

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Abelson (Abl) family nonreceptor tyrosine kinases are essential regulators of cell morphogenesis in developing metazoan organisms. Mutant animals that lack Abl kinases exhibit defects in epithelial and neuronal morphogenesis. In cultured cells, the vertebrate Abl and Abl-related gene (Arg) proteins promote formation of actin-based protrusive structures, such as filopodia and lamellipodia. Abl family kinases act as relays that coordinate changes in cytoskeletal structure in response growth factors and adhesive adhesion receptor activation. These cytoskeletal rearrangements are achieved through the ability of these kinases to control the Rho and Rac GTPases and to stimulate assembly of protein complexes that activate nucleation of actin filaments by the Arp2/3 complex. Abl and Arg also contain extended C-termini that bind directly to F-actin and microtubules and may mediate interactions between these cytoskeletal networks in cells. Arg, for instance, can promote the cooperative assembly of an F-actin-rich scaffold in cells, which may serve as a base for the elaboration of actin-rich protrusive structures in cells.
Introduction
Changes in cell shape and migration are powered by dynamic rearrangements of the actin cytoskeleton. These changes are coordinated in space and time by protein machines that regulate actin filament polymerization/depolymerization, organize actin filaments into bundles or networks, and push or pull on these actin superstructures. These actin-based machines are controlled by signals from cell surface receptors that provide continuous updates on the extracellular physicochemical environment.
Abelson (Abl) family nonreceptor tyrosine kinases are essential regulators of cytoskeletal rearrangements in developing metazoan organisms. Abl family kinases act as relays that coordinate changes in cytoskeletal structure in response to discrete external cues. In this chapter, I review the cellular and developmental processes regulated by Abl family kinases and discuss the molecular mechanisms by which Abl family kinases control these processes.
Abl Family Kinases: Basic Anatomy
The N-Terminal Half of Abl Family Kinases Is Highly Conserved
The Abl kinase family is composed of the Abl and Arg (Abl-related gene, aka Abl2) kinases in vertebrates,1,2 the Abl kinase in flies,3 and the Abl-1 kinase in worms.4 Following a short variable region, Abl family kinases contain tandem Src homology (SH) 3, SH2, and tyrosine kinase domains (Fig. 1). The SH3 and SH2 domains regulate kinase activity. In the inactive state, the SH3 and SH2 domains form an inhibitory scaffold along the back face of the kinase domain that holds it in the “off ” state.5,6 Upon activation, the molecule undergoes a conformational change in which the SH3 and SH2 are relieved from their inhibitory conformation and promote kinase activity by facilitating interactions with substrates.7,8 The Chapter by Hantschel and Superti-Furga contains an excellent review of Abl kinase regulation.
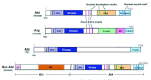
The C-Terminal Halves of Abl Family Kinases Contain Actin and Microtubule-Binding Domains
Abl family kinases share less sequence conservation downstream of the tyrosine kinase domain. The functional features of the C-terminal half are most well-characterized in Abl and Arg. Immediately C-terminal to the kinase domain, Abl and Arg contain three conserved Pro-X-X-Pro (PXXP) motifs that serve as binding sites for SH3-domain-containing proteins9-13 (Fig. 1). The C-terminal halves of Abl family kinases are unique among nonreceptor tyrosine kinases because they contain multiple domains that interact directly with actin and microtubules. Both Abl and Arg share C-terminal calponin homology F-actin-binding domains14-16 (Fig. 1). A globular (G-) actin-binding domain precedes this F-actin-binding domain in Abl,15 whereas in Arg, it is preceded by an I/LWEQ homology F-actin-binding domain and a microtubule-binding domain.16,17 Based on sequence comparisons, the fly Abl and Abl-1 proteins in worms are likely to contain an F-actin binding domain at their C-termini, but the actin-binding properties of these family members have not yet been examined.
Abl Contains Nuclear Localization and Export Sequences
In addition to the features that Abl shares with Arg, the C-terminal half of Abl also contains a DNA-binding domain with 3 HMG-like motifs,18 3 nuclear localization sequences,19 and a nuclear export sequence20 (Fig. 1). These features appear to be important for Abl to function in the cell nucleus. The Chapter by Wang, Minami, and Zhu contains an excellent review of Abl's nuclear functions.
The Bcr-Abl Oncoprotein
Mutant forms of Abl cause chronic myelogenous leukemia (CML) and acute lymphocytic leukemia (ALL) in humans. Oncogenic activation of Abl is most commonly associated with a chromosomal translocation that fuses sequences encoding a portion of the Bcr protein to an N-terminal part of Abl resulting in a hybrid gene encoding the Bcr-Abl oncoprotein.21 A coiled-coil motif in Bcr promotes oligomerization of the Bcr-Abl fusion protein22,23 (Fig. 1). This oligomerization promotes kinase autophosphorylation and activates Abl tyrosine kinase activity,24-26 leading to the activation of anti-apoptotic and mitogenic signaling pathways.27-29 Oligomerization also promotes binding of Bcr-Abl to F-actin stress fibers,24 which may contribute to the alterations in cytoskeletal structure30 and cell adhesion systems in Bcr-Abl-transformed cells.31
Roles for Abl Family Kinases in Cellular Morphogenesis
Genetic studies reveal that Abl family kinases are essential for normal epithelial and neuronal cell morphogenesis during development. Studies of Abl family kinase function in cultured cells support a model in which Abl family kinases relay information from adhesion receptors and growth factor receptors to direct specific changes in cytoskeletal structure and function. This Chapter will not include Abl family kinase regulation of neuronal morphogenesis and synaptic function, which is reviewed in an excellent Chapter by Thompson and Van Vactor.
Abl and Arg Promote Cell Protrusions in Response to Growth Factors or Adhesive Cues
The first evidence that Abl family kinases regulate actin-based protrusions of the cell membrane came from observations of Bcr-Abl-transformed cells.30 Salgia and colleagues showed that Bcr-Abl-transformed NIH3T3 fibroblasts exhibit more filopodia, lamellipodia, and membrane ruffles than their untransformed counterparts.30 In contrast to the normal smooth round appearance of BaF3 hematopoietic cells, Bcr-Abl-expressing BaF3 cells extended numerous pseudopodia and were hypermotile on various adhesive surfaces.30 Despite this clear demonstration that an activated form of Abl could promote changes in cytoskeletal structure, it was unclear how this activity related to the normal functions of Abl or Arg.
Subsequent studies of Abl- and/or Arg-deficient fibroblasts have clearly demonstrated that Abl and Arg act downstream of growth factor and adhesion receptors to promote cytoskeletal rearrangements. Application of platelet-derived growth factor (PDGF) to fibroblasts leads to dramatic actin-based ruffling of the cell periphery.32,33 The finding that PDGF induces Abl kinase activity led Plattner and Pendergast and colleagues to examine a role for Abl in membrane ruffling (Fig. 2A). Abl-deficient fibroblasts exhibit dramatically reduced membrane ruffling in response to PDGF, but this response can be restored by Abl reexpression.33 These studies were the first to demonstrate that a normal function of Abl is to coordinate morphogenetic responses to a specific extracellular cue. Subsequent studies have further confirmed that Abl family kinases are required for membrane ruffling following cell exposure to PDGF and epithelial growth factor (EGF).34 Although the function of these growth factor-induced ruffles is unknown, they may promote cell migration by helping cells sample three-dimensional space.
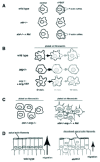
Abl family kinases also mediate cytoskeletal rearrangements in response to integrin-mediated adhesion to extracellular matrix proteins. Wild type fibroblasts are highly dynamic as they attach and spread on fibronectin-coated surfaces, frequently extending and retracting actin-based lamellipodial and filopodial protrusions.17 Kymographic analysis reveals that Arg-deficient fibroblasts exhibit significantly fewer lamellipodial protrusions and retractions than wild type fibroblasts during adhesion and spreading on fibronectin (Fig. 2B). Reexpression of a functional Arg-yellow fluorescent protein (Arg-YFP) fusion restores normal lamellipodial behavior to these cells.17 Importantly, Arg-YFP colocalizes with F-actin in these protrusive lamellipodial structures.16,17 In a similar vein, abl-/-arg-/- fibroblasts, which have regular smooth peripheries as they adhere to and spread on fibronectin,35 form multiple actin-based filopodial microspikes on fibronectin when Abl is reexpressed in these cells35 (Fig. 2C). Abl localizes to the tips of these filopodia,36 where it may regulate their protrusion, retraction, or adhesion to the extracellular matrix.
