

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Skeletal muscle and adipose tissue are the major sites of postprandial glucose disposal. The insulin-regulated transport of glucose into these tissues is a multi-step process that begins with the binding of insulin to its cell surface receptor. Once activated, the insulin receptor generates multiple intracellular signaling cascades, some of which induce the rapid redistribution of the GLUT4 facilitative glucose transporter from intracellular compartments to the plasma membrane. Although probably best known for its role in glucose homeostasis, insulin regulates a variety of metabolic, mitogenic, and anti-apoptotic processes in specific tissues. Moreover, in addition to insulin several other hormones and growth factors can also activate signaling targets that function downstream of the insulin receptor. For example, phosphatidylinositol-3'-kinase (PI3K), a key enzyme in the signaling pathway leading to insulin-stimulated glucose uptake, can be activated by many extracellular signals. However, only insulin and highly related hormones such as IGF-I efficiently stimulate acute glucose transport. These observations suggest that cellular mechanisms have evolved for maintaining specificity among signaling pathways mediated by various hormones and growth factors. Elucidating the underlying mechanisms for this specificity is a current challenge engaging the attention of many researchers, and will require a thorough understanding of the signaling pathways responsible for each cellular response. To this end, recent work suggests that the coordination of multiple pathways occurs in part through the intracellular compartmentalization of key signaling molecules. Indeed, subcellular compartmentalization plays a critical role in maintaining the specificity of insulin signaling and the fidelity of GLUT4 vesicle trafficking.
Introduction
The elevated levels of blood sugar and amino acids that occur following a meal signal pancreatic beta cells to release insulin into the bloodstream. Once in the vascular system, circulating insulin markedly enhances glucose transport into skeletal muscle and adipose tissue, the peripheral sites responsible for the majority of postprandial glucose disposal. In response to insulin, glucose enters muscle and fat cells through aqueous pores formed by the glucose transporter 4 (GLUT4) protein. GLUT4 is the fourth of 13 members of a family of facilitative sugar transporters and is the only isoform that is widely accepted as being insulin-responsive. Like other GLUT family members, GLUT4 is a 12 transmembrane protein; unlike most other isofoms, GLUT4 is predominantly localized to intracellular compartments in the basal state.1-4 However, far from being static, GLUT4 slowly cycles between intracellular compartments and the cell surface, even in the absence of insulin.5,6 Since the rate of GLUT4 vesicle endocytosis is significantly higher than its rate of exocytosis, the vast majority of GLUT4 is excluded from the cell surface under basal conditions. In contrast, activation of the insulin receptor triggers a large increase in the rate of GLUT4 vesicle exocytosis and a concomitant decrease in the rate of endocytosis. This insulin-dependent shift in GLUT4 vesicle trafficking results in a net increase of GLUT4 protein at the cell surface, thus allowing glucose to enter target cells.6 Once circulating insulin returns to basal levels, GLUT4 is rapidly internalized through clathrin-coated pits and recycled back to its intracellular storage compartments. This regulated transition in the relative rates of GLUT4 vesicle endo- versus exocytosis is orchestrated by a series of vesicular trafficking processes, beginning with the selection of GLUT4 molecules in donor membrane compartments. Subsequent steps include vesicle budding, trafficking, tethering, docking, and fusion with acceptor membrane compartments.7,8 Importantly, these dynamic processes are regulated by intracellular signals that originate with the activated insulin receptor.
The Insulin Receptor and Its Immediate Downstream Substrate Proteins
Insulin regulates biological responsiveness by activating the intrinsic tyrosine kinase of its receptor.9 Unlike most other receptor tyrosine kinases, the insulin receptor is composed of two extracellular a-subunits and two transmembrane β-subunits disulfide linked into a α2β2 heterotetrameric complex.10 Insulin binds to the α-subunits and induces a conformational change that activates the intrinsic tyrosine kinase domain of its intracellular β-subunits. This results in a series of trans-autophosphorylation reactions on specific tyrosine residues within the two cytoplasmic tails of the receptor.11,12 Following activation, most receptor tyrosine kinases directly recruit downstream effectors to the phosphotyrosine residues present in their intracellular tails. In contrast, the insulin receptor primarily recruits scaffolding proteins that lack intrinsic enzymatic activity. These substrates are phosphorylated at multiple tyrosine residues, thus providing a range of potential docking sites for downstream signaling molecules. Since these proximal substrate proteins are released from the activated insulin receptor, each can recruit a distinct subset of signaling molecules to the cell surface, including certain microdomains of the plasma membrane, as well as intracellular vesicular or cytoplasmic compartments.
