

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Members of the tumor necrosis factor (TNF) family govern many diverse physiological and cellular responses including cellular proliferation, differentiation, and apoptosis. Ligands of this family interact through a distinct set of specific receptors that lack enzymatic activity and therefore are dependent on the association of adaptor molecules. One receptor/ligand pair known as receptor activator of nuclear factor-kappa B (RANK) and RANK ligand (RANKL) regulates bone remodeling, mammary gland development, and lymph node organogenesis. RANK interacts with five members of the TNF receptor-associated factor (TRAF) family, of which TRAF6 is indispensable for its signaling capability. An accumulation of evidence from various research laboratories indicates TRAFs, but more importantly TRAF6, is the key to understanding how RANKL links cytoplasmic signaling to the nuclear transcriptional program.
Introduction
Bone remodeling is a dynamic and continuing process of degradation of old bone by the resorption activity of the osteoclast and deposition of new bone by the osteoblast. The osteoclast is a fully differentiated, multi-nucleated cell originating from the hematopoietic monocyte-macrophage linage. The physiological importance of maintaining a balance of the osteoclast and osteoblast is underscored by diseases related to increased osteoclast activity such as postmenopausal osteoporosis, Paget's disease, rheumatoid arthritis, and tumor-induced osteolytic bone destruction.1-4 Recent evidence has indicated that RANKL, a member of the tumor necrosis factor family, and its receptor RANK are essential regulators of osteoclast differentiation and activation.1-4
In mice, targeted disruption of the genes for RANKL or RANK leads to a severe defect in bone resorption due to the lack of multi-nucleated osteoclasts, which is indicated by severe osteopetrosis.5-9 In contrast, mice lacking osteoprotegerin (OPG), a soluble decoy receptor for RANKL, develop severe osteoporosis. Thus, the regulation of differentiation and activation of this specialized cell by RANKL, OPG, and RANK emphasizes the physiological significance of these molecules in bone homeostasis. Therefore, the precise identification of the regulatory network controlled by the signaling of RANK is essential to understanding the molecular mechanism of osteoclast differentiation and may lead to the development of novel therapeutic agents to treat bone diseases. We understand that RANKL functions in other cellular and biological systems such as mammary gland development and dendritic-cell/T-cell communication, but most the signaling by RANKL and RANK has been uncovered in the context of the osteoclast. Thus, we will focus this review to signal transduction identified through the efforts of many investigators in the area of bone biology.
TRAF Interaction Motifs in the Cytoplasmic Domain of RANK
TRAFs constitute a family of seven known adaptor proteins and most of them participate in activation of the transcription factor NF-κB and members of the mitogen-activated protein (MAP) kinase family including MAPK, c-Jun N-terminal kinase (JNK), and p38.10-12 Several TRAF proteins interact directly with the intracellular regions of various members of the TNF receptor family, including CD27, CD30, CD40, TNFR2, lymphotoxin beta-receptor, and the Herpes virus entry mediator. All TRAF proteins have a highly conserved motif at the C terminus, termed the TRAF domain, which mediates its interaction with the receptor. In contrast, the N-terminal domain of the TRAFs is less well conserved, but consists of Zn-finger motifs and in some TRAFs a RING (Really Interesting New Gene) domain, which has E3 ubiquitin ligase activity as discussed below.
Like most other members of the TNF receptor family transient expression of RANK in mammalian cells or stimulation of RANK expressing cells with RANKL leads to the activation of signaling pathways including NF-κB, JNK, p38, and MAPK. Since these pathways are most likely regulated by RANK interacting with TRAF adaptor molecules, various groups attempted to determine which TRAFs interact with RANK and which regions of RANK are responsible for binding to the TRAFs by using a variety of experimental approaches.13-19 We first reported the interaction of TRAF2, TRAF5, and TRAF6 with RANK and demonstrated that RANK could activate both the NF-κB and JNK pathways.14 Following our initial report others also demonstrated the interaction of TRAF2, TRAF5, and TRAF6 and additionally TRAF1 and TRAF3 with RANK.16-19 Subsequently, through a detailed deletion analysis approach, we identified a novel TRAF6-binding motif in RANK that is distinct from that of the binding sites for TRAF2 and TRAF5.15 In addition, an identical TRAF6-binding motif in CD40 was described using a combinatorial peptide library approach.20 Taken together, the cytoplasmic domain of RANK could interact with TRAF1, TRAF2, TRAF3, TRAF5, and TRAF6, however, only TRAF2, TRAF5, and TRAF6 are functionally competent to activate signaling pathways.