These studies demonstrate that Abl and Arg can each coordinate cell protrusions downstream of integrin adhesion receptors. The degree to which Abl and Arg overlap in the regulation of filopodia and lamellipodia is currently unclear. Simultaneous localization of fluorescently-labeled Abl and Arg in fibroblasts should reveal which protrusive structures contain which kinases. The analysis of abl-/-arg-/- cells reconstituted with Abl or Arg alone or both kinases should clarify which protrusions depend on which kinases.
Abl Family Kinases Regulate Epithelial Morphogenesis
Genetic studies in flies and mice have revealed a conserved function for Abl family kinases in the regulation of epithelial cell morphogenesis.
The dorsal epidermis in flies is created through the convergent migration and joining of epithelial sheets, a process known as dorsal closure. Cells at the leading edge of the sheet uniformly elongate and assemble a regular actomyosin “purse-string” at their leading edges. The uniform constriction of this purse-string is believed to coordinate the drawing together of epithelial sheets. The Enabled (Ena) protein, a regulator of actin filament length, is enriched at the adherens junctions of these leading edge cells, where it likely helps coordinate the deposition and integrity of the actin purse-string.
Flies that lack both maternal and zygotic sources of fly Abl (ablMZ mutants) exhibit defects in dorsal epithelial closure.37ablMZ epithelial cells do not elongate uniformly perpendicular to the closure (Fig. 2D). Ena localization is less uniform in the ablMZ epithelial cells, with some cells containing larger concentrations of Ena than their neighbors. Cells with more Ena have more F-actin, leading to an uneven organization of the purse string. As a result of these defects, dorsal closure proceeds more slowly and irregularly in ablMZ embryos and in some embryos, holes in the epidermis remain. Consistent with a possible alteration in adherens junction structure and function, ablMZ mutants express reduced levels of α-catenin and β-catenin.
Formation of the neural tube, the precursor to the vertebrate brain and spinal cord has many similarities to dorsal closure in flies. The central nervous system begins as a flat sheet of ectoderm along the dorsomedial surface of the developing vertebrate embryo. The cells first undergo microtubule-dependent elongation to form the columnar epithelium of the neural plate.38-40 Subsequent to this thickening, a contractile actomyosin latticework is formed at the apical surface of these neuroepithelial cells. Contraction of these filaments contributes to the polarized wedge shape that is essential for formation and maintenance of the neural tube.38,40,41
Abl and Arg are expressed in the mouse neuroepithelium, where they colocalize with the contractile apical actin latticework. abl-/-arg-/- embryos suffer from severe defects in neural tube formation.41a Closure of the neural tube is delayed and even incomplete in some embryos. Upon closure, the neural tube of abl-/- arg-/- embryos collapses into the central lumen of the neural tube. abl-/- arg-/- neuroepithelial cells exhibit patchy disruptions of the apical actin latticework and ectopic actin-rich contractile structures at the basolateral surface. The fact that these defects are never observed in abl-/- or arg-/- single mutant embryos, suggests that Abl and Arg play redundant roles neuroepithelial morphogenesis.
Abl Regulates Cellularization in Fly Embryos
ablMZ mutant flies have a high number of multinucleate cells.42 This phenotype can be traced to defects in the formation of the pseudocleavage and cellularization furrows during the late syncytial stages of development.
In the late syncytial stages, nuclei undergo a coordinated localization to the embryo cortex where they continue through 4 coordinated rounds of cell division. During division, the nuclei become separate by an actin-rich pseudocleavage furrow that forms an inverted cup around the metaphase plate. After the final division, a cellularization furrow extends down and engulfs the entire nucleus with membrane.
The formation of pseudocleavage and cellularization furrows is controlled by dynamic rearrangements of the actin cytoskeleton. Actin forms a cap above interphase nuclei, but relocalizes and forms a pseudocleavage furrow during metaphase. Immunohistochemical staining showed that Abl is concentrated at apical junctions and localization extends down into cleavage furrows during these events.43 Some of these pseudocleavage furrows disintegrate in abl mutants, allowing two nuclei to share the same compartment. These defects correlate with increased staining for Ena, Diaphanous, the Arp2/3 complex, and F-actin at the apical surface and reduced F-actin in the pseudocleavage and cellularization furrows. A reduction in Ena levels suppresses the cellularization defects in abl mutants. Together, these studies suggest that Abl acts at the apical surface of the cell to manage Ena localization or activity.
Pathogens Exploit Abl-Regulated Pathways to Infect and Traffic within and between Cells
The ability of Abl family kinases to promote cell protrusions has been hijacked by some viruses and intracellular bacteria as a means to infect and move within and between cells.
The pathogenic bacterium Shigella flexneri infects nonphagocytic cells of the colonic mucosal lining. Contact between Shigella and a target cell induces actin-based protrusive structures that surround and eventually engulf the bacterium.44 Abl and Arg localize to bacterial entry sites45 and their kinase activities are induced by exposure to Shigella. This induction of kinase activity is critical: abl-/- arg-/- cells or wild type cells treated with the Abl/Arg kinase inhibitor STI571 are resistant to Shigella infection. Activation of Abl/Arg by Shigella infection leads to increased phosphorylation of the Crk adaptor protein and is associated with downstream activation of the Rac and Cdc42 GTPases.45 Activation of these downstream pathways is likely to underlie the complex actin-based rearrangements required for Shigella internalization.45 In support of this, expression of a mutant Crk with a nonphosphorylatable substitution at the Abl/Arg phosphorylation site can inhibit Shigella infection. Once inside cells, Shigella escapes the vacuole and propels itself through the cytoplasm on a “comet tail” of polymerized actin. The IcsA coat protein on Shigella nucleates actin comet formation by recruiting and activating the N-WASP protein to promote Arp2/3 complex-dependent actin polymerization. It is not clear whether Abl family kinases are involved in the formation of the Shigella actin comet tail.
Vaccinia virus, a member of the poxvirus family, also utilizes cellular cytoskeletal systems to traffic within and between cells. Following replication and viral particle assembly in the cytoplasm, some particles become coated with membrane. These intracellular enveloped virions (IEVs) traffic to the cell surface along microtubules using the kinesin motor protein.46,47 Once at the plasma membrane, the virus induces actin tail formation, which is believed to allow release of cell-associated enveloped virus (CEV) by propelling it away from the cell surface. Src family kinases localize to viral particles where they phosphorylate the vaccinia A36R protein, and recruit the Grb2 and Nck adaptors, N-WASP, and the Arp2/3 complex to promote formation of the actin comet tail.48
A recent study also shows that Abl and Arg localize to vaccinia virus comet tails.49 Analysis of comet tail formation in mutant cell lines or in cells treated with different kinase inhibitors suggests that comet tail formation may require either Abl family or Src family kinases. Treatment with the selective Abl/Arg inhibitor STI571 (Gleevec) reduced the release of the enveloped virus.49 Treatment of animals with STI571 also blocked the spread of vaccinia virus, although it remains possible that the inhibitor may be inhibiting Src or other kinases under the concentrations at which it was used. Nevertheless, this study introduces the possibility of using kinase inhibitors as adjuvants to vaccination to control poxvirus infection.
Mechanisms by Which Abl Family Kinases Regulate Cytoskeletal Structure
Abl family kinases respond to signals from cell surface receptors by directing changes in cytoskeletal structure. The past several years have seen a great increase in our understanding of the molecular mechanisms by which Abl family kinases act to control cytoskeletal rearrangements. A dominant theme is that Abl family kinases regulate the cytoskeletal structure by controlling the activity of the Rho family GTPases Rho and Rac. New studies also suggest that Abl family kinases can act directly on regulators of the Arp2/3 complex to promote actin polymerization. Finally, several recent experiments suggest that the C-terminus of Abl family kinases can act in a kinase-independent manner to regulate the structure of the F-actin and microtubule cytoskeletons.
A Primer: Rho Family GTPases Are Master Regulators of Cytoskeletal Rearrangements
Rho family GTPases, such as Rho, Rac, and Cdc42 act as molecular switches that regulate cytoskeletal rearrangements by cycling between an inactive GDP-bound form and an active GTP-bound form. In their active forms, Rho family GTPases interact with various effectors that regulate actin polymerization, F-actin severing, F-actin bundling, and actomyosin contractility.50-52 Different Rho family GTPases interface with different effectors to produce different cytoskeletal structures, such as actin stress fibers, filopodia, or lamellipodia.53-56
Signals originating from cell surface receptors control the activity of Rho family GTPases by acting on two classes of regulatory molecules: guanine nucleotide exchange factors (GEFs) that activate Rho family GTPases by promoting the exchange of GTP for GDP and GTPase-activating proteins (GAPs) that inhibit Rho family GTPases by stimulating them to hydrolyze bound GTP. In some cases, the GEFs and GAPs are controlled directly by cell surface receptors. Alternatively, GEFs and GAPs can be controlled through the activity of a variety of protein kinases and cellular binding partners.