The activated insulin receptor phosphorylates a variety of downstream targets, including members of the insulin receptor substrate (IRS1,2,3,4), Gab1 (Grb2-associated binder 1), p53/ 58-IRS, APS (adaptor protein containing PH domain and SH2 domain), Cbl, Shc, and SIRPs (signal regulatory proteins) families.13 Among these various substrates, the IRS proteins are the most extensively characterized.9,14 The tyrosine phosphorylation of IRS proteins provides binding sites for a range of proteins that contain SH2 domains, including the p85 regulatory subunit of the type 1A PI 3-kinase, the protein tyrosine phosphatase SHP2, the nonreceptor tyrosine kinases Fyn and Csk, and the adaptor proteins Grb2 and Nck. These SH2 adaptor proteins often contain SH3 domains that bind proline-rich sequences harboring the consensus sequence PXXP, and provide further protein-protein interactions with additional downstream effectors. This combinatorial assembly of multiple signaling molecules on the IRS scaffolding proteins may impart one level of specificity to insulin receptor signaling cascades.
The PI 3-Kinase Is Necessary for Insulin-Stimulated GLUT4 Translocation
Among the downstream signaling molecules recruited to tyrosine-phosphorylated IRS proteins, it has been well established that the type 1A PI 3-kinase is crucial for a wide variety of insulin's metabolic actions. This enzyme catalyzes the phosphorylation at the D3 position of the inositol ring of PI 4,5P2, producing PI 3,4,5P3 in the plasma membrane. The formation of 3' phosphoinositides (PI3,4,5P3 and PI3,4P2) creates recognition sites for a number of proteins containing pleckstrin homology (PH) domains. These include the serine/threonine kinases phosphoinositide-dependent protein kinase (PDK1) and protein kinase B (PKB/Akt),15 as well as the guanine nucleotide exchange factor ARNO.16 Once PI 3,4,5P3 is generated at the cell surface by insulin stimulation, these PH domain-containing proteins are efficiently recruited to the inner surface of the plasma membrane, where they function in the propagation of additional signals. Not surprisingly, the experimental inhibition of PI 3-kinase activity, and thus the formation of PI3,4,5P3, completely abrogates glucose uptake and GLUT4 translocation in muscle and fat cells.17,18 Following the termination of insulin receptor activity, PI 3,4,5P3 is rapidly dephosporylated by the 3' phosphoinositide phosphatase PTEN (phosphatase and tensin homolog), which releases the PH-domain containing proteins back to the cytoplasm.19,20 This regulated transition in the spatial compartmentalization of PH-domain proteins between the cytoplasm and the plasma membrane provides a key regulatory mechanism for the PI 3-kinase signaling pathway.
As mentioned above, ARNO is a PH-domain containing protein that is recruited to the cell surface through interactions with PI 3,4,5P3. ARNO serves as an exchange factor for a subset of ARF (ADP ribosylation factor) proteins, including ARF6.21 ARFs are members of the Ras superfamily of small GTP binding proteins that function during the early stages of vesicle formation.22 In the GDP-bound state ARFs are cytosolic or only weakly associated with membranes. However, upon binding GTP they form strong interactions with target membranes and recruit vesicle coat proteins to the donor compartment. The coat proteins interact with cargo molecules and deform the membrane surface into a nascent vesicle. ARF6 is structurally divergent from other ARF isoforms and has been implicated in several vesicular trafficking processes, including the recycling of endosomal compartments at the cell surface,23 the calcium-regulated exocytosis of dense-core granules,24 and insulin-regulated GLUT4 translocation.25 In addition, ARF6 is insensitive to Brefeldin A, a fungal metabolite that inhibits a subset of ARF isoforms.26 This is significant because GLUT4 translocation is not blocked by BFA treatment. Moreover, phospholipase D (PLD), a downstream target of ARF6, was shown to colocalize with GLUT4 vesicles and to potentiate the effects of insulin on GLUT4 translocation.27-29 PLD catalyzes the hydrolysis of phosphatidylcholine, producing phosphatidic acid (PA). PA is involved in a variety of signal transduction and membrane trafficking processes.30 In addition, a recent study demonstrated the insulin-dependent activation of PLD and ARF6 through ARNO in insulin receptor-expressing Rat1 fibroblasts.31 Thus, the insulin-stimulated recruitment of ARNO to the cell surface could lead to ARF6 and PLD activation and the subsequent production of PA. Although a precise functional role for these molecules in insulin-stimulated GLUT4 translocation remains unclear, PLD activity has recently been shown to potentiate the fusion of GLUT4 vesicles with the plasma membrane.29
On the other hand, substantial evidence has demonstrated that two serine/threonine kinases, PDK1 and PKB (also called Akt), play indispensable roles in the insulin-stimulated GLUT4 translocation process. PKB isoforms are a subgroup of the AGC family of protein kinases that possesses two critical regulatory phosphorylation sites, the first of which is a threonine residue (Thr308) located within the activation loop of the kinase domain. This residue is phosphorylated by PDK1.32 The full activation of PKB also requires phosphorylation on serine 473 (Ser473), which may result from the activity of another putative kinase, or perhaps via autophosphorylation.15 Although the details remain to be elucidated, the PI 3,4,5P3-dependent redistribution of these kinases is a key step for propagating insulin receptor signals leading to a wide array of insulin