human vascular endothelial cells. Blood. 1994;84:2578–2590. [PubMed: 7919375]
- 54.
- Schmid EF, Binder K, Grell M, Scheurich P, Pfizenmaier K. Both tumor necrosis factor receptors, TNFR60 and TNFR80, are involved in signaling endothelial tissue factor expression by juxtracrine tumor necrosis factor alpha. Blood. 1995;86:1836–1841. [PubMed: 7544644]
- 55.
- Del Prete G, de Carli M, Lammel RM, D'Elios MM, Daniel KC, Giusti B. et al. Th1 and Th2 T-helper cells exert opposite effects on procoagulant activity and tissue factor production by human monocytes. Blood. 1995;86:250–257. [PubMed: 7795230]
- 56.
- Osnes LT, Westvik AB, Joo GB, Okkenhaug C, Kierulf P. Inhibition of IL-1 induced tissue factor (TF) synthesis and procoagulant activity (PCA) in purified human monocytes by IL-4, IL-10 and IL-13. Cytokine. 1996;8:822–827. [PubMed: 9047078]
- 57.
- Ernofsson M, Tenno T, Siegbahn A. Inhibition of tissue factor surface expression in human peripheral blood monocytes exposed to cytokines. Br J Haematol. 1996;95:249–257. [PubMed: 8904877]
- 58.
- Schecter AD, Rollins BJ, Zhang YJ, Charo IF, Fallon JT, Rossikhina M. et al. Tissue factor induced by monocyte chemoattractant protein-1 in human aortic smooth muscle and THP-1 cells. J Biol Chem. 1997;272:28568–28573. [PubMed: 9353321]
- 59.
- Neumann FJ, Ott I, Marx N, Luther T, Kenngott S, Gawaz M. et al. Effect of human recombinant interleukin-6 and interleukin-8 on monocyte procoagulant activity. Arterioscler Thromb Vasc Biol. 1997;17:3399–3405. [PubMed: 9437185]
- 60.
- Meisel SR, Shimon I, Edgington TS, Melmed S, Cercek B, Shah PK. Leukemia inhibitory factor enhances tissue factor expression in human monocyte-derived macrophages: A gp130-mediated mechanism. Br J Haematol. 1999;107:747–755. [PubMed: 10606879]
- 61.
- Veltrop MH, Beekhuizen H, Thompson J. Bacterial species- and strain-dependent induction of tissue factor in human vascular endothelial cells. Infect Immun. 1999;67:6130–6138. [PMC free article: PMC97002] [PubMed: 10531276]
- 62.
- Pradier O, Gérard C, Delvaux A, Lybin M, Abramowicz D, Capel P. et al. Interleukin-10 inhibits the induction of monocyte procoagulant activity by bacterial lipopolysaccharide. Eur J Immunol. 1993;23:2700–2703. [PubMed: 8405069]
- 63.
- Ramani M, Khechai F, Ollivier V, Ternisien C, Bridey F, Hakim J. et al. Interleukin-10 and pentoxifylline inhibit C-reactive protein-induced tissue factor gene expression in peripheral human blood monocytes. FEBS Letters. 1994;356:86–88. [PubMed: 7988727]
- 64.
- Wharram BL, Fitting K, Kunkel SL, Remick DG, Merritt SE, Wiggins RC. Tissue factor expression in endothelial cell/monocyte cocultures stimulated by lipopolysaccharide and/or aggregated IgG. Mechanisms of cell: Cell communication. J Immunol. 1991;146:1437–1445. [PubMed: 1993838]
- 65.
- Lo SK, Cheung A, Zheng Q, Silverstein RL. Induction of tissue factor on monocytes by adhesion to endothelial cells. J Immunol. 1995;154:4768–4777. [PubMed: 7536780]
- 66.
- Van der Poll T, Büller HR, ten Cate H, Wortel CH, Bauer KA, Van Deventer S J H. et al. Activation of coagulation after administration of tumor necrosis factor to normal subjects. N Engl J Med. 1990;322:1622–1627. [PubMed: 2188129]
- 67.
- Bauer KA, ten Cate H, Barzegar S, Spriggs DR, Sherman ML, Rosenberg RD. Tumor necrosis factor infusions have a procoagulant effect on the hemostatic mechanism of humans. Blood. 1989;74:165–172. [PubMed: 2752108]
- 68.
- Van der Poll T, Jansen PM, van Zee KJ, Welborn M B I I I, de Jong I, Hack CE. et al. Tumor necrosis factor induces activation of coagulation and fibrinolysis in baboons through an exclusive effect on the p55 receptor. Blood. 1996;88:922–927. [PubMed: 8704250]
- 69.