Regulation of the Rac GTPase by Abl Family Kinases
The Rac GTPase is a central regulator of lamellipodial formation and membrane ruffling in response to growth factor receptor signaling.52,54 Active Rac promotes the recruitment and assembly of complexes containing WASp/WAVE-family proteins which activate the Arp2/3 complex to nucleate new actin filaments.57
Several observations in diverse experimental systems have implicated Abl family kinases in the regulation of Rac activity. Rac, Abl, and Arg all become activated in fibroblasts following PDGF treatment and both Rac and Abl activities are essential for PDGF-induced membrane ruffling.33,58,59 Rac is also activated by Bcr-Abl and expression of a dominant-negative Rac can significantly delay disease onset in a mouse model of Bcr-Abl-induced leukemogenesis.60 Similarly, dominant-negative Rac blocks some of the mitogenic effects of the v-Abl oncoprotein in fibroblasts.61 Rac and Cdc42 are both activated upon cellular exposure to Shigella in wild type cells, but this activation is not observed in abl-/- arg-/- cells.45 Thus, Abl family kinases control Rac activation in a wide variety of contexts.
Growth-Factor-Induced Rac Activation Requires Abl Phosphorylation of Sos-1
The murine Son-of-Sevenless 1 (Sos-1) can act as a GEF for both Ras and Rac, but acts as a Rac GEF when complexed with the Eps8 and Abi1 proteins following growth factor stimulation.62 Exposure of cells to EGF or PDGF leads to increased phosphorylation of Sos-134 (Fig. 3). This growth factor-induced phosphorylation of Sos-1 is blocked in abl-/- arg-/- cells or in wild type cells treated with the Abl/Arg kinase inhibitor STI571. Sos-1 phosphorylation correlates with increased Rac GEF activity and this GEF activity is reduced by treatment with alkaline phosphatase. Growth factor-induced-Rac activation is reduced in abl-/- arg-/- cells, but can be increased upon reexpression of Abl. The reduced Rac activation has functional consequences, as abl-/- and abl-/- arg-/- cells exhibit a reduced ruffling response to PDGF.33,34 These data demonstrate that Abl (and possibly Arg) contribute to Rac activation downstream of growth factor stimulation.

Interestingly, while Abl phosphorylation stimulates Sos-1 Rac GEF activity, its Ras GEF activity is high irrespective of its phosphorylation status. The basis of this differential requirement for phosphorylation awaits further biochemical studies to elucidate the molecular mechanisms by which Abl phosphorylation regulates Sos-1.
Abl Modulates Rac Activation Downstream of Integrin-Mediated Adhesion
The CAS (Crk-associated substrate) protein becomes heavily phosphorylated in response to integrin-mediated adhesion (see refs. 63 ,64 and references therein). Tyrosine phosphorylation of CAS promotes its assembly into a Rac-activating complex that contains the adaptor protein CrkII (Crk), Elmo, and the Rac GEF Dock180.63,64 The Crk adaptor contains an SH2 domain followed by two SH3 domains. Crk uses these domains to hold together the Rac activation complex: the Crk SH2 domain binds to phosphorylated CAS,65 while the first Crk SH3 domain associates with Dock180.66
A number of observations suggest that Abl family kinases regulate the formation of the CAS/Crk/Dock180/Elmo complex. CrkII binds Abl and Arg and serves as their substrate in vitro and in vivo,9,67,68 with Abl and Arg phosphorylating Crk at a single tyrosine residue (Y221) in a linker region between its two SH3 domains.9,67 The Crk SH2 domain can bind to its own tyrosine-phosphorylated linker region, and therefore this phosphorylation has been proposed to inhibit Crk binding to other phosphotyrosine-containing proteins, including CAS.67 Abl kinase activity levels do negatively correlate with Crk binding to CAS in vivo. For example, whereas abl-/- arg-/- fibroblasts have high levels of Crk:CAS containing-complexes, formation of these complexes is reduced in cells reexpressing Abl.69
It is difficult to reconcile a simple model in which Abl phosphorylation of Crk inhibits the formation Crk:CAS complexes with the numerous examples (cited above) in which increased Abl kinase activity is associated with Rac activation. The more likely scenario is that phosphorylation of Crk represents an intermediate in the assembly of Crk/CAS/Elmo/Dock180 complexes (Fig. 3). Chodniewicz and Klemke propose a model in which Abl family kinases form complexes with Crk and help relocate it to the plasma membrane.64 Once at the membrane the Crk:Abl complex may be dissociated by phosphatases, leaving Crk available to associate with CAS and Dock180. Mutation of tyrosine 221 in Crk to phenylalanine promotes increased complex formation with Abl70 and blocks activation of Rac by Abl family kinases.45 This residue may play an important role in the dissociating Crk from Abl and promoting its association with CAS and Dock180.
Arg Is Required for Integrin-Dependent Inhibition of Rho
The RhoA (Rho) GTPase promotes the formation of actin stress fibers and focal adhesions in response to growth factor receptor signaling.53 Active Rho activates Rho kinase, thereby promoting actomyosin contractility necessary for focal adhesion and stress fiber formation.52
Arg is particularly abundant in developing brain tissue and analysis of phosphotyrosine-containing proteins during postnatal brain development revealed that the 190 kD GTPase-activating protein for Rho (p190RhoGAP) has reduced tyrosine phosphorylation in arg-/- brain. Arg phosphorylates p190RhoGAP directly on tyrosine 1105 (Y1105), thereby stimulating its ability to inhibit Rho.71 Integrin-mediated adhesion to fibronectin promotes p190RhoGAP phosphorylation in wild type fibroblasts. This adhesion-dependent phosphorylation of p190RhoGAP is absent from arg-/- fibroblasts, but can be restored by reexpression of a functional Arg-YFP fusion protein. Arg-deficient fibroblasts have larger, more numerous focal adhesions and larger stress fibers, a phenotype that appears to result from hyperactive Rho in these cells (A.L. Miller and A.J. Koleske, unpublished data). These observations suggest that Arg acts on p190RhoGAP to inhibit Rho following integrin-mediated cell attachment and spreading (Fig. 3).
Treatment of cultured neurons with the Abl/Arg inhibitor STI571 leads to simplification of neurite structure35,72,73 that correlates with an increase in Rho activity.72 Inhibition of Rho suppresses the effects of STI571 treatment on neurite structure.72 This finding supports a model in which Abl family kinases act to inhibit Rho.
Phosphorylation of Y1105 promotes formation of a complex between p190RhoGAP and the 120 kD GTPase activating protein for Ras (p120RasGAP).74,75 This interaction is mediated in part by binding of one of two SH2 domains in p120RasGAP to the phosphorylated Y1105 in p190RhoGAP.71,75 Increased p190RhoGAP phosphorylation and increased formation of the p190RhoGAP:p120RasGAP complex correlates with increased disassembly of actin stress fibers in Src-overexpressing or EGF-treated cells.76,77 These data suggest that Arg-mediated phosphorylation activates p190RhoGAP by promoting its binding to p120RasGAP.
Previous studies had shown that p190RhoGAP is a major cellular target of Src family kinases.78 Adhesion-dependent suppression of Rho activity does not occur in fibroblasts that lack the Src, Yes, and Fyn kinases (SYF fibroblasts).79 An adhesion-dependent increase in p190RhoGAP phosphorylation is also not observed in these cells. Interestingly, Src family kinases phosphorylate p190RhoGAP at Y1105, the same site targeted by Arg.75
Arg and Src family kinases might represent alternative pathways leading from integrin receptors to p190RhoGAP phosphorylation. However, this model would not explain why elimination of either Arg or Src/Fyn/Yes eliminates all adhesion-dependent p190RhoGAP phosphorylation. A more likely scenario is that Arg and Src family kinases are in the same cascade, but only one of the kinases actually phosphorylates p190RhoGAP. Several studies have shown that Src is required for Abl kinase activation upon growth factor receptor stimulation.33,80 Src family kinases activate Abl family kinases through phosphorylation of an activation loop tyrosine in the Abl/Arg kinase domain.33,80-82 Arg phosphorylates p190RhoGAP in vitro with a KM of 130 nM. Src can also phosphorylate p190RhoGAP in vitro, but it is unclear how its efficiency compares to that of Arg.75 These observations suggest a model in which integrin engagement activates Src to phosphorylate Arg, which, in turn, phosphorylates and activates p190RhoGAP. Alternatively, Arg or Src might be required to set up an appropriate cellular microenvironment to allow phosphorylation of p190RhoGAP by the other kinase.
Abl Family Kinases also Regulate Actin Structure Independently of Rho GTPases
In addition to their ability to regulate Rho and Rac, Abl family kinases interact with several actin regulatory complexes to control cytoskeletal rearrangements.