Crystallographic analysis of TRAF6 in a complex with the TRAF6-binding peptide derived from either RANK or CD4021 revealed the molecular basis for recognition of the TRAF6-binding peptide. Interestingly, these results suggested that TRAF6 recognizes a consensus motif consisting of Pro-Xaa-Glu-Xaa-Xaa-Ar/Ac (where Xaa represents any amino acid and Ar is an aromatic and Ac an acidic residue)21 that was different from the known TRAF2 consensus motif of Pro/Ser/Thr/ Ala-Xaa-Gln/Glu-Glu.22,23 Furthermore, the structure of TRAF2 in complex with its binding peptide indicated that similar surface residues of TRAF2 are conserved in TRAF1, TRAF3, and TRAF5 implying that these TRAFs also recognize the same TRAF2 consensus binding domain.23 Therefore, further inspection of the entire cytoplasmic domain of RANK indicated nine potential TRAF binding sites, six TRAF2-like binding sites (PTM1-6)19,24 and three TRAF6-like binding sites (BSI-III)21 (Fig. 1). Taken collectively with all of the published data, PTM5 and PTM6 most likely contribute to the interaction of TRAF2 and TRAF5 with RANK, while sites depicted by PTM1-4 presumably represent nonfunctional TRAF binding sites. Although not experimentally confirmed, TRAF1 and TRAF3 could potential interact with PTM5 and PTM6 because they also recognize the same motif as TRAF2. In contrast, all three TRAF6-binding sites in RANK appear to interact with TRAF6, but BSI seems to be the major site.
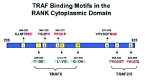
This structure based analysis of TRAF6 confirmed that there are distinct differences in peptide binding to TRAF6 and to the other TRAFs, which may provide the specificity of TRAF6 and its biological function. As stated above, inspection of the sequence of RANK indicated two more additional TRAF6-binding motifs denoted by BSII and BSIII (Fig. 1). While TRAF6 apparently binds to BSI with the highest affinity, TRAF6 also interacts with BSII and BSIII albeit with a 10-fold lower affinity.21,25 The binding specificity of TRAF6 provided the means to determine whether the RANK-TRAF6 interaction is required for RANKL signaling. We employed a novel approach by constructing a cell-permeable TRAF6 decoy peptide (T6DP) derived from BSI and BSII to investigate the effects of blocking the RANK-TRAF6 interaction. Due to the difference in peptide binding to the TRAFs, we could specifically block the RANK-TRAF6 interaction without hindering the interaction of the other TRAFs to RANK. Results from these studies indicated that T6DPs significantly blocked RANKL-mediated activation of NF-κB, JNK, and osteoclast differentiation, however T6DP-I was more effective than T6DP-II.21 In support of our hypothesis, Gohda et al25 constructed a CD40-RANK chimeric receptor with different combinations of mutants in the three TRAF6-binding sites. Their data corroborated our hypothesis that TRAF6 binds with highest affinity to BSI and that this site alone is sufficient for osteoclast differentiation. Although BSIII is capable of binding to TRAF6, a mutant chimeric receptor having only intact BSIII failed to cause calcium oscillations and activation of nuclear factor of activated T cells (NFAT) c1, which are critical for osteoclast differentiation. Furthermore, this hypothesis was supported by a similar report indicating that at least two of the three TRAF6-binding sites have the potential to induce osteoclast differentiation.26 So, why does RANK have more than one TRAF6-binding site? The occurrence of several TRAF6-binding sites may indicate a need of cooperation in order to amplify the TRAF6 signal more efficiently than having only a single TRAF6-binding site. This type of amplification of the TRAF6 signal appears to be required for efficient osteoclastogenesis and for the establishment of normal bone remodeling in vivo.
Whereas the significance of TRAF6 in RANK signaling is clear based on the phenotype of the TRAF6 deficient mice,27,28 the functional significance of RANK interacting with TRAF1, TRAF2, TRAF3, and TRAF5 remains elusive. In order to potentially identify a function of these TRAFs in RANK signaling, we used a similar approach described earlier21 with cell-permeable decoy peptides derived from the TRAF2 and TRAF5 binding sites in RANK. To date, we have failed to identify a function of TRAF2 and TRAF5 in RANK signaling by this approach, since incubation of these decoy peptides with cells appears not to interfere with any RANKL signaling pathway (A. T. P. and B. G. D., unpublished observations). Nonetheless, further studies are required to unravel the significance of RANK interacting with TRAF molecules other than TRAF6.