actions, including GLUT4 translocation. Indeed, when constitutively targeted to the cell surface through the incorporation of an N-terminal myristoylation signal, PKB was found to induce GLUT4 translocation.33-36 In contrast, the experimental inhibition of PKB activity, by introducing blocking antibodies or by expressing dominant-interfering mutants, prevented insulin-stimulated GLUT4 translocation.34,37 In other studies, the insulin-dependent association of PKB with GLUT4-containing membrane compartments was observed.38-40 More recently, the use of siRNA to reduce the expression levels of PKB isoforms showed that knockdown of PKBβ (Akt2) prevented insulin-stimulated GLUT4 translocation,41,42 whereas reduction of PKBα (Akt1) had no significant effect. Similarly, genetic ablation of PKBβ in mice caused insulin-resistance by preventing glucose uptake and GLUT4 translocation.43,44 In contrast, loss of PKBa resulted in growth retardation but no obvious defects in glucose homeostasis.45,46
PKB was first implicated in insulin-stimulated GLUT4 translocation in 1996,33 however the identification of downstream substrates involved in glucose uptake has proven challenging. Recently, a screen was conducted based upon the consensus PKB phosphorylation motif, RXRXXS/T, where X is any amino acid. Using an antibody directed against the phosphorylated form of this motif, Kane et al (2002) were able to immunoprecipitate potential PKB substrates from insulin-stimulated adipocytes.47 One interesting candidate resulting from this screen was AS160, which contains six consensus PKB phosphorylation sites as well as a Rab GAP domain.48 Rab proteins comprise the largest branch of the Ras superfamily of small GTP-binding proteins.49 Rab proteins regulate membrane transport between organelles and may contribute to the specificity of membrane trafficking processes.50 Like other small GTPases, Rabs oscillate between a GDP-bound (“off ”) and a GTP-bound (“on”) state. In the GTP-bound state, Rabs interact with effector proteins and are thought to regulate several steps of membrane transport, including vesicle budding, motility, tethering, and fusion.51 Following a round of vesicle fusion, the Rab protein is returned to its GDP-bound state. Insulin-responsive cells express several Rab isoforms, and to date Rabs 4, 5, and 11 have been implicated in GLUT4 trafficking processes.
Of the six consensus PKB phosphorylation sites present in AS160, five were phosphorylated in response to insulin stimulation: Ser318, Ser570, Ser588, Thr642, and Thr571.48 In addition, when 4 of these five sites were mutated to alanine, the resulting construct (designated AS160-4P) behaved in a dominant-interfering manner and significantly inhibited GLUT4 translocation when over-expressed in adipocytes. As mentioned above, a myristoylated form of PKB induces GLUT4 translocation in the absence of insulin. This provides a method to directly test whether AS160 functions downstream of PKB, and Zeigerer et al (2004) recently demonstrated that expression of AS160-4P blocks the ability of myristoylated PKB to induce GLUT4 translocation.52 These results support the hypothesis that AS160 functions downstream of PKB, although its precise role remains to be elucidated. Indeed, it is not yet known which Rab isoforms are regulated by the GAP domain of AS160. Nevertheless, these results suggest that insulin-stimulated GLUT4 translocation may require an active, GTP-bound Rab. A model for how AS160 could function in GLUT4 translocation is as follows: In the basal state, the GAP activity of AS160 could maintain a Rab protein in the inactive, GDP-bound form. Insulin stimulation could then result in the phosphorylation and inhibition of AS160 GAP activity, thus allowing the conversion of the Rab protein to the active GTP-bound form. Once in the active GTP-bound state, the Rab protein can then participates in one or more steps of GLUT4 vesicle exocytosis. Although many questions remain regarding the function of AS160, the identification of an insulin-stimulated PKB substrate that has the potential to regulate Rab protein function could provide a possible mechanism for linking insulin signaling to GLUT4 vesicle trafficking.
Another recently identified PKB substrate involved in GLUT4 translocation is the SNARE (soluble N-ethylmaleimide-sensitive fusion (NSF) attachment protein (SNAP) receptors) associated protein synip.53 Synip was originally identified in a yeast two-hybrid screen using the cytosolic domain of syntaxin 4 as bait.54 A multidomain protein, synip has an N-terminal PDZ domain, central EF and coiled-coiled domains, and a C-terminal WW motif. In the basal state, synip binds syntaxin 4 and blocks the ability of VAMP2 to interact with syntaxin 4. This may prevent promiscuous fusions between GLUT4 vesicles and the cell surface. However, insulin causes synip to dissociate from syntaxin 4, thus allowing productive SNARE pairing between syntaxin 4 and VAMP2. In addition, recent work has identified an unusual potential PKBβ phosphorylation site within synip (RxKxRS97xS99).53 Interestingly, serine 99 appears to be a specific substrate of PKBβ, but not PKBα or PKBγ. Insulin stimulation resulted in the phosphorylation of synip at serine 99, and this lead to the dissociation of the synip-syntaxin 4 complex. Moreover, mutation of serine 99 to phenylalanine prevented the dissociation of synip from syntaxin 4 and also inhibited GLUT4 translocation in a dominant interfering manner. Thus, the insulin-dependent phosphorylation of synip by PKBβ may provide a mechanism for insulin to regulate the docking/fusion of GLUT4 vesicles with the cell surface.