Abl Interactor (Abi) Proteins Mediate Interactions between Abl Family Kinases and Arp2/3 Regulatory Complexes
The Abi-1 and Abi-2 proteins were identified in two-hybrid screens for proteins that interact with Abl and Arg.11-13 Abi-1 localizes to filopodial and lamellipodial tips, sites of active actin polymerization.83 Abi-1 and Abi-2 have been identified in protein complexes containing WAVE proteins, which promote Arp2/3 complex-dependent actin polymerization. These complexes also contain the Nap1 and Pir120 proteins.84-88 Together, these studies raise the possibility that Abi-1 and Abi-2 mediate interactions with Abl family kinases and WAVE proteins. Like Abi-1, Abl and Arg have been localized to sites of actin polymerization at lamellipodial protrusions and filopodial tips. Abl was recently shown to be part of a complex containing WAVE2 and Abi-1.89 Adhesion to fibronectin stimulates translocation of these proteins to the cell periphery where they colocalize with F-actin-rich lamellipodia. Activation of Abl kinase activity promotes tyrosine phosphorylation of Abi-1 and WAVE2. Mutation of a single tyrosine residue (Y150) in WAVE2 reduced tyrosine phosphorylation of WAVE2 and assembly of complexes containing WAVE2 and Abi-1. Abl also can stimulate the ability of purified Abi-1/WAVE2 to activate Arp2/3 complex-mediated actin polymerization in vitro. Together, these findings suggest that Abl mediates the formation of an actin polymerization regulatory complex containing in response to integrin-mediated adhesion (Fig. 4A). It is unclear at present whether the Abi-1:Abl:WAVE2 complexes also contain other proteins (e.g., PIR121, Nap1, HSPC300) that have been described in WAVE-containing complexes.
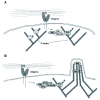
The Arg-interacting protein nArgBP2 was also recently shown to bind to WAVE1 and WAVE2.90 nArgBP2 might serve as an alternative to Abi proteins to mediate interactions between Abl and WAVE family proteins.
Abl Phosphorylation of Dok Family Proteins Promotes Filopodia Formation
The Downstream of kinase (Dok1) protein was originally identified as a protein that was heavily phosphorylated in Bcr-Abl-transformed cells.91,92 Subsequent studies have identified 5 family members (Dok1-5), each containing a pleckstrin homology (PH) domain, a phosphotyrosine binding (PTB) and a C-terminal tail with multiple potential tyrosine phosphorylation sites. Dok1 and Dok2 can serve as essential downstream effectors of Abl in filopodium/actin microspike formation.
Dok1 becomes tyrosine phosphorylated following integrin-mediated adhesion. This phosphorylated Dok1 binds avidly to the Abl SH2 domain.36 Phosphorylation occurs on a single site (Y361) located in the C-terminal tail. In addition to having several potential phosphorylation sites, the Dok2 tail contains a PMMP motif that can interact selectively with the Abl SH3 domain.93 Mutation of this motif abrogates Dok2 association with Abl when the two proteins are coexpressed. Abl expression also promotes Dok2 phosphorylation, which elevates Abl kinase activity, most likely by stabilizing interactions between the two proteins.
Expression of Dok-1 or Dok-2 with Abl leads to increased formation of F-actin microspikes.36,93 Phosphorylation of Y361 in the Dok1 tail serves as a binding site for the Nck adaptor protein, which can bind to and activate cytoskeletal regulators such as WASp,94 N-WASP,95 and PAK.96 These findings lead to a model in which Abl phosphorylation promotes the assembly of an Abl:Dok1:Nck complex that promotes actin polymerization into microspikes/filopodia (Fig. 4B). In support of this model, Abl and Dok1 both localize to filopodial tips. Cells that lack Dok-1 or both Nck1 and Nck2 form fewer actin-rich filopodia when plated on fibronectin. Ena/VASP family proteins are also found at the filopodial tips where they may regulate the actin filament elongation.97
Abl Family Kinases Interact Functionally with Ena/VASP Proteins
Ena/VASP proteins, which include the fly Enabled (Ena) protein, Unc-34/Enabled in worms, and the mammalian Enabled (Mena), VASP (vasodilator-stimulated phosphoprotein), and EVL (Ena/VASP-like protein), regulate the elongation of actin filaments in a number of different cellular structures.98 Despite the identification of the enabled gene over 15 years ago as a dosage-dependent modifier of the abl mutant fly phenotypes,99 we still do not have a clear mechanistic picture of how Abl family kinases interact at a molecular level with Ena/VASP family proteins.
Genetic studies in flies suggest that Abl may regulate the localization of Ena/VASP family proteins. In the late syncytial divisions and during cellularization in wild type embryos, Ena is distributed at moderate levels at the apical cortex, with weaker levels throughout the cytoplasm.42 In abl mutant flies, Ena is strongly enriched at apical surface in association with the increased F-actin structures. This observation suggests that Abl acts to inhibit Ena localization at the apical surface, possibly by interfering with the binding of Ena to one of its partners.
Ena/VASP proteins contain a central proline-rich domain (PRD) that can bind to the Abl SH3 domain. Binding of Abl to the PRD could prevent Ena from binding to other proteins via this domain. Moreover, phosphorylation of specific residues in the PRD of Ena, EVL, and VASP abolishes their ability to bind to SH3-domain-containing proteins.100 These phosphorylations appear to regulate Ena/VASP protein interactions in response to discrete extracellular cues. For example, adhesion-dependent phosphorylation of VASP by protein kinase A prevents its binding to Abl during initial cell spreading.100 Abl phosphorylation of Ena in vitro prevents its subsequent binding to the Abl SH3 domain. Vertebrate Abl and fly Abl can promote Mena and Ena phosphorylation respectively when they are coexpressed in cultured cells and this phosphorylation is enhanced by expression of Abi-1 or fly Abi, respectively.101,102 These data suggest that Abi proteins might stabilize interactions between Ena/VASP proteins and Abl family kinases to allow for Ena/VASP phosphorylation. NABP Future studies should clarify whether this event occurs as part of a response to physiological cues.
Abl Family Kinases Interact Directly with F-Actin and Microtubules
Abl family kinases are unique among nonreceptor tyrosine kinases in having extended C-terminal halves that contain domains that bind to actin, and, in the case of Arg, microtubules (see Fig. 1). In addition to their interactions with cytoskeletal regulatory proteins, Abl family kinases can influence cytoskeletal structure through these direct interactions with cytoskeletal components.
Arg Bundles F-Actin and May Form an F-Actin Scaffold to Recruit Cytoskeletal Regulators
Arg contains two distinct F-actin binding domains: a calponin homology (CH) F-actin binding domain at its C-terminus and an I/LWEQ (talin-like) F-actin binding domain located midway between its kinase domain and the CH domain.16 The region in Abl corresponding to the CH domain in Arg can be separated into two distinct regions that bind to G- and F-actin.15 An internal domain in Abl corresponding to the I/LWEQ domain can also bind weakly to F-actin (Maithreyi Krishnaswami and A.J.K. unpublished data).
Arg can assemble F-actin into tight bundles in vitro and this activity requires both the I/LWEQ and CH domains.16 Reconstruction of electron microscopic images of short segments of Arg:F-actin complexes revealed that Arg could bind to F-actin in several modes.103 Analysis of Arg mutants lacking either the I/LWEQ or CH domains revealed that the different modes resulted from binding via the different F-actin binding domains. The CH domain bound to subdomain 1 (SD1) of actin and induced a tilt in the actin filament. The I/LWEQ could bind in two modes to either SD1 or SD4. Importantly, only one mode of F-actin binding was found in each Arg:F-actin segment. These data showed that Arg cannot use both domains to bind simultaneously to the same filament. All of the binding modes leave the other domain available to bind an adjacent filament for bundle formation.
Arg binds cooperatively to F-actin in vitro during bundle formation.16 This property may help concentrate Arg locally within cells, as local Arg binding to F-actin would promote additional binding of Arg to F-actin locally. Electron microscopic structural studies of Arg bound to individual actin filaments show that Arg binding induces structural changes to the actin filament, changing both the structure of actin subunits and their helical pitch.103 These structural changes are highly cooperative and propagate along patches of unoccupied actin filament. Importantly, some of the changes in actin filament structure resemble the F-actin conformation found in Arg:F-actin complexes. These findings suggest Arg-induced structural alterations to the actin filament could promote Arg binding to F-actin at sites distal to the initial binding site.