- DeLa Cadena RA, Majluf-Cruz A, Stadnicki A, Tropea M, Reda D, Agosti JM. et al. Recombinant tumor necrosis factor receptor p75 fusion protein (TNFR:Fc) alters endotoxin-induced activation of the kinin, fibrinolytic, and coagulation systems in normal humans. Thromb Haemost. 1998;80:114–118. [PubMed: 9684796]
- 70.
- Jansen PM, Boermeester MA, Fischer E, de Jong IW, van der Poll T, Moldawer LL. et al. Contribution of interleukin-1 to activation of coagulation and fibrinolysis, to neutrophil degranulation and the release of sPLA2 in sepsis. Studies in non-human primates following interleukin-1α administration and during lethal bacteremia. Blood. 1995;86:1027–1034. [PubMed: 7620156]
- 71.
- Stouthard J M L, Levi M, Hack CE, Veenhof C H N, Romijn JA, Sauerwein HP. et al. Interleukin-6 stimulates coagulation, not fibrinolysis, in humans. Thromb Haemost. 1996;76:738–742. [PubMed: 8950783]
- 72.
- Van der Poll T, Levi M, Hack CE, ten Cate H, Van Deventer S J H, Eerenberg A J M. et al. Elimination of interleukin 6 attenuates coagulation activation in experimental endotoxemia in chimpanzees. J Exp Med. 1994;179:1253–1259. [PMC free article: PMC2191443] [PubMed: 8145042]
- 73.
- Lauw FN, Dekkers P E P, te Velde AA, Speelman P, Levi M, Kurimoto M. et al. Interleukin 12 induces sustained activation of multiple host inflammatory mediator systems in chimpanzees. J Infect Dis. 1999;179:646–652. [PubMed: 9952371]
- 74.
- Baars JW, de Boer JP, Wagstaff J, Roem D, Eerenberg-Belmer AJ, Nauta J. et al. Interleukin-2 induces activation of coagulation and fibrinolysis: Resemblance to the changes seen during endotoxaemia. Br J Haematol. 1992;82:295–301. [PubMed: 1419810]
- 75.
- Pajkrt D, van der Poll T, Levi M, Cutler DL, Affrime MB, van den Ende A. et al. Interleukin 10 inhibits activation of coagulation and fibrinolysis during human endotoxemia. Blood. 1997;89:2701–2705. [PubMed: 9108387]
- 76.
- Van der Poll T, Jansen PM, Montegut WJ, Braxton CC, Calvano SE, Stackpole SA. et al. Effects of IL-10 on systemic inflammatory responses during sublethal primate endotoxemia. J Immunol. 1997;158:1971–1975. [PubMed: 9029140]
- 77.
- Esmon CT. New potential therapeutic modalities: aPC. Sepsis. 1999;3:161–172.
- 78.
- Stearns-Kurosawa DJ, Kurosawa S, Mollica JS, Ferrell GL, Esmon CT. The endothelial cell protein C receptor augments protein C activation by the thrombin-thrombomodulin complex. Proc Natl Acad Sci USA. 1996;93:10212–10216. [PMC free article: PMC38363] [PubMed: 8816778]
- 79.
- Regan LM, Stearns-Kurosawa DJ, Kurosawa S, Mollica J, Fukudome K, Esmon CT. The endothelial cell protein C receptor: inhibition of activated protein C anticoagulant function without modulation of reaction with proteinase inhibitors. J Biol Chem. 1996;271:17499–17503. [PubMed: 8663474]
- 80.
- Taylor F B Jr., Chang A, Esmon T, D'Angelo A, Vigano-D'Angelo S, Blick KE. Protein C prevents the coagulopathic and lethal effects of Escherichia coli infusion in the baboon. J Clin Invest. 1987;79:918–925. [PMC free article: PMC424237] [PubMed: 3102560]
- 81.
- Taylor F B Jr, Stearns-Kurosawa J, Kurosawa S. et al. The endothelial cell protein C receptor aids in host defense against Escherichia coli sepsis. Blood. 2000;95:1680–1686. [PubMed: 10688824]
- 82.
- Taylor F B Jr., Chang A, Ferrell G, Mather T, Blick K, Esmon CT. C4b-binding protein exacerbates the host response to Escherichia coli. Blood. 1991;78:357–363. [PubMed: 1829967]
- 83.
- Lentz SR, Tsiang M, Sadler JE. Regulation of thrombomodulin by tumor necrosis factor-α: Comparison of transcriptional and posttranscriptional mechanism. Blood. 1991;77:542–550. [PubMed: 1846763]
- 84.