TRAF6 – The Critical Adaptor for RANK Signaling
Many RING domain proteins have been shown to function as E3 ubiquitin ligases that mediate polyubiquitination of target proteins, which are subsequently degraded by the 26S proteasome.29,30 The N-terminus of TRAF6 contains a conserved RING domain that is common to many ubiquitin E3 ligases. Indeed, the copurification of a dimeric E2 ubiquitin-conjugating enzyme consisting of Ubc13 and Uev1A with TRAF6 provided the initial proof that TRAF6 may function as an E3 ubiquitin ligase.31 Unlike most E3 ligases, the primary function of TRAF6 is not to target proteins for degradation, but to activate downstream kinase cascades. In fact, the TRAF6-Ubc13-Uev1A complex catalyses lysine 63 (Lys63)-linked polyubiquitin chains to mediate the activation of TGF-β-activated kinase 1 (TAK1), that further activates the MAPK kinase 6 (MKK6) and Inhibitor of NF-κB alpha (IκBα) kinase (IKK) complexes.31,32 A single point mutation of the highly conserved cysteine residue in the RING domain of TRAF6 abolishes the ubiquitin-conjugating activity of TRAF6 and its ability to activate NF-κB, suggesting that the NF-κB-inducing activity of TRAF6 is linked to its ubiquitin-conjugating activity31 (B. L. and B. G. D., unpublished observations). Notably, the auto-ubiquitination of TRAF6 via a Lys63-linked polyubiquitin chain mediates the recruitment of the Zn-finger domain of TAK1 binding protein 2 (TAB2), resulting in the activation of TAK1 and in turn the phosphorylation of the active-site loop in MKK6 and IKK.33 While Mizukami et al34 demonstrated that RANKL induces the formation of a complex consisting of RANK-TRAF6-TAB2-TAK1-TAB1, the authors did not explore the biochemical mechanism for this complex interaction. Nonetheless, these results support the importance of a TRAF6-TAB2-TAK1-TAB1 complex in mediating RANKL signaling.
The functional role of the E3 ubiquitin ligase activity of TRAF6 remains largely unknown in RANKL and RANK signaling. Indeed, preliminary evidence from our laboratory indicates RANKL stimulates the Lys63-linked polyubiquitination of TRAF6 and that the E3 ubiquitin ligase activity is required for RANK signaling and osteoclast formation (B. L. and B. G. D., unpublished observations). In somewhat contrast to our data, Kobayashi et al35 suggested that the RING domain is not required for the formation of multinucleated, tartrate resistant acid phosphatase (TRAP) positive osteoclasts, but is required for the formation of the actin ring and maturation of osteoclasts. The discrepancy between these results could reflect the differences in experimental design and future studies are required to resolve these inconsistencies.
Importantly, not only is ubiquitination necessary for signaling by TRAF6, but also its de-ubiquitination is essential for controlling its activity. A20, a molecule that was discovered more than 10 years ago and shown to interact with TRAF2 and TRAF6, negatively regulates NF-κB activation induced by TNF, IL-1, and LPS but its mechanism of action remained largely unknown.36,37 However, recent evidence suggests that A20 might be related to the ubiquitin pathway because reexamination of its sequence has revealed an N-terminal ovarian tumor (OTU) domain that has de-ubiquitinating activity.38-41 In confirmation of its de-ubiquitinating activity, A20 is capable of de-ubiquitinating Lys63-linked polyubiquitinated TRAF640 (B. L. and B. G. D., unpublished observations). Additionally, A20 is able to negatively regulate RANKL-mediated signaling (B. L. and B. G. D., unpublished observations), which suggests that ubiquitin editing functions of TRAF6 and A20 may in fact regulate RANKL signaling. Thus, further investigation of the E3 ligase activity of TRAF6 and the role of de-ubiquitinating enzymes is needed to understand how the RING domain of TRAF6 influences signaling by RANK.