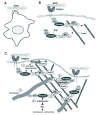
An important unresolved issue is whether Arg organizes F-actin into bundles or higher-order structures within cells. In fibroblasts attaching to fibronectin, Arg localizes to the periphery where it promotes the formation of F-actin-rich protrusive lamellipodial structures. Abl can also bundle F-actin in vitro, and is found in association with F-actin-rich structures in a variety of cell types.36,104 An Arg C-terminal fragment (Arg688-1182) containing the two F-actin-binding domains is sufficient to form F-actin-rich structures at the cell periphery. These observations suggest that Arg can organize actin filaments into bundles or other higher order structures in vivo. However, while Arg induces highly dynamic F-actin-rich structures that protrude and retract, the F-actin-rich structures induced by Arg688-1182 are largely static. These findings suggest that Arg uses its 688-1182 fragment to localize to the periphery and locally organize F-actin structure, but likely requires other domains to recruit and/or regulate WASp/WAVE proteins and Rho family GTPases to further elaborate these actin-based scaffolds into dynamic structures (Fig. 5). Thus, Arg could simultaneously act as both a building block of an actin bundle scaffold and a regulator of the addition and dynamics of actin filaments arising from this scaffold. Future studies using fluorescence, video and electron microscopy should reveal the cytoskeletal ultrastructure and distribution of regulatory proteins in the Arg-containing protrusive structures.
Abl May Be Subject to Feedback Inhibition by F-Actin
Abl kinase activity is inhibited in fibroblasts that are held in suspension, and is induced upon attachment of cells to fibronectin.35,105,106 An increased amount of F-actin coimmunoprecipitates with Abl from suspended cells relative to adherent cells. Treatment of cells with Latrunculin A, which reduces cellular F-actin concentration by blocking actin polymerization, leads to a potent induction of Abl kinase activity, even in suspended cells. Together, these observations strongly suggest that Abl kinase activity is influenced by differences in F-actin abundance or conformation in suspended versus adherent cells.
It has been proposed that F-actin inhibits Abl kinase activity by stabilizing an inhibited conformation of the enzyme.107 Purified F-actin inhibits the ability of Abl to phosphorylate a peptide derived from C-terminal domain (CTD) of RNA polymerase II in vitro.106 Both the F-actin-binding and SH2 domains of Abl are required for effect. However, F-actin does not inhibit the ability of Abl to phosphorylate its substrate Crk in vitro.108 One possible explanation for this selective inhibitory effect is that F-actin competes with the CTD for binding to the Abl SH2 or F-actin-binding domains. The CTD binds with low micromolar affinity to an Abl fragment containing the F-actin binding domain.109 Abl mutants lacking the SH2 or F-actin-binding domains do not bind to F-actin in vitro and therefore would not be inhibited by F-actin. Abl phosphorylation of Crk is not inhibited because Crk does not interact with the SH2 or F-actin binding domains.
A more conventional scenario is that Abl exists in an inactive conformation in unattached cells and integrin-mediated adhesion promotes activation of Abl kinase activity. If this is the case, how does Latrunculin A treatment lead to activation of Abl kinase activity in the absence of adhesion? Latrunculin A treatment of suspended fibroblasts might activate adhesion receptors, mobilize calcium, or activate Rho family GTPases as it has been shown to do in other cell types.110,111 One or more of these pathways could activate Abl kinase activity.
Abl Family Kinases Mediate Interactions between the F-Actin and Microtubule Networks
Polarized cell migration requires dynamic interactions between the actin and microtubule cytoskeletons.112,113 MTs assume a polarized orientation in migrating cells, with their plus ends pointed toward the leading edge.112,113 Microtubule extension into the periphery is required for lamellipodial protrusion at the leading edge.114 Recent studies suggest that Abl family kinases might mediate interactions between F-actin and MTs at the leading edge.
Arg has a microtubule-binding domain located just between its two F-actin-binding domains. 17 Arg can bind MTs with high affinity and crosslink F-actin bundles to MTs in vitro.17 These data suggest that Arg might mediate interactions between F-actin and MTs in cells (Fig. 5). In adhering fibroblasts, Arg localizes to the cell periphery and promotes the formation of F-actin-rich lamellipodial protrusions. MTs concentrate and insert into these structures.17 Elimination of MTs with nocodazole disrupts Arg localization to the periphery, suggesting that Arg:MT interactions help concentrate Arg at protrusive sites. An Arg mutant lacking part of the MT-binding domain does not promote lamellipodial protrusions, even though it can still concentrate at peripheral sites. These data suggest that Arg:MT interactions are essential for Arg to promote lamellipodial protrusion. Arg might serve as part of a peripheral target for MTs.
The microtubule plus end binding protein CLASP binds to a subset of microtubules that interact with the actin-rich periphery in Xenopus growth cones.115 Interestingly, its ortholog Orbit/MAST was identified as a regulator of midline axon guidance in flies. Importantly, genetic studies indicate that Orbit/MAST function is required for axon guidance defects resulting from Abl overexpression in the intersegmental nerve. These data indicate that a critical mediator or MT:F-actin interactions in the growth cone periphery acts downstream of Abl. Future studies should indicate how Abl modulates Orbit/MAST activity during growth cone guidance.
Conclusion
Abundant evidence implicates Abl family kinases as important links between cell surface receptors and the cellular machinery that regulates cytoskeletal rearrangements. In addition to the signaling partners of Abl family kinases discussed here, future studies are likely to identify additional pathways by which these kinases promote changes in cytoskeletal structure.
One big remaining challenge is to understand how Abl family kinases interface with their various effectors to produce cytoskeletal changes. Having identified many components of Abl kinase signaling pathways, we need to rigorously characterize their biochemical properties and examine how these properties are affected by interactions with Abl. We also need to pinpoint these activities in the cell and measure how they regulate F-actin and MT rearrangements. These efforts will benefit from ongoing advances in microscopy techniques, including both FRET-based probes to monitor biochemical activities and protein interactions and fluorescent speckle microscopy to examine actin and MT dynamics in live cells. The ultimate goal should be to understand Abl-controlled cytoskeletal structures well enough to model them in silico and make testable predictions about their properties.
In addition to activating mitogenic and anti-apoptotic pathways, oncogenic forms of Abl family kinases lead to derangement of cytoskeletal and adhesion pathways. A second challenge is to understand how Bcr-Abl-induced cytoskeletal changes contribute to disease phenotypes. These processes can be examined using mouse model systems of Bcr-Abl-positive leukemias (see chapter by Ren). Deletion of the F-actin binding domain reduces the oncogenic properties of the p190 form of Bcr-Abl. This observation suggests that disruption of cytoskeletal control pathways might be a useful strategy to treat Bcr-Abl-positive leukemias. The goal here will be to test whether these cytoskeletal pathways can be targeted for therapeutic purposes. Although Abl kinase inhibitors show tremendous promise for leukemia treatment, this treatment will probably be most useful in combination with other drugs or therapies.
Acknowledgements
I thank Ed Egelman, Vitoly Galkin, Mark Mooseker, Mark Peifer, David Van Vactor, for many helpful discussions and Stefanie Lapetina, Matt Miller, and Justin Peacock for comments on this Chapter. Work in my laboratory is supported by grants from the PHS (NS39475), the Kavli Institute for Neuroscience at Yale, and a Scholar Award from the Leukemia and Lymphoma Society of America.
References
- 1.
- Wang JY. et al. The mouse c-abl locus: Molecular cloning and characterization. Cell. 1984;36(2):349–56. [PubMed: 6319018]
- 2.
- Kruh GD. et al. The complete coding sequence of arg defines the Abelson subfamily of cytoplasmic tyrosine kinases. Proc Natl Acad Sci USA. 1990;87(15):5802–6. [PMC free article: PMC54416] [PubMed: 2198571]
- 3.
- Henkemeyer MJ. et al. DNA sequence, structure, and tyrosine kinase activity of the Drosophila melanogaster Abelson proto-oncogene homolog. Mol Cell Biol. 1988;8(2):843–53. [PMC free article: PMC363215] [PubMed: 2832740]
- 4.
- Deng X. et al. Caenorhabditis elegans ABL-1 antagonizes p53-mediated germline apoptosis after ionizing irradiation. Nat Genet. 2004;36(8):906–12. [PubMed: 15273685]
- 5.
- Barila D, Superti-Furga G. An intramolecular SH3-domain interaction regulates c-Abl activity. Nat Genet. 1998;18(3):280–2. [PubMed: 9500553]
- 6.
- Nagar B. et al. Structural basis for the autoinhibition of c-Abl tyrosine kinase. Cell. 2003;112(6):859–71. [PubMed: 12654251]
- 7.
- Mayer BJ, Baltimore D. Mutagenic analysis of the roles of SH2 and SH3 domains in regulation of the Abl tyrosine kinase. Mol Cell Biol. 1994;14(5):2883–94. [PMC free article: PMC358656] [PubMed: 8164650]
- 8.
- Mayer BJ, Hirai H, Sakai R. Evidence that SH2 domains promote processive phosphorylation by protein-tyrosine kinases. Curr Biol. 1995;5(3):296–305. [PubMed: 7780740]
- 9.