- Grey ST, Csizmadia V, Hancock WW. Differential effect of tumor necrosis factor-a on thrombomodulin gene expression by human monocytoid (THP-1) cells versus endothelial cells. Int J Hematol. 1998;67:53–62. [PubMed: 9594445]
- 85.
- Raife TJ, Demetroulis EM, Lentz SR. Regulation of thrombomodulin expression by all-trans retinoic acid and tumor necrosis factor-a: Differential responses in keratinocytes and endothelial cells. Blood. 1996;88:2043–2049. [PubMed: 8822923]
- 86.
- Fukudome K, Esmon CT. Identification, cloning and regulation of a novel endothelial cell protein C/activated protein C receptor. J Biol Chem. 1994;269:26486–26491. [PubMed: 7929370]
- 87.
- Xu J, Qu D, Esmon NL, Esmon CT. Metalloproteolytic release of endothelial cell protein C receptor. J Biol Chem. 2000;275:6038–6044. [PubMed: 10681599]
- 88.
- Redl H, Schlag G, Schiesser A, Davies J. Thrombomodulin release in baboon sepsis: its dependence on the dose of Escherichia coli and the presence of tumor necrosis factor. J Infect Dis. 1995;171:1522–1527. [PubMed: 7769287]
- 89.
- Yamamoto K, Shimokawa T, Kojima T, Loskutoff DJ, Saito H. Regulation of murine protein C gene expression in vivo: Effects of tumor necrosis factor-α, interleukin-1 and transforming growth factor-β Thromb Haemost. 1999;82:1297–1301. [PubMed: 10544917]
- 90.
- De Boer JP, Creasy AA, Chang A, Roem D, Brouwer MC, Eerenberg A J M. et al. Activation patterns of coagulation and fibrinolysis in baboons following infusion with lethal or sublethal dose of Escherichia coli. Circ Shock. 1993;39:59–67. [PubMed: 7683256]
- 91.
- Schleef RR, Bevilaqua MP, Sawdey M, Gimbrone M A Jr., Loskutoff DJ. Cytokine activation of vascular endothelium. Effects on tissue-type plasminogen activator and type 1 plasminogen activator inhibitor. J Biol Chem. 1988;263:5797–5803. [PubMed: 3128548]
- 92.
- Sawdey M, Podor TJ, Loskutoff DJ. Regulation of type 1 plasminogen activator inhibitor gene expression in cultured bovine aortic endothelial cells. Induction by transforming growth factor-β, lipopolysaccharide, and tumor necrosis factor-a. J Biol Chem. 1989;264:10396–10401. [PubMed: 2499579]
- 93.
- Van Hinsbergh VW, van den Berg EA, Fiers W, Dooijewaard G. Tumor necrosis factor induces the production of urokinase-type plasminogen activator by human endothelial cells. Blood. 1990;75:1991–1998. [PubMed: 2140060]
- 94.
- Gallicchio M, Hufnagl P, Wojta J, Tipping P. IFN-γ inhibits thrombin- and endotoxin-induced plasminogen activator inhibitor type 1 in human endothelial cells. J Immunol. 1996;157:2610–2617. [PubMed: 8805664]
- 95.
- Arnman V, Stemme S, Rymo L, Risberg B. Interferon-γ modulates the fibrinolytic response in cultured human endothelial cells. Thromb Res. 1995;77:431–440. [PubMed: 7778058]
- 96.
- Takahashi K, Uwabe Y, Sawasaki Y, Kiguchi T, Nakamura H, Kashiwabara K. et al. Increased secretion of urokinase-type plasminogen activator by human lung microvascular endothelial cells. Am J Physiol. 1998;275:L47–L54. [PubMed: 9688934]
- 97.
- Niedbala MJ, Picarella MS. Tumor necrosis factor induction of endothelial cell urokinase-type plasminogen activator mediated proteolysis of extracellular matrix and its antagonism by gamma-interferon. Blood. 1992;79:678–687. [PubMed: 1732009]
- 98.
- Wojta J, Gallicchio M, Zoellner H, Filonzi EL, Hamilton JA, McGrath K. Interleukin-4 stimulates expression of urokinase-type-plasminogen activator in cultured human foreskin microvascular endothelial cells. Blood. 1993;81:3285–3292. [PubMed: 8507866]
- 99.
- de Boer JP, Abbink JJ, Brouwer MC, Meijer C, Roem D, Voorn GP. et al. PAI-1 synthesis in the human hepatoma cell line HepG2 is increased by cytokines—Evidence that the liver contributes to acute phase behaviour of PAI-1. Thromb Haemost. 1991;65:181–185. [PubMed: 1711245]
- 100.