Recent evidence indicates that the activation of NFATc1 is critical for RANKL-mediated osteoclast differentiation.42,43 NFATc1 expression is dependent on both the TRAF6 and c-Fos pathways,43 but how RANKL induces the expression of c-Fos still remains unclear.44 RANKL-induced recruitment of TRAF6 mobilizes intracellular calcium, by an unknown mechanism, which results in the activation of calcineurin that directly de-phosphorylates NFATc1 allowing for its rapid translocation into the nucleus. While activated NFATc1 induces a number of genes involved in cell differentiation, NFATc1 also regulates itself to amplify the transcriptional program for terminal osteoclast differentiation.1 Furthermore, RANKL also regulates cytoskeleton reorganization during osteoclast differentiation, which has been hypothesized to originate from the activation of phosphatidylinositol-3-kinase (PI3K) through a TRAF6-Src complex.45 Collectively, RANK induces a series of signals initiated by the E3 ubiquitin ligase activity of TRAF6 acting through a unique Lys63-linked polyubiquitin chain to activate kinases and phosphatases, which subsequently trigger a distinct set of genes required for osteoclast differentiation and function (Fig. 2).
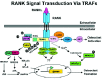
TRAFs in Osteoclast Differentiation
Signaling through RANK is essential for osteoclast formation, maturation and survival. A lack of RANK or its ligand RANKL in mice leads to osteopetrosis due to a complete absence of osteoclasts.6,8,46 Since the deletion of TRAF6 in mice leads to severe osteopetrosis with defects in bone remodeling and tooth eruption, TRAF6 appears to have a dominant, nonredundant role in osteoclast function. These phenotypes are attributed to a lack of osteoclast function, but conflicting results have been reported from two independent groups that have generated TRAF6 knockout mice. Naito et al28 showed a significant reduction in osteoclast numbers in bone sections of TRAF6 deficient mice and the inability of TRAF6-/- splenocytes to differentiate into functional osteoclasts. In contrast, Lomega et al27 observed a comparable number of osteoclasts in wild-type and TRAF6 deficient mice; however, the osteoclasts lacked contact with bone surfaces and were unable to resorb bone suggesting that TRAF6 is important for osteoclast activation rather than differentiation. The discrepancy between these two reports has yet to be resolved, which might have been caused by different deletion strategies. Nonetheless, in vitro differentiation of osteoclast progenitors with monocyte/macrophage-colony stimulating factor (M-CSF) and RANKL is completely abolished in the absence of TRAF6. Although TRAF6 interaction with RANK is essential for osteoclast cytoskeletal organization and resorption,13,21,45 overexpression of TRAF6 in osteoclast progenitor cells leads to RANKL-independent osteoclast formation.25,26 (B. L. and B. G. D., unpublished observations), underlying an important role of TRAF6 in early osteoclast differentiation.
The role of TRAF5 in osteoclastogenesis is not as clear as for TRAF6. Mice lacking TRAF5 are healthy and do not show any obvious bone phenotype under physiological conditions,47 arguing against an important function of TRAF5 in osteoclast function. However, Kanazawa et al48 reported impaired acute osteoclastogenesis in TRAF5 deficient mice after parathyroid hormone (PTH)-induced hypercalcemia and a decrease in osteoclast differentiation from bone marrow-derived monocytes, suggesting that TRAF5 is at least important for acute, stress-induced osteoclastogenesis. An explanation for the mild phenotype could be a redundancy between TRAF5 and TRAF2, therefore masking the importance of TRAF5 in osteoclastogenesis.
Unveiling the role of TRAF2 in osteoclastogenesis is hindered by the fact that loss of TRAF2 leads to embryonic and neonatal lethality.49 Interestingly, RANKL treatment of fetal liver-derived monocytes from TRAF2 knockout mice only indicated a 20% decrease in osteoclast formation, but osteoclast formation by TNF was completely ablated, suggesting only a minor role of TRAF2 in RANKL induced osteoclastogenesis.50
Conclusions and Future Directions
The discovery of RANKL and RANK has provided insights into normal physiological bone homeostasis and novel therapeutic targets for the treatment of diseases associated with increased bone resorption. Targeted deletion of each TRAF molecule in the mouse has provided a link to their physiological function and importantly to the role of TRAF6 in bone maintenance and RANK signaling. The identification of the TRAF interaction motifs in the RANK cytoplasmic domain appear to have been well described, however the functional significance of RANK interacting with TRAF1, TRAF2, TRAF3, and TRAF5 remains elusive at this time. Do these TRAFs serve an alterative function in RANK signaling in other tissues beside bone? RANKL stimulation also induces the phosphorylation of MAPK most likely through TRAF6, but the mechanism is not clear. The evidence supporting the E3 ubiquitin ligase activity of TRAF6 in IL-1 and Toll receptor signaling is strong, but little is known about its function in RANK signaling. Specifically what are the molecular targets for Lys63-linked polyubiquitination that is facilitated by TRAF6 in RANK signaling? What roles do de-ubiquitinating enzymes play in terminating RANKL signaling? TRAF6 recruits Src to the receptor complex, but how does TRAF6 induce the activity of Src in the context of osteoclast differentiation and function? What is the molecular mechanism by which TRAF6 regulates calcium oscillations during osteoclast differentiation? The importance of research in bone diseases will continue due to the high prevalence of osteoporosis and other metabolic bone disorders. Thus, the elucidation of how TRAF molecules regulate the impact of RANKL signaling will provide a further understanding of normal physiological bone homeostasis.