- Ren R, Ye ZS, Baltimore D. Abl protein-tyrosine kinase selects the Crk adapter as a substrate using SH3-binding sites. Genes Dev. 1994;8(7):783–95. [PubMed: 7926767]
- 10.
- Pendergast AM. The Abl family kinases: Mechanisms of regulation and signaling. Adv Cancer Res. 2002;85:51–100. [PubMed: 12374288]
- 11.
- Shi Y, Alin K, Goff SP. Abl-interactor-1, a novel SH3 protein binding to the carboxy-terminal portion of the Abl protein, suppresses v-abl transforming activity. Genes Dev. 1995;9(21):2583–97. [PubMed: 7590237]
- 12.
- Dai Z, Pendergast AM. Abi-2, a novel SH3-containing protein interacts with the c-Abl tyrosine kinase and modulates c-Abl transforming activity. Genes Dev. 1995;9(21):2569–82. [PubMed: 7590236]
- 13.
- Wang B. et al. Identification of ArgBP1, an Arg protein tyrosine kinase binding protein that is the human homologue of a CNS-specific Xenopus gene. Oncogene. 1996;12(9):1921–9. [PubMed: 8649853]
- 14.
- McWhirter JR, Wang JY. An actin-binding function contributes to transformation by the Bcr-Abl oncoprotein of Philadelphia chromosome-positive human leukemias. Embo J. 1993;12(4):1533–46. [PMC free article: PMC413366] [PubMed: 8467803]
- 15.
- Van Etten RA. et al. The COOH terminus of the c-Abl tyrosine kinase contains distinct F- and G-actin binding domains with bundling activity. J Cell Biol. 1994;124(3):325–40. [PMC free article: PMC2119935] [PubMed: 8294516]
- 16.
- Wang Y. et al. The Abl-related gene (Arg) nonreceptor tyrosine kinase uses two F-actin-binding domains to bundle F-actin. Proc Natl Acad Sci USA. 2001;98(26):14865–70. [PMC free article: PMC64950] [PubMed: 11752434]
- 17.
- Miller AL. et al. The Abl-related gene (Arg) requires its F-actin-microtubule cross-linking activity to regulate lamellipodial dynamics during fibroblast adhesion. J Cell Biol. 2004;165(3):407–19. [PMC free article: PMC2172189] [PubMed: 15138293]
- 18.
- Miao YJ, Wang JY. Binding of A/T-rich DNA by three high mobility group-like domains in c-Abl tyrosine kinase. J Biol Chem. 1996;271(37):22823–30. [PubMed: 8798460]
- 19.
- Wen ST, Jackson PK, Van EttenRA. The cytostatic function of c-Abl is controlled by multiple nuclear localization signals and requires the p53 and Rb tumor suppressor gene products. Embo J. 1996;15(7):1583–95. [PMC free article: PMC450068] [PubMed: 8612582]
- 20.
- Taagepera S. et al. Nuclear-cytoplasmic shuttling of C-ABL tyrosine kinase. Proc Natl Acad Sci USA. 1998;95(13):7457–62. [PMC free article: PMC22649] [PubMed: 9636171]
- 21.
- Melo JV. BCR-ABL gene variants. Baillieres Clin Haematol. 1997;10(2):203–22. [PubMed: 9376660]
- 22.
- McWhirter JR, Galasso DL, Wang JY. A coiled-coil oligomerization domain of Bcr is essential for the transforming function of Bcr-Abl oncoproteins. Mol Cell Biol. 1993;13(12):7587–95. [PMC free article: PMC364830] [PubMed: 8246975]
- 23.
- Zhao X. et al. Structure of the Bcr-Abl oncoprotein oligomerization domain. Nat Struct Biol. 2002;9(2):117–20. [PubMed: 11780146]
- 24.
- McWhirter JR, Wang JY. Activation of tyrosinase kinase and microfilament-binding functions of c-abl by bcr sequences in bcr/abl fusion proteins. Mol Cell Biol. 1991;11(3):1553–65. [PMC free article: PMC369443] [PubMed: 1705008]
- 25.
- Muller AJ. et al. BCR first exon sequences specifically activate the BCR/ABL tyrosine kinase oncogene of Philadelphia chromosome-positive human leukemias. Mol Cell Biol. 1991;11(4):1785–92. [PMC free article: PMC359845] [PubMed: 2005881]
- 26.
- Smith KM, Yacobi R, Van EttenRA. Autoinhibition of Bcr-Abl through its SH3 domain. Mol Cell. 2003;12(1):27–37. [PMC free article: PMC7128750] [PubMed: 12887890]
- 27.
- Raitano AB, Whang YE, Sawyers CL. Signal transduction by wild-type and leukemogenic Abl proteins. Biochim Biophys Acta. 1997;1333(3):F201–16. [PubMed: 9426204]
- 28.
- Chopra R, Pu QQ, Elefanty AG. Biology of BCR-ABL. Blood Rev. 1999;13(4):211–29. [PubMed: 10741897]
- 29.
- Ren R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer. 2005;5(3):172–83. [PubMed: 15719031]
- 30.
- Salgia R. et al. BCR/ABL induces multiple abnormalities of cytoskeletal function. J Clin Invest. 1997;100(1):46–57. [PMC free article: PMC508164] [PubMed: 9202056]
- 31.
- Weisberg E. et al. Role of focal adhesion proteins in signal transduction and oncogenesis. Crit Rev Oncog. 1997;8(4):343–58. [PubMed: 9622054]
- 32.
- Wennstrom S. et al. Membrane ruffling and chemotaxis transduced by the PDGF beta-receptor require the binding site for phosphatidylinositol 3' kinase. Oncogene. 1994;9(2):651–60. [PubMed: 8290276]
- 33.
- Plattner R. et al. c-Abl is activated by growth factors and Src family kinases and has a role in the cellular response to PDGF. Genes Dev. 1999;13(18):2400–11. [PMC free article: PMC317022] [PubMed: 10500097]
- 34.
- Sini P. et al. Abl-dependent tyrosine phosphorylation of Sos-1 mediates growth-factor-induced Rac activation. Nat Cell Biol. 2004;6(3):268–74. [PubMed: 15039778]
- 35.
- Woodring PJ. et al. Modulation of the F-actin cytoskeleton by c-Abl tyrosine kinase in cell spreading and neurite extension. J Cell Biol. 2002;156(5):879–92. [PMC free article: PMC2173320] [PubMed: 11864995]
- 36.
- Woodring PJ. et al. c-Abl phosphorylates Dok1 to promote filopodia during cell spreading. J Cell Biol. 2004;165(4):493–503. [PMC free article: PMC2172353] [PubMed: 15148308]
- 37.
- Grevengoed EE. et al. Abelson kinase regulates epithelial morphogenesis in Drosophila. J Cell Biol. 2001;155(7):1185–98. [PMC free article: PMC2199330] [PubMed: 11756472]
- 38.
- Burnside B. Microtubules and microfilaments in newt neurulation. Dev Biol. 1971;26:416–441. [PubMed: 5118745]
- 39.
- Burnside B. Microtubules and microfilaments in amphibian neurulation. Amer Zool. 1973;13:989–1006.
- 40.
- Karfunkel P. The activity of microtubules and microfilaments in neurulation in the chick. J Exp Zool. 1971;181:289–302. [PubMed: 4341108]
- 41.
- Baker PC, Schroeder TE. Cytoplasmic filaments and morphogenetic movement in the amphibian neural tube. Dev Biol. 1967;15:432–450. [PubMed: 6032487]
- 41a.
- Koleske AJ, Gifford AM, Scott ML. et al. Essential roles for the Abl and Arg tyrosine kinases in neurulation. Neuron. 1998;21:1259–1272. [PubMed: 9883720]
- 42.
- Grevengoed EE. et al. Balancing different types of actin polymerization at distinct sites: Roles for Abelson kinase and Enabled. J Cell Biol. 2003;163(6):1267–79. [PMC free article: PMC2173720] [PubMed: 14676307]
- 43.
- Bennett RL, Hoffmann FM. Increased levels of the Drosophila Abelson tyrosine kinase in nerves and muscles: Subcellular localization and mutant phenotypes imply a role in cell-cell interactions. Development. 1992;116(4):953–66. [PubMed: 1295746]
- 44.
- Adam T. et al. Cytoskeletal rearrangements and the functional role of T-plastin during entry of Shigella flexneri into HeLa cells. J Cell Biol. 1995;129(2):367–81. [PMC free article: PMC2199910] [PubMed: 7721941]
- 45.
- Burton EA, Plattner R, Pendergast AM. Abl tyrosine kinases are required for infection by Shigella flexneri. Embo J. 2003;22(20):5471–9. [PMC free article: PMC213767] [PubMed: 14532119]
- 46.