- Healy AM, Gelehrter TD. Induction of plasminogen activator inhibitor-1 in HepG2 human hepatoma cells by mediators of the acute phase response. J Biol Chem. 1994;269:19095–19100. [PubMed: 8034668]
- 101.
- Seki T, Gelehrter TD. Interleukin-1 induction of type-1 plasminogen activator inhibitor (PAI-1) gene expression in the mouse hepatocyte line, AML 12. J Cell Physiol. 1996;168:648–656. [PubMed: 8816919]
- 102.
- Busso N, Nicodeme E, Chesne C, Guillouzo A, Belin D, Hyafil F. Urokinase and type 1 plasminogen activator inhibitor production by normal hepatocytes: modulation by inflammatory agents. Hepatology. 1994;20:186–190. [PubMed: 8020888]
- 103.
- Suffredini AF, Harpel PC, Parrillo JE. Promotion and subsequent inhibition of plasminogen activation after administration of intravenous endotoxin to normal subjects. N Engl J Med. 1989;320:1165–1172. [PubMed: 2496309]
- 104.
- Van der Poll T, Levi M, Büller HR, Van Deventer S J H, De Boer JP, Hack CE. et al. Fibrinolytic response to tumor necrosis factor in healthy subjects. J Exp Med. 1991;174:729–732. [PMC free article: PMC2118940] [PubMed: 1714936]
- 105.
- Van Hinsbergh VW, Bauer KA, Kooistra T, Kluft C, Dooijewaard G, Sherman ML. et al. Progress of fibrinolysis during tumor necrosis factor infusions in humans. Concomitant increase in tissue-type plasminogen activator, plasminogen activator inhibitor type-1, and fibrin(ogen) degradation products. Blood. 1990;76:2284–2289. [PubMed: 1701665]
- 106.
- Logan TF, Virji MA, Gooding WE, Bontempo FA, Ernstoff MS, Kirkwood JM. Plasminogen activator and its inhibitor in cancer patients treated with tumor necrosis factor. J Natl Cancer Inst. 1992;84:1802–1810. [PubMed: 1433370]
- 107.
- Fearns C, Loskutoff DJ. Induction of plasminogen activator inhibitor type 1 gene expression in murine liver by lipopolysaccharide. Cellular localization and role of endogenous tumor necrosis factor-α Am J Pathol. 1997;150:579–590. [PMC free article: PMC1858291] [PubMed: 9033272]
- 108.
- Seki T, Healy AM, Fletcher DS, Noguchi T, Gelehrter TD. IL-1β mediates induction of hepatic type I plasminogen activator inhibitor in response to local tissue injury. Am J Physiol. 1999;277:G801–G809. [PubMed: 10516146]
- 109.
- Mileno MD, Margolis NH, Clark BD, Dinarello CA, Burke JF, Gelfand JA. Coagulation of whole blood stimulates interleukin-1β gene expression. J Infect Dis. 1995;172:308–311. [PubMed: 7797938]
- 110.
- Jones A, Geczy CL. Thrombin and factor Xa enhance the production of interleukin-1. Immunology. 1990;71:236–241. [PMC free article: PMC1384310] [PubMed: 2228024]
- 111.
- Johnson K, Aarden LA, Choi Y, De Groot E, Creasey A. The proinflammatory cytokine response to coagulation and endotoxin in whole blood. Blood. 1996;87:5051–5060. [PubMed: 8652818]
- 112.
- Johnson K, Choi Y, De Groot E, Samuels I, Creasey A, Aarden LA. Potential mechanisms for a proinflammatory vascular cytokine response to coagulation activation. J Immunol. 1998;160:5130–5135. [PubMed: 9590265]
- 113.
- Suk K, Cha SH. Thrombin-induced interleukin-8 production and its regulation by interferon-γ and prostaglandin E2 in human monocytic U937 cells. Immunol Lett. 1999;67:223–227. [PubMed: 10369130]
- 114.
- Sower LE, Froelich CJ, Fenton JW, Klimpel GR. Thrombin induces IL-6 production in fibroblasts and epithelial cells. Evidence for the involvement of the seven-transmembrane domain (STD) receptor for alpha thrombin. J Immunol. 1995;155:895–901. [PubMed: 7608566]
- 115.
- Qi J, Goralnick S, Kreutzer DL. Fibrin regulation of interleukin-8 gene expression in human vascular endothelial cells. Blood. 1997;90:3595–3602. [PubMed: 9345043]
- 116.