In addition to PKB, a number of studies have suggested that atypical PKCs (PKCλ/ζ) may also function as downstream targets for the IRS-PI 3-kinase signaling pathway leading to GLUT4 translocation.55-57 Although atypical PKC isoforms lack PH domains, they are recruited to the plasma membrane where they undergo PDK1-dependent activation in response to insulin stimulation.58,59 Numerous reports have suggested the involvement of aPKCs in various metabolic actions of insulin and their possible dysfunction in insulin-resistant states.60 For example, the expression of constitutively active PKCλ/ζ mutants increase, whereas dominant-interfering mutants and blocking antibodies inhibit, insulin-induced GLUT4 translocation.56,57 Moreover, recent work has provided a functional link between PKCλ, Rab4, and the microtubule motor protein KIF3.61 Imamura et al (2003) found that insulin caused GTP loading of Rab4, an effect efficiently blocked by PI3K inhibitors and a dominant-interfering PKCλ mutant.61 Furthermore, Rab4 was found to interact with KIF3, and blocking antibodies against KIF3 significantly inhibited GLUT4 translocation. In addition, insulin enhanced the association between KIF3 and microtubules whereas this response was blocked by PI3K inhibitors and a dominant-negative PKCλ mutant. However, other recent work has implicated the microtubule motor KIF5B in GLUT4 translocation.62 Using DNA microarrays, these authors found that KIF5B is the predominant kinesin expressed in 3T3L1 adipocytes, and that dominant-interfering mutants of this isoform effectively blocked insulin-stimulated GLUT4 translocation.
Is There a Second Signaling Pathway Required for Insulin-Stimulated Glucose Uptake?
While it has been well established that activation of PI 3-kinase and subsequent generation of 3'-phosphoinositides through the IRS signaling pathway are essential for insulin-stimulated GLUT4 translocation, an important open question is the basis for the remarkable specificity of insulin action. For example, the PI 3-kinase signaling pathway is widely activated by a range of hormones and growth factors, yet only insulin evokes specific metabolic activities. For example, activation of integrin receptor signaling induces PI 3-kinase and PI3,4,5P3 production in adipocytes, but does not stimulated glucose uptake or GLUT4 translocation.63,64 Moreover, a cell-permeable analog of PI3,4,5P3 did not stimulate glucose uptake when added alone to cells.65 However, in the presence of wortmannin, which completely blocks endogenous PI 3-kinase activity, the coadministration of the PI3,4,5P3 analog together with insulin resulted in enhanced glucose uptake by adipocytes. Subsequent studies revealed that the PI3,4,5P3 analog induced GLUT4 translocation, although the transporter was not functional, suggesting the presence of a PI 3-kinase independent glucose transport activation pathway.66,67 Together, these data provide compelling evidence that the IRS-PI 3-kinase pathway is necessary, but not sufficient for insulin-dependent glucose transport.
Biological responses often reflect the integration of outputs from multiple downstream pathways activated by a given receptor. Indeed, even when downstream signaling molecules are shared by several different receptors, specificity can result from unique wiring circuitries or distinct combinations of signaling intermediates. In the context of insulin-induced GLUT4 translocation in adipocytes, Cbl, a substrate for insulin receptor tyrosine kinase, triggers a second insulin receptor pathway that functions in concert with the IRS-PI 3-kinase signaling pathway.6 In addition to multiple tyrosine residues that can be phosphorylated by the insulin receptor kinase, the Cbl protein also contains SH2, zinc ring finger, proline-rich, and leucine zipper domains. Importantly, the stimulation of Cbl phosphorylation by insulin is observed in fully differentiated adipocytes that can display 10-20 fold increases in glucose uptake in response to insulin. In contrast, fibroblasts overexpressing the insulin receptor are incapable of stimulating Cbl phosphorylation.68
Fully differentiated adipocytes express two Cbl adaptor proteins, the adaptor protein containing a PH and SH2 domain (APS) and the Cbl associated protein (CAP); both adaptors are required for insulin-induced tyrosine phosphorylation of Cbl.69-72 The activated insulin receptor recruits APS via its SH2 domain. Structural analysis of the insulin receptor kinase domain/APS SH2 domain complex revealed that the APS SH2 domain forms a homodimer, with each partner binding to a different subunit of the insulin receptor heterodimer.73 The recruitment of APS to the insulin receptor then results in phosphorylation of a carboxyl-terminal tyrosine residue, which in turn serves as a binding site for the SH2 domain of Cbl.74 Cbl also interacts with CAP, and is recruited to APS together with CAP.69,75 Intriguingly, CAP is under the transcriptional control of the nuclear receptor PPARγ, and its expression is stimulated by a class of insulin-sensitizing PPARγ agonists, the thiazolidinediones (TZDs). CAP expression correlates well with both insulin stimulation of Cbl phosphorylation and insulin sensitivity in 3T3L1 adipocytes and in mice.76,77 Consistent with these findings, dominant negative forms of APS, Cbl, and CAP inhibited insulin-stimulated glucose uptake and GLUT4 translocation.74,78-80 In addition, the use of RNAi to reduce the protein levels of Cbl or APS in 3T3L1 adipocytes also abrogated glucose uptake,79 although some data appear to be at variance with these results.81
At present, the preponderance of evidence supports the existence of a second, tissue-specific insulin signaling pathway that is necessary for GLUT4 translocation. This pathway, defined by APS-CAP-Cbl, provides an important conceptual framework for our understanding of insulin's metabolic actions, especially with regard to its highly restricted tissue specificity. Thus, insulin-induced GLUT4 translocation in adipocytes results from two independent insulin receptor signals, one being the CAP-Cbl signaling pathway that is activated only in fully differentiated adipocytes, and the other being the IRS-PI 3-kinase signaling pathway that is relatively broadly activated by a number of hormones and growth factors (Fig. 1).