References
- 1.
- Takayanagi H. Mechanistic insight into osteoclast differentiation in osteoimmunology. J Mol Med. 2005;83(3):170–179. [PubMed: 15776286]
- 2.
- Teitelbaum SL, Ross FP. Genetic regulation of osteoclast development and function. Nat Rev Genet. 2003;4(8):638–649. [PubMed: 12897775]
- 3.
- Quinn JM, Gillespie MT. Modulation of osteoclast formation. Biochem Biophys Res Commun. 2005;328(3):739–745. [PubMed: 15694408]
- 4.
- Blair HC, Robinson LJ, Zaidi M. Osteoclast signalling pathways. Biochem Biophys Res Commun. 2005;328(3):728–738. [PubMed: 15694407]
- 5.
- Bucay N, Sarosi I, Dunstan CR. et al. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998;12(9):1260–1268. [PMC free article: PMC316769] [PubMed: 9573043]
- 6.
- Kong YY, Yoshida H, Sarosi I. et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397(6717):315–323. [PubMed: 9950424]
- 7.
- Mizuno A, Amizuka N, Irie K. et al. Severe osteoporosis in mice lacking osteoclastogenesis inhibitory factor/osteoprotegerin. Biochem Biophys Res Commun. 1998;247(3):610–615. [PubMed: 9647741]
- 8.
- Li J, Sarosi I, Yan XQ. et al. RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc Natl Acad Sci USA. 2000;97(4):1566–1571. [PMC free article: PMC26475] [PubMed: 10677500]
- 9.
- Dougall WC, Glaccum M, Charrier K. et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999;13(18):2412–2424. [PMC free article: PMC317030] [PubMed: 10500098]
- 10.
- Kobayashi T, Walsh MC, Choi Y. The role of TRAF6 in signal transduction and the immune response. Microbes Infect. 2004;6(14):1333–1338. [PubMed: 15555541]
- 11.
- Chung JY, Park YC, Ye H. et al. All TRAFs are not created equal: Common and distinct molecular mechanisms of TRAF-mediated signal transduction. J Cell Sci. 2002;115(Pt 4):679–688. [PubMed: 11865024]
- 12.
- Wu H, Arron JR. TRAF6, a molecular bridge spanning adaptive immunity, innate immunity and osteoimmunology. Bioessays. 2003;25(11):1096–1105. [PubMed: 14579250]
- 13.
- Armstrong AP, Tometsko ME, Glaccum M. et al. A RANK/TRAF6-dependent signal transduction pathway is essential for osteoclast cytoskeletal organization and resorptive function. J Biol Chem. 2002;277(46):44347–44356. [PubMed: 12185073]
- 14.
- Darnay BG, Haridas V, Ni J. et al. Characterization of the intracellular domain of receptor activator of NF-kappaB (RANK). Interaction with tumor necrosis factor receptor-associated factors and activation of NF-kappaB and c-Jun N-terminal kinase. J Biol Chem. 1998;273(32):20551–20555. [PubMed: 9685412]
- 15.
- Darnay BG, Ni J, Moore PA. et al. Activation of NF-kappaB by RANK requires tumor necrosis factor receptor-associated factor (TRAF) 6 and NF-kappaB-inducing kinase. Identification of a novel TRAF6 interaction motif. J Biol Chem. 1999;274(12):7724–7731. [PubMed: 10075662]
- 16.
- Galibert L, Tometsko ME, Anderson DM. et al. The involvement of multiple tumor necrosis factor receptor (TNFR)-associated factors in the signaling mechanisms of receptor activator of NF-kappaB, a member of the TNFR superfamily. J Biol Chem. 1998;273(51):34120–34127. [PubMed: 9852070]
- 17.