- Senden N M H, Jeunhomme T M A A, Heemskerk J W M, Wagenvoord R, Van't Veer C, Hemker HC. et al. Factor Xa induces cytokine production and expression of adhesion molecules by human umbilical vein endothelial cells. J Immunol. 1998;161:4318–4324. [PubMed: 9780208]
- 117.
- Ueno A, Murakami K, Yamanouchi K, Watanabe M, Kondo T. Thrombin stimulates production of interleukin-8 in human endothelial cells. Immunology. 1996;88:76–81. [PMC free article: PMC1456474] [PubMed: 8707354]
- 118.
- Anrather D, Millan MT, Palmetshofer A, Robson SC, Geczy C, Ritchie AJ. et al. Thrombin activates nuclear factor-κB and potentiates endothelial cell activation by TNF. J Immunol. 1997;159:5620–5628. [PubMed: 9548505]
- 119.
- Hancock WW, Tsuchida A, Hau H, Thomson NM, Salem HH. The anticoagulants protein C and protein S display potent anti-inflammatory and immunosuppressive effects relevant to transplant biology and therapy. Transplant Proc. 1992;24:2302–2303. [PubMed: 1413069]
- 120.
- Grey ST, Tsuchida A, Hau H, Orthner CL, Salem HH, Hancock WW. Selective inhibitory effects of the anticoagulant activated protein C on the responses of human mononuclear phagocytes to LPS, IFN-γ, or phorbol ester. J Immunol. 1994;153:3664–3672. [PubMed: 7523500]
- 121.
- Murakami K, Okajima K, Uchiba M, Johno M, Nakagaki T, Okabe H. et al. Activated protein C attenuates endotoxin-induced pulmonary vascular injury by inhibiting activated leukocytes in rats. Blood. 1996;87:642–647. [PubMed: 8555486]
- 122.
- Hancock WW, Bach FH. Immunobiology and therapeutic applications of protein C/protein S/thrombomodulin in human and experimental allotransplantation and xenotransplantation. Trends Cardiovasc Med. 1997;7:174–183. [PubMed: 21235882]
- 123.
- Hooper WC, Phillips DJ, Renshaw MA, Evatt BL, Benson JM. The up-regulation of IL-6 and IL-8 in human endothelial cells by activated protein C. J Immunol. 1998;161:2567–2573. [PubMed: 9725257]
- 124.
- Robson SC, Saunders R, Kirsch RE. Monocyte-macrophage release of IL-1 is inhibited by type-1 plasminogen activator inhibitor. J Clin Lab Immunol. 1990;33:83–90. [PubMed: 1967070]
- 125.
- Sitrin RG, Shollenberger SB, Strieter RM, Gyetko MR. Endogenously produced urokinase amplifies tumor necrosis factor-a secretion by THP-1 mononuclear phagocytes. J Leukoc Biol. 1996;59:302–311. [PubMed: 8604004]
- 126.
- Parmely MJ, Sterner KE, Gale A, Zhou WW. U937 cells can utilize plasminogen activator to regulate human interferon-γ J Interferon Res. 1993;13:397–406. [PubMed: 8151133]
- 127.
- Taylor F B Jr., Chang A C K, Peer GT, Mather T, Blick K, Catlett R. et al. DEGR-factor Xa blocks disseminated intravascular coagulation initiated by Escherichia coli without preventing shock or organ damage. Blood. 1991;78:364–368. [PubMed: 2070073]
- 128.
- Uchiba M, Okajima K, Murakami K, Nawa K, Okabe H, Takatsuki K. Recombinant human soluble thrombomodulin reduces endotoxin-induced pulmonary vascular injury via protein C activation in rats. Thromb Haemost. 1995;74:1265–1270. [PubMed: 8607107]
- 129.
- Taoka Y, Okajima K, Uchiba M, Murakami K, Harada N, Johno M. et al. Activated protein C reduces the severity of compression-induced spinal cord injury in rats by inhibiting activation of leukocytes. J Neurosci. 1998;18:1393–1398. [PMC free article: PMC6792717] [PubMed: 9454848]
- Introduction
- Production of Cytokines
- Role of Cytokines in Organ Toxicity in Systemic Infection and Inflammation
- Effect of Cytokines on Tissue Factor and Coagulation Activation
- Effect of Cytokines on the Protein C-Protein S-Thrombomodulin System
- Effect of Cytokines on the Fibrinolytic System
- Interplay Between Cytokines and Mediators of Hemostasis
- Conclusion
- References
- Cytokines as Regulators of Coagulation - Madame Curie Bioscience DatabaseCytokines as Regulators of Coagulation - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...