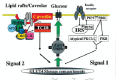
The APS-CAP-Cbl Pathway Is Compartmentalized Within Plasma Membrane Microdomains
In addition to an activation mechanism that relies on signaling molecules expressed exclusively in adipocytes, the second insulin signaling pathway is also compartmentalized within specialized lipid raft/caveolae microdomains of the plasma membrane. Lipid raft microdomains are defined by their distinct lipid compositions. They are highly enriched in cholesterol and sphingolipids. A subset of these domains contains the structural protein caveolin, which forms small invaginations of the membrane termed caveolae. Caveolae are abundant in fat, muscle, and endothelial cells. Lipid rafts/caveolae are enriched in a number of proteins involved in signaling, including heterotrimeric Gαq, H-Ras, endothelial Nitric Oxide Synthase (eNOS), growth factor receptors and the src family tyrosine kinases Fyn and Lyn.82 The insulin receptor has also been reported to segregate into these domains, and to catalyze the tyrosine phosphorylation of caveolin.83-85
The stimulation of Cbl tyrosine phosphorylation by insulin appears to occur primarily in lipid raft subdomains, due mainly to the associated protein CAP. CAP is a multifunctional adaptor protein with three adjacent SH3 domains in the carboxyl terminus, which directly binds the proline-rich domain of Cbl, and a Sorbin Homology (SoHo) domain in the amino terminus, which binds flotillin, a lipid raft/caveolar protein.78,85 This latter interaction stabilizes the CAP/Cbl complex in rafts. Once tyrosine-phosphorylated, Cbl recruits a signaling complex containing the adaptor protein CrkII and the guanine nucleotide exchange factor C3G to lipid rafts. C3G acts on TC10, a Rho family small GTP-binding protein. The activation of TC10 by insulin via the CAP-Cbl-CrkII-C3G signaling complex appears to be necessary for GLUT4 translocation, since prevention of its proper activation by over-expression of either TC10 or CAP mutants significantly inhibits insulin-induced GLUT4 translocation in adipocytes.78,86 Moreover, this signaling pathway must occur in lipid rafts, since cholesterol removal from the plasma membrane by methyl-β-cyclodextrin or by overexpression of mutant forms of caveolin3 also markedly inhibits insulin activation of TC10 mediated through the CAP-Cbl signaling pathway.87
TC10 Generates Spatially Compartmentalized Signals That Contribute to the Specificity of Insulin Action
TC10 is a member of the Rho family of GTP-binding proteins and has a high degree of sequence similarity with Rac, Rho, and Cdc42, well known actin regulators in various cell types.88-91 Like other Rho family members, TC10 operates as a molecular switch cycling between inactive GDP-bound and active GTP-bound conformational states.89 The active GTP-bound TC10 can bind numerous potential effector molecules possessing a Cdc42/Rac interactive binding (CRIB) domain such as p21-activated protein kinase (PAK), the Borg family of interacting proteins, the mammalian partition-defective homologue Par6, and the N-WASP isoform of the Wiscott-Aldrich Syndrome protein,88,92-94 as well as others without such domains, including Exo70, CIP4, and PIST.95-97
One interesting morphological feature of the adipocyte plasma membrane is the presence of both multiple individual caveolae and clusters of caveolae organized into large ring-shape structures (caveolae-rosettes) that are visualized at both the electron and light microscopic levels.87,98,99 Intriguingly, TC10 also colocalizes with these caveolin-positive caveolae-rosette structures. Most Rho members contain a single carboxyl-terminal cysteine residue in the appropriate sequence context for geranylgeranylation, and interact with guanine nucleotide dissociation inhibitors.90 In contrast, TC10 contains a sequence similar to that of H-Ras, encoding for both farnesylation and palmitoylation. These post-translational modifications target TC10 persistently to lipid raft/caveolae microdomains in adipocytes.87,100 Furthermore, the localization of TC10 in lipid rafts is necessary for its activation by insulin. Indeed, when experimentally mis-targeted to nonraft regions of the plasma membrane, TC10 failed to undergo insulin-dependent activation.