- Hollinshead M. et al. Vaccinia virus utilizes microtubules for movement to the cell surface. J Cell Biol. 2001;154(2):389–402. [PMC free article: PMC2150758] [PubMed: 11470826]
- 47.
- Rietdorf J. et al. Kinesin-dependent movement on microtubules precedes actin-based motility of vaccinia virus. Nat Cell Biol. 2001;3(11):992–1000. [PubMed: 11715020]
- 48.
- Newsome TP, Scaplehorn N, Way M. SRC mediates a switch from microtubule- to actin-based motility of vaccinia virus. Science. 2004;306(5693):124–9. [PubMed: 15297625]
- 49.
- Reeves PM. et al. Disabling poxvirus pathogenesis by inhibition of Abl-family tyrosine kinases. Nat Med. 2005 [PubMed: 15980865]
- 50.
- Kaibuchi K, Kuroda S, Amano M. Regulation of the cytoskeleton and cell adhesion by the Rho family GTPases in mammalian cells. Annu Rev Biochem. 1999;68:459–86. [PubMed: 10872457]
- 51.
- Ridley AJ. Rho family proteins: Coordinating cell responses. Trends Cell Biol. 2001;11(12):471–7. [PubMed: 11719051]
- 52.
- Burridge K, Wennerberg K. Rho and Rac take center stage. Cell. 2004;116(2):167–79. [PubMed: 14744429]
- 53.
- Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70(3):389–99. [PubMed: 1643657]
- 54.
- Ridley AJ. et al. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70(3):401–10. [PubMed: 1643658]
- 55.
- Kozma R. et al. The Ras-related protein Cdc42Hs and bradykinin promote formation of peripheral actin microspikes and filopodia in Swiss 3T3 fibroblasts. Mol Cell Biol. 1995;15(4):1942–52. [PMC free article: PMC230420] [PubMed: 7891688]
- 56.
- Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81(1):53–62. [PubMed: 7536630]
- 57.
- Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003;112(4):453–65. [PubMed: 12600310]
- 58.
- Nobes CD. et al. Activation of the small GTP-binding proteins rho and rac by growth factor receptors. J Cell Sci. 1995;108(Pt 1):225–33. [PubMed: 7738099]
- 59.
- Plattner R. et al. Bidirectional signaling links the Abelson kinases to the platelet-derived growth factor receptor. Mol Cell Biol. 2004;24(6):2573–83. [PMC free article: PMC355852] [PubMed: 14993293]
- 60.
- Skorski T. et al. BCR/ABL-mediated leukemogenesis requires the activity of the small GTP-binding protein Rac. Proc Natl Acad Sci USA. 1998;95(20):11858–62. [PMC free article: PMC21730] [PubMed: 9751755]
- 61.
- Renshaw MW, Lea-Chou E, Wang JY. Rac is required for v-Abl tyrosine kinase to activate mitogenesis. Curr Biol. 1996;6(1):76–83. [PubMed: 8805224]
- 62.
- Scita G. et al. EPS8 and E3B1 transduce signals from Ras to Rac. Nature. 1999;401(6750):290–3. [PubMed: 10499589]
- 63.
- DeMali KA, Wennerberg K, Burridge K. Integrin signaling to the actin cytoskeleton. Curr Opin Cell Biol. 2003;15(5):572–82. [PubMed: 14519392]
- 64.
- Chodniewicz D, Klemke RL. Regulation of integrin-mediated cellular responses through assembly of a CAS/Crk scaffold. Biochim Biophys Acta. 2004;1692(2-3):63–76. [PubMed: 15246680]
- 65.
- Klemke RL. et al. CAS/Crk coupling serves as a “molecular switch” for induction of cell migration. J Cell Biol. 1998;140(4):961–72. [PMC free article: PMC2141747] [PubMed: 9472046]
- 66.
- Matsuda M. et al. Interaction between the amino-terminal SH3 domain of CRK and its natural target proteins. J Biol Chem. 1996;271(24):14468–72. [PubMed: 8662907]
- 67.
- Feller SM, Knudsen B, Hanafusa H. c-Abl kinase regulates the protein binding activity of c-Crk. Embo J. 1994;13(10):2341–51. [PMC free article: PMC395099] [PubMed: 8194526]
- 68.
- Wang B. et al. Proline-rich sequences mediate the interaction of the Arg protein tyrosine kinase with Crk. Oncogene. 1996;13(7):1379–85. [PubMed: 8875975]
- 69.
- Kain KH, Klemke RL. Inhibition of cell migration by Abl family tyrosine kinases through uncoupling of Crk-CAS complexes. J Biol Chem. 2001;276(19):16185–92. [PubMed: 11279004]
- 70.
- Escalante M. et al. Phosphorylation of c-Crk II on the negative regulatory Tyr222 mediates nerve growth factor-induced cell spreading and morphogenesis. J Biol Chem. 2000;275(32):24787–97. [PubMed: 10825157]
- 71.
- Hernandez SE, Settleman J, Koleske AJ. Adhesion-dependent regulation of p190RhoGAP in the developing brain by the Abl-related gene tyrosine kinase. Curr Biol. 2004;14(8):691–6. [PubMed: 15084284]
- 72.
- Jones SB, Lu HY, Lu Q. Abl tyrosine kinase promotes dendrogenesis by inducing actin cytoskeletal rearrangements in cooperation with Rho family small GTPases in hippocampal neurons. J Neurosci. 2004;24(39):8510–21. [PMC free article: PMC6729913] [PubMed: 15456825]
- 73.
- Moresco EM. et al. Integrin-mediated dendrite branch maintenance requires Abelson (Abl) family kinases. J Neurosci. 2005;25(26):6105–18. [PMC free article: PMC6725048] [PubMed: 15987940]
- 74.
- Hu KQ, Settleman J. Tandem SH2 binding sites mediate the RasGAP-RhoGAP interaction: A conformational mechanism for SH3 domain regulation. Embo J. 1997;16(3):473–83. [PMC free article: PMC1169651] [PubMed: 9034330]
- 75.
- Roof RW. et al. Phosphotyrosine (p-Tyr)-dependent and -independent mechanisms of p190 RhoGAP-p120 RasGAP interaction: Tyr 1105 of p190, a substrate for c-Src, is the sole p-Tyr mediator of complex formation. Mol Cell Biol. 1998;18(12):7052–63. [PMC free article: PMC109287] [PubMed: 9819392]
- 76.
- Chang JH. et al. c-Src regulates the simultaneous rearrangement of actin cytoskeleton, p190RhoGAP, and p120RasGAP following epidermal growth factor stimulation. J Cell Biol. 1995;130(2):355–68. [PMC free article: PMC2199934] [PubMed: 7542246]
- 77.
- Haskell MD. et al. Phosphorylation of p190 on Tyr1105 by c-Src is necessary but not sufficient for EGF-induced actin disassembly in C3H10T1/2 fibroblasts. J Cell Sci. 2001;114(Pt 9):1699–708. [PubMed: 11309200]
- 78.
- Brouns MR, Matheson SF, Settleman J. p190 RhoGAP is the principal Src substrate in brain and regulates axon outgrowth, guidance and fasciculation. Nat Cell Biol. 2001;3(4):361–7. [PubMed: 11283609]
- 79.
- Arthur WT, Petch LA, Burridge K. Integrin engagement suppresses RhoA activity via a c-Src-dependent mechanism. Curr Biol. 2000;10(12):719–22. [PubMed: 10873807]
- 80.
- Dorey K. et al. Phosphorylation and structurebased functional studies reveal a positive and a negative role for the activation loop of the c-Abl tyrosine kinase. Oncogene. 2001;20(56):8075–84. [PubMed: 11781820]
- 81.
- Tanis KQ. et al. Two distinct phosphorylation pathways have additive effects on Abl family kinase activation. Mol Cell Biol. 2003;23(11):3884–96. [PMC free article: PMC155218] [PubMed: 12748290]
- 82.
- Schindler T. et al. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science. 2000;289(5486):1938–42. [PubMed: 10988075]
- 83.
- Stradal T. et al. The Abl interactor proteins localize to sites of actin polymerization at the tips of lamellipodia and filopodia. Curr Biol. 2001;11(11):891–5. [PubMed: 11516653]
- 84.
- Eden S. et al. Mechanism of regulation of WAVE1-induced actin nucleation by Rac1 and Nck. Nature. 2002;418(6899):790–3. [PubMed: 12181570]
- 85.
- Echarri A. et al. Abl interactor 1 (Abi-1) wave-binding and SNARE domains regulate its nucleocytoplasmic shuttling, lamellipodium localization, and wave-1 levels. Mol Cell Biol. 2004;24(11):4979–93. [PMC free article: PMC416433] [PubMed: 15143189]
- 86.