- Hsu H, Lacey DL, Dunstan CR. et al. Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc Natl Acad Sci USA. 1999;96(7):3540–3545. [PMC free article: PMC22329] [PubMed: 10097072]
- 18.
- Kim HH, Lee DE, Shin JN. et al. Receptor activator of NF-kappaB recruits multiple TRAF family adaptors and activates c-Jun N-terminal kinase. FEBS Lett. 1999;443(3):297–302. [PubMed: 10025951]
- 19.
- Wong BR, Josien R, Lee SY. et al. The TRAF family of signal transducers mediates NF-kappaB activation by the TRANCE receptor. J Biol Chem. 1998;273(43):28355–28359. [PubMed: 9774460]
- 20.
- Pullen SS, Dang TT, Crute JJ. et al. CD40 signaling through tumor necrosis factor receptor-associated factors (TRAFs). Binding site specificity and activation of downstream pathways by distinct TRAFs. J Biol Chem. 1999;274(20):14246–14254. [PubMed: 10318845]
- 21.
- Ye H, Arron JR, Lamothe B. et al. Distinct molecular mechanism for initiating TRAF6 signalling. Nature. 2002;418(6896):443–447. [PubMed: 12140561]
- 22.
- Park YC, Burkitt V, Villa AR. et al. Structural basis for self-association and receptor recognition of human TRAF2. Nature. 1999;398(6727):533–538. [PubMed: 10206649]
- 23.
- Ye H, Park YC, Kreishman M. et al. The structural basis for the recognition of diverse receptor sequences by TRAF2. Molecular Cell. 1999;4(3):321–330. [PubMed: 10518213]
- 24.
- Liu W, Xu D, Yang H. et al. Functional identification of three receptor activator of NF-kappa B cytoplasmic motifs mediating osteoclast differentiation and function. J Biol Chem. 2004;279(52):54759–54769. [PubMed: 15485878]
- 25.
- Gohda J, Akiyama T, Koga T. et al. RANK-mediated amplification of TRAF6 signaling leads to NFATc1 induction during osteoclastogenesis. EMBO J. 2005;24(4):790–799. [PMC free article: PMC549623] [PubMed: 15678102]
- 26.
- Kadono Y, Okada F, Perchonock C. et al. Strength of TRAF6 signalling determines osteoclastogenesis. EMBO Rep Feb. 2005;6(2):171–176. [PMC free article: PMC1299255] [PubMed: 15678156]
- 27.
- Lomaga MA, Yeh WC, Sarosi I. et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 1999;13(8):1015–1024. [PMC free article: PMC316636] [PubMed: 10215628]
- 28.
- Naito A, Azuma S, Tanaka S. et al. Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis in TRAF6-deficient mice. Genes Cells. 1999;4(6):353–362. [PubMed: 10421844]
- 29.
- Pickart CM, Cohen RE. Proteasomes and their kin: Proteases in the machine age. Nat Rev Mol Cell Biol. 2004;5(3):177–187. [PubMed: 14990998]
- 30.
- Pickart CM, Eddins MJ. Ubiquitin: Structures, functions, mechanisms. Biochim Biophys Acta. 2004;1695(1-3):55–72. [PubMed: 15571809]
- 31.
- Deng L, Wang C, Spencer E. et al. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103(2):351–361. [PubMed: 11057907]
- 32.
- Wang C, Deng L, Hong M. et al. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412(6844):346–351. [PubMed: 11460167]
- 33.
- Kanayama A, Seth RB, Sun L. et al. TAB2 and TAB3 activate the NF-kappaB pathway through binding to polyubiquitin chains. Mol Cell. 2004;15(4):535–548. [PubMed: 15327770]
- 34.
- Mizukami J, Takaesu G, Akatsuka H. et al. Receptor activator of NF-kappaB ligand (RANKL) activates TAK1 mitogen-activated protein kinase kinase kinase through a signaling complex containing RANK, TAB2, and TRAF6. Mol Cell Biol. 2002;22(4):992–1000. [PMC free article: PMC134634] [PubMed: 11809792]
- 35.
- Kobayashi N, Kadono Y, Naito A. et al. Segregation of TRAF6-mediated signaling pathways clarifies its role in osteoclastogenesis. Embo J. 2001;20(6):1271–1280. [PMC free article: PMC145527] [PubMed: 11250893]
- 36.