Recent results have demonstrated that the caveolae-rosette domains are directly involved in the organization of a unique filamentous actin structure in 3T3L1 adipocytes.99 In most cell types, F-actin forms stress fibers, lamellipodia and fillopodia. However, the actin cytoskeleton is dramatically changed during the differentiation of fibroblast-like preadipocytes into adipocytes. Although preadipocytes contain well defined stress fibers following differentiation into adipocytes, this F-actin converts to cortical actin lining the inner face of the cell surface membrane. 91,101 At the same time, the levels of caveolin mRNA and protein expression increase 20 fold, and the number of caveolae increases 10 fold.98,102 This marked induction of caveolin is accompanied by the clustering of individual caveolae (50-80nm) into higher-order caveolae-rosettes structures that can be visualized by fluorescent microscopy.87,98,99 Intriguingly, fully differentiated 3T3L1 adipocytes display patches of punctate F-actin that emanate from the organized caveolae-rosette structure. This unique arrangement of caveolae and actin has been designated caveolin-associated F-actin (Cav-actin).99
Currently, the molecular basis underlying the conversion from stress fiber type F-actin to the Cav-actin structures in adipocytes remains unknown. However, this dramatic structural change suggests that actin modulators and their regulatory mechanisms are essential for adipocyte function. In this regard, treatment of adipocytes with a variety of agents that perturb actin turnover, including cytochalasin D, latrunculin A or B, jasplakinolide, or swinholide all inhibit insulin-induced GLUT4 translocation.101,103,104 Furthermore, insulin stimulates dynamic actin remodeling at both the inner surface of the plasma membrane and in the perinuclear region that is sensitive to C. difficile toxin B.101 Moreover, over-expression of TC10 mutants completely disrupts the cortical F-actin including Cav-actin structures, and abolishes insulin-induced actin remodeling that results in an inhibition of GLUT4 translocation.99,101 Interestingly, recent work has suggested that the blockade of GLUT4 translocation caused by actin-disrupting agents can be overcome by the expression of an active, myristoylated form of PKB.105 These results suggest that actin may be critical for maintaining the intracellular organization of signaling complexes, rather than being required for the GLUT4 exocytotic process per se.
The observation that Cdc42, a very close relative of TC10, has no impact on either Cav-actin or GLUT4 translocation clearly indicates that the compartmentalization of TC10 to the caveolae-rosette structures is necessary to maintain and control the adipocyte actin structure. In addition, the dynamic remodeling of actin by TC10 is required for insulin-stimulated GLUT4 translocation. Although the actin regulatory mechanism and effector molecules governed by TC10 in adipocytes are still unclear, available evidence indicates an involvement of N-WASP in the dynamic actin remodeling and GLUT4 translocation process, since the dominant-interfering N-WASP mutant, partially but significantly inhibits both events.106,107
Downstream Targets of TC10
The data described above suggest that lipid raft/caveolae microdomains function as signaling platforms for the regulation of specific biological actions of insulin such as GLUT4 translocation, via the localization of TC10 effectors (Fig. 2). The exocyst complex consists of eight proteins: Sec3, Sec5, Sec6, Sec8, Sec10, Sec15, Exo70 and Exo84 that were originally identified in yeast.108 This octameric complex is involved in the tethering or docking of exocytotic vesicles.108 In 3T3L1 adipocytes, it was found that the exocyst component Exo70 can bind active GTP-bound TC10, in the process mediating translocation or assembly of the complex at the plasma membrane. 97
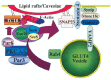
Overexpression of full length Exo70 in 3T3L1 adipocytes resulted in the potentiation of insulin stimulated glucose uptake. In contrast, a carboxyl-terminal-deletion mutant of Exo70 (C-truncated Exo70) markedly inhibited glucose uptake without any obvious inhibition of GLUT4 translocation to the plasma membrane. By using an exofacial Myc epitope-tagged GLUT4 reporter protein, the C-truncated-Exo70 mutant was found to specifically prevent the tethering and/or docking of GLUT4-containing vesicles, a necessary prerequisite for the final plasma membrane fusion. However, the C-truncated-Exo70 mutant did not affect the intracellular trafficking of GLUT4 from storage compartments to the plasma membrane, as GLUT4 vesicles were observed to accumulate underneath the plasma membrane.97 Together, these findings suggest that the recruitment or assembly of the exocyst complex to the lipid raft microdomain through the interaction between TC10 and Exo70 is required for the final steps in the GLUT4 exocytosis process.