- Kunda P. et al. Abi, Sra1, and Kette control the stability and localization of SCAR/WAVE to regulate the formation of actin-based protrusions. Curr Biol. 2003;13(21):1867–75. [PubMed: 14588242]
- 87.
- Innocenti M. et al. Abi1 is essential for the formation and activation of a WAVE2 signalling complex. Nat Cell Biol. 2004;6(4):319–27. [PubMed: 15048123]
- 88.
- Steffen A. et al. Sra-1 and Nap1 link Rac to actin assembly driving lamellipodia formation. Embo J. 2004;23(4):749–59. [PMC free article: PMC380996] [PubMed: 14765121]
- 89.
- Leng Y. et al. Abelson-interactor-1 promotes WAVE2 membrane translocation and Abelson-mediated tyrosine phosphorylation required for WAVE2 activation. Proc Natl Acad Sci USA. 2005;102(4):1098–103. [PMC free article: PMC545868] [PubMed: 15657136]
- 90.
- Cestra G. et al. The Abl/Arg substrate ArgBP2/nArgBP2 coordinates the function of multiple regulatory mechanisms converging on the actin cytoskeleton. Proc Natl Acad Sci USA. 2005;102(5):1731–6. [PMC free article: PMC547834] [PubMed: 15659545]
- 91.
- Yamanashi Y, Baltimore D. Identification of the Abl- and rasGAP-associated 62 kDa protein as a docking protein, Dok. Cell. 1997;88(2):205–11. [PubMed: 9008161]
- 92.
- Carpino N. et al. p62(dok): A constitutively tyrosine-phosphorylated, GAP-associated protein in chronic myelogenous leukemia progenitor cells. Cell. 1997;88(2):197–204. [PubMed: 9008160]
- 93.
- Master Z. et al. Dok-R binds c-Abl and regulates Abl kinase activity and mediates cytoskeletal reorganization. J Biol Chem. 2003;278(32):30170–9. [PubMed: 12777393]
- 94.
- Rivero-Lezcano OM. et al. Wiskott-Aldrich syndrome protein physically associates with Nck through Src homology 3 domains. Mol Cell Biol. 1995;15(10):5725–31. [PMC free article: PMC230823] [PubMed: 7565724]
- 95.
- Rohatgi R. et al. Nck and phosphatidylinositol 4,5-bisphosphate synergistically activate actin polymerization through the N-WASP-Arp2/3 pathway. J Biol Chem. 2001;276(28):26448–52. [PubMed: 11340081]
- 96.
- Zhao ZS, Manser E, Lim L. Interaction between PAK and nck: A template for Nck targets and role of PAK autophosphorylation. Mol Cell Biol. 2000;20(11):3906–17. [PMC free article: PMC85736] [PubMed: 10805734]
- 97.
- Svitkina TM. et al. Mechanism of filopodia initiation by reorganization of a dendritic network. J Cell Biol. 2003;160(3):409–21. [PMC free article: PMC2172658] [PubMed: 12566431]
- 98.
- Krause M. et al. Ena/VASP proteins: Regulators of the actin cytoskeleton and cell migration. Annu Rev Cell Dev Biol. 2003;19:541–64. [PubMed: 14570581]
- 99.
- Gertler FB, Doctor JS, Hoffmann FM. Genetic suppression of mutations in the Drosophila abl proto-oncogene homolog. Science. 1990;248(4957):857–60. [PubMed: 2188361]
- 100.
- Comer AR. et al. Phosphorylation of Enabled by the Drosophila Abelson tyrosine kinase regulates the in vivo function and protein-protein interactions of Enabled. Mol Cell Biol. 1998;18(1):152–60. [PMC free article: PMC121469] [PubMed: 9418863]
- 101.
- Tani K. et al. Abl interactor 1 promotes tyrosine 296 phosphorylation of mammalian enabled (Mena) by c-Abl kinase. J Biol Chem. 2003;278(24):21685–92. [PubMed: 12672821]
- 102.
- Juang JL, Hoffmann FM. Drosophila abelson interacting protein (dAbi) is a positive regulator of abelson tyrosine kinase activity. Oncogene. 1999;18(37):5138–47. [PubMed: 10498863]
- 103.
- Galkin VE. et al. The Arg nonreceptor tyrosine kinase modifies F-actin structure. J Mol Biol. 2005;346(2):565–75. [PubMed: 15670605]
- 104.
- Koleske AJ. et al. Essential roles for the Abl and Arg tyrosine kinases in neurulation. Neuron. 1998;21(6):1259–72. [PubMed: 9883720]
- 105.
- Lewis JM. et al. Integrin regulation of c-Abl tyrosine kinase activity and cytoplasmic-nuclear transport. Proc Natl Acad Sci USA. 1996;93(26):15174–9. [PMC free article: PMC26376] [PubMed: 8986783]
- 106.
- Woodring PJ, Hunter T, Wang JY. Inhibition of c-Abl tyrosine kinase activity by filamentous actin. J Biol Chem. 2001;276(29):27104–10. [PubMed: 11309382]
- 107.
- Wang JY. Controlling Abl: Auto-inhibition and coinhibition? Nat Cell Biol. 2004;6(1):3–7. [PubMed: 14704671]
- 108.
- Woodring PJ, Hunter T, Wang JY. Mitotic phosphorylation rescues Abl from F-actin-mediated inhibition. J Biol Chem. 2005;280(11):10318–25. [PubMed: 15632178]
- 109.
- Baskaran R, Chiang GG, Wang JY. Identification of a binding site in c-Ab1 tyrosine kinase for the C-terminal repeated domain of RNA polymerase II. Mol Cell Biol. 1996;16(7):3361–9. [PMC free article: PMC231330] [PubMed: 8668151]
- 110.
- Lim D, Lange K, Santella L. Activation of oocytes by latrunculin A. Faseb J. 2002;16(9):1050–6. [PubMed: 12087066]
- 111.
- Zimmerman AW. et al. Cytoskeletal restraints regulate homotypic ALCAM-mediated adhesion through PKCalpha independently of Rho-like GTPases. J Cell Sci. 2004;117(Pt 13):2841–52. [PubMed: 15169840]
- 112.
- Vasiliev JM. Polarization of pseudopodial activities: Cytoskeletal mechanisms. J Cell Sci. 1991;98(Pt 1):1–4. [PubMed: 2055949]
- 113.
- Bershadsky AD, Vaisberg EA, Vasiliev JM. Pseudopodial activity at the active edge of migrating fibroblast is decreased after drug-induced microtubule depolymerization. Cell Motil Cytoskeleton. 1991;19(3):152–8. [PubMed: 1878985]
- 114.
- Waterman-Storer CM. et al. Microtubule growth activates Rac1 to promote lamellipodial protrusion in fibroblasts. Nat Cell Biol. 1999;1(1):45–50. [PubMed: 10559863]
- 115.
- Lee H. et al. The microtubule plus end tracking protein Orbit/MAST/CLASP acts downstream of the tyrosine kinase Abl in mediating axon guidance. Neuron. 2004;42(6):913–26. [PubMed: 15207236]
- 116.
- Hernandez SE. et al. How do Abl family kinases regulate cell shape and movement? Trends Cell Biol. 2004;14(1):36–44. [PubMed: 14729179]
- Introduction
- Abl Family Kinases: Basic Anatomy
- Roles for Abl Family Kinases in Cellular Morphogenesis
- Mechanisms by Which Abl Family Kinases Regulate Cytoskeletal Structure
- Abl Family Kinases also Regulate Actin Structure Independently of Rho GTPases
- Abl Family Kinases Interact Directly with F-Actin and Microtubules
- Conclusion
- Acknowledgements
- References
- Regulation of Cytoskeletal Dynamics and Cell Morphogenesis by Abl Family Kinases...Regulation of Cytoskeletal Dynamics and Cell Morphogenesis by Abl Family Kinases - Madame Curie Bioscience Database
- Dynamic Interaction between PARP-1, PCNA and p21waf1/cip1 - Madame Curie Bioscie...Dynamic Interaction between PARP-1, PCNA and p21waf1/cip1 - Madame Curie Bioscience Database
- DNA: Alternative Conformations and Biology - Madame Curie Bioscience DatabaseDNA: Alternative Conformations and Biology - Madame Curie Bioscience Database
- NMD in Drosophila: A Snapshot into the Evolution of a Conserved mRNA Surveillanc...NMD in Drosophila: A Snapshot into the Evolution of a Conserved mRNA Surveillance Pathway - Madame Curie Bioscience Database
- A Prelude to Translational (Re)Initiation - Madame Curie Bioscience DatabaseA Prelude to Translational (Re)Initiation - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...