- Beyaert R, Heyninck K, Van HuffelS. A20 and A20-binding proteins as cellular inhibitors of nuclear factor-kappa B-dependent gene expression and apoptosis. Biochem Pharmacol. 2000;60(8):1143–1151. [PubMed: 11007952]
- 37.
- Lee EG, Boone DL, Chai S. et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289(5488):2350–2354. [PMC free article: PMC3582399] [PubMed: 11009421]
- 38.
- Balakirev MY, Tcherniuk SO, Jaquinod M. et al. Otubains: A new family of cysteine proteases in the ubiquitin pathway. EMBO Rep. 2003;4(5):517–522. [PMC free article: PMC1319179] [PubMed: 12704427]
- 39.
- Evans PC, Ovaa H, Hamon M. et al. Zinc-finger protein A20, a regulator of inflammation and cell survival, has de-ubiquitinating activity. Biochem J. 2004;378(Pt 3):727–734. [PMC free article: PMC1224040] [PubMed: 14748687]
- 40.
- Boone DL, Turer EE, Lee EG. et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol. 2004;5(10):1052–1060. [PubMed: 15334086]
- 41.
- Wertz IE, O'Rourke KM, Zhou H. et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430(7000):694–699. [PubMed: 15258597]
- 42.
- Ishida N, Hayashi K, Hoshijima M. et al. Large scale gene expression analysis of osteoclastogenesis in vitro and elucidation of NFAT2 as a key regulator. J Biol Chem. 2002;277(43):41147–41156. [PubMed: 12171919]
- 43.
- Takayanagi H, Kim S, Koga T. et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3(6):889–901. [PubMed: 12479813]
- 44.
- Ikeda F, Nishimura R, Matsubara T. et al. Critical roles of c-Jun signaling in regulation of NFAT family and RANKL-regulated osteoclast differentiation. J Clin Invest. 2004;114(4):475–484. [PMC free article: PMC503767] [PubMed: 15314684]
- 45.
- Wong BR, Besser D, Kim N. et al. TRANCE, a TNF family member, activates Akt/PKB through a signaling complex involving TRAF6 and c-Src. Mol Cell. 1999;4(6):1041–1049. [PubMed: 10635328]
- 46.
- Kim N, Odgren PR, Kim DK. et al. Diverse roles of the tumor necrosis factor family member TRANCE in skeletal physiology revealed by TRANCE deficiency and partial rescue by a lymphocyte-expressed TRANCE transgene. Proc Natl Acad Sci USA. 2000;97(20):10905–10910. [PMC free article: PMC27122] [PubMed: 10984520]
- 47.
- Nakano H, Sakon S, Koseki H. et al. Targeted disruption of Traf5 gene causes defects in CD40- and CD27-mediated lymphocyte activation. Proc Natl Acad Sci USA. 1999;96(17):9803–9808. [PMC free article: PMC22291] [PubMed: 10449775]
- 48.
- Kanazawa K, Azuma Y, Nakano H. et al. TRAF5 functions in both RANKL- and TNFalpha-induced osteoclastogenesis. J Bone Miner Res. 2003;18(3):443–450. [PubMed: 12619928]
- 49.
- Yeh WC, Shahinian A, Speiser D. et al. Early lethality, functional NF-kappaB activation, and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity. 1997;7(5):715–725. [PubMed: 9390694]
- 50.
- Kanazawa K, Kudo A. TRAF2 is essential for TNF-alpha-induced osteoclastogenesis. J Bone Miner Res. 2005;20(5):840–847. [PubMed: 15824857]
- TRAFs in RANK Signaling - Madame Curie Bioscience DatabaseTRAFs in RANK Signaling - Madame Curie Bioscience Database
- Keratins As Targets in and Modulators of Liver Diseases - Madame Curie Bioscienc...Keratins As Targets in and Modulators of Liver Diseases - Madame Curie Bioscience Database
- Embryonic Salivary Gland Branching Morphogenesis - Madame Curie Bioscience Datab...Embryonic Salivary Gland Branching Morphogenesis - Madame Curie Bioscience Database
- Earthworm Immunity - Madame Curie Bioscience DatabaseEarthworm Immunity - Madame Curie Bioscience Database
- Overview of Intracellular Compartments and Trafficking Pathways - Madame Curie B...Overview of Intracellular Compartments and Trafficking Pathways - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...