At the final stage of exocytosis, the lipid bilayers of the plasma membrane and vesicle membrane fuse in a reaction catalyzed by the interactions between integral membrane proteins that are present in the target membrane (t-SNAREs) and the exocytotic vesicle membrane (v-SNAREs).109 The formation of a stable ternary complex between these SNARE proteins brings the exocytotic vesicle and target membrane into close proximity, and eventually leads to their fusion. In adipocytes, the v-SNARE VAMP-2 (or synaptobrevin 2) in the GLUT4-containing vesicles, and the t-SNAREs syntaxin4 and SNAP23 in the plasma membrane play a crucial role in the final fusion step of GLUT4-containing vesicles.110 Among these SNARE proteins, SNAP23 is known to be palmitoylated and is targeted to the detergent-insoluble region of the plasma membrane.111 Interestingly, the exocyst complex has been reported to be required for exocytotic vesicle targeting and docking at specific areas of the plasma membrane such as sites of polarized exocytosis.108 In the context of insulin-induced GLUT4 translocation in adipocytes, this might be equivalent to the spatially restricted microdomains of the plasma membrane. In any case, it should also be recognized that in addition to these interactions, there are several accessory proteins involved in the exocytosis of GLUT4-containing vesicles including Munc18c, Tomosyn, and Synip.54,112,113
Another downstream effector of TC10, TCGAP, was recently identified in a yeast two-hybrid screen.114 TCGAP is a multi-domain protein with N-terminal PX and SH3 domains, a central Rho-GAP domain, and several C-terminal proline-rich regions. TCGAP interacts with Cdc42 and TC10β via its GAP domain, although GAP activity towards these proteins was not detected in vivo. Nevertheless, TCGAP translocated to the plasma membrane in response to insulin, and overexpression of either the full-length or the C-terminal region of TCGAP inhibited insulin-stimulated GLUT4 translocation and glucose uptake. Although the PX domain was found to interact specifically with PI4,5P2, the binding partners for the SH3 and proline-rich domains of TCGAP remain to be determined. Thus, TCGAP appears to function downstream of TC10 in the insulin-regulated translocation of GLUT4, although its precise functional role remains to be elucidated and is an active area of current investigation.
Recent studies have also suggested that the atypical protein kinase C isoforms lambda and zeta (PKCλ/ζ) are downstream targets for the IRS-PI 3-kinase signaling pathway, since these enzymes are direct substrate for PDK1.115,116 However, PKCλ/ζ also form a quaternary complex with Par6 and Par3/ASIP.93,117 The Par proteins were originally identified as molecules involved in asymmetric cell division and polarized growth in C. elegans development.118 Par6 contains a PDZ (PSD-95/Dlg/ZO-1) domain downstream of a motif that is similar to a CRIB domain, and both are apparently required for the association of Par6 and Cdc42, a Rho family member structurally close to TC10, with exception of its carboxy-terminal CAAX motif.87,100 Par6 and PKCλ/ζ both contain PB1 (Phox and Bem1) domains that are responsible for forming heterodimeric complexes.119 Par3, also termed ASIP, contains three PDZ domains and specifically binds to both Par6 and PKCλ/ζ.120 Thus Par6 and Par3 proteins appear to serve as scaffolding molecules, linking PKCλ/ζ and the Rho family small GTP-binding proteins.93,121
In fully differentiated 3T3L1 adipocytes, expression of either constitutively active TC10 mutants or C3G, which activates endogenous TC10, results in the recruitment of PKCλ/ζ to the plasma membrane through the Par6-Par3 complex. Furthermore, PKCλ/ζ translocates to the plasma membrane with Par proteins in a manner dependent on insulin activation of TC10.94 More specifically, PKCλ/ζ appears to translocate to caveolae-rosette structures and undergoes the phosphorylation of Thr402/410 at the activation loop. Because insulin activates PDK1 and induces Thr402/Thr410 phosphorylation, it has been assumed that PKCλ/ζ is recruited to the plasma membrane by PDK1. However, the activation of the PI 3-kinase pathway by expression of the constitutively active membrane targeted p110 (catalytic subunit of the PI 3-kinase) failed to recruit PKCλ/ζ. In contrast, insulin-induced recruitment of PKCλ/ζ was prevented by C. difficile toxin B and methyl-β-cyclodextrin pretreatments. Moreover, PKCλ/ζ recruitment was also prevented by overexpression of the dominant-interfering Par6 mutant lacking CRIB domain. Thus, these data provide an important connection between two independent insulin receptor signaling pathways by demonstrating that PKCλ/ζ is a convergent downstream target of both the IRS-PI 3-kinase and Cbl-TC10 signaling cascades. Importantly, only the TC10 pathway results in the recruitment of PKCλ/ζ to caveolae in fully differentiated 3T3L1 adipocytes.
Although the physiological role of the recruited PKCλ/ζ remains to be determined, emerging evidence suggests that the proper compartmentalization of PKCλ/ζ to caveolae may provide an important clue to explain the basis for signaling specificity. For example, PKC is a relatively promiscuous serine/threonine kinase and PKCλ/ζ-dependent phosphorylation sites are highly degenerate (RXS, RXXS, or RXXSXS), compared to consensus site for the related kinase PKB/Akt (RXRXXS).122 Thus, many proteins are potentially PKC substrates and are phosphorylated in vitro, however, only a subset of these is actually phosphorylated in vivo. The spatial compartmentalization of PKCλ/ζ to the caveolae may therefore allow the enzyme to phosphorylate substrate proteins only in the restricted region that could lead to signaling specificity.
Sorting GLUT4 In to and Out of the Insulin-Responsive Storage Compartment
Exit of GLUT4 from the Insulin-Responsive Compartment
Regulated exocytosis requires that target proteins be compartmentalized in a way that renders them highly responsive to specific extracellular signals.123 The most studied examples of this process include the secretion of soluble proteins such as hormones and neurotransmitters. In these cases, the membrane compartment itself is mobilized to the cell surface and the vesicular contents are released from the cell following the fusion event. In contrast, GLUT4 is an integral membrane protein that continually cycles between the cell surface and intracellular compartments, even in the absence of insulin.6 A key question concerns the mechanism by which muscle and fat cells efficiently skew the distribution of GLUT4 toward intracellular compartments under basal conditions. In one current model, GLUT4-containing vesicles are sequestered intracellularly through direct interactions between the transporter and a retention receptor. Using a novel functional screening approach, Bogan et al (2003) have recently identified a candidate GLUT4-interacting partner that may serve as an intracellular tether for this transporter.124 This interacting partner, termed TUG (Tether, containing a UBX domain, for GLUT4), interacts specifically with GLUT4, but not with GLUT1. In addition, insulin appears to disassemble the TUG-GLUT4 complex, as detected by an insulin-stimulated reduction in the amount of TUG coimmunoprecipitated with GLUT4 in 3T3L1 adipocytes. The C-terminal region of TUG, from residues 463-550, was necessary for the efficient sequestration of GLUT4, and expression of a C-terminal fragment that encompassed this region inhibited the fold-stimulation of GLUT4 translocation in response to insulin. In part, this occurred through an increase in the basal translocation of GLUT4. In contrast, expression of full-length TUG resulted in a more rapid response to insulin together with a greater extent of translocation. These results suggest that TUG may function as a tether that efficiently sequesters GLUT4 within intracellular compartment(s) in the absence of insulin. Presumably TUG also interacts with other, as yet unidentified proteins, but in any event insulin stimulation appears to release GLUT4 from TUG, allowing the transporter to traffic to the cell surface. In the future, it will be interesting to reduce the levels of the TUG protein, perhaps by RNA interference or targeted gene disruption, as this would be predicted to cause increased basal GLUT4 translocation.
In addition to the ‘retention receptor’ model described above for retaining GLUT4 intracellularly, another mechanism, termed ‘dynamic retention’ has also been proposed.125,126 In this latter model, GLUT4 continually undergoes budding and fusion processes between the GLUT4 storage compartments, endosomes, and the plasma membrane. This model is based in part on the observation that GLUT4 is about five times more likely to fuse with an endosome compartment than with the plasma membrane in the basal state. Indeed, using a GLUT4 reporter construct with an engineered exofacial myc-epitope, Karylowski et al (2004) showed that GLUT4 was equally distributed between endosomes and the insulin-responsive storage compartment under basal conditions.125 In contrast, the transferrin receptor (TfR) was confined to the general endosome population and was excluded from the specialized GLUT4 storage compartment. Based on these and other observations, the authors propose that GLUT4 is in dynamic equilibrium with the endosome compartments and that specific sorting processes allow the continuous retrieval of GLUT4, but not general endosome markers such as TfR, back to the insulin-responsive compartment. The authors further propose that the large increase in GLUT4 exocytosis induced by insulin results from the mobilization of GLUT4 from both the endosomal and the specialized insulin-responsive storage compartments. Although the details remain to be elucidated, it should be noted that the ‘retention receptor’ and ‘dynamic retention’ models are not mutually exclusive.
Entry of Newly-Synthesized GLUT4 into the Insulin-Responsive Compartment
GLUT4 is a fairly stable protein with a half-life of ˜50 h in 3T3L1 adipocytes.127 Since GLUT4 continually cycles to and from the cell surface, there must be a sorting process within endosomes such that GLUT4 is returned, directly or indirectly, to the insulin-responsive compartment.5,128 Indeed, using various exofacial labeling techniques, the internalization of GLUT4 from the cell surface has been studied by several groups.5 Following endocytosis, GLUT4 is routed through endosomes and equilibrates with the unlabeled intracellular GLUT4 population. Importantly, this internalized GLUT4 was able to undergo subsequent rounds of insulin-stimulated translocation, indicating that it was returned to the insulin-responsive compartment.2,129-132 Although numerous studies have examined the trafficking dynamics of GLUT4 following endocytosis from the cell surface, much less attention has been given to the sorting events responsible for targeting newly-synthesized GLUT4 into the insulin-responsive compartment.
Like other membrane proteins, GLUT4 is inserted into membranes of the endoplasmic reticulum following its initial biosynthesis. GLUT4 then traffics to the Golgi complex, presumably en mass with other membrane proteins. At the trans-Golgi network (TGN), many proteins undergo sorting processes that allow them to traffic to the cell surface or to specific intracellular compartments.133 In the case of newly-synthesized GLUT4, it is possible that this transporter is sorted directly into the insulin-responsive compartment from the TGN, or alternatively that GLUT4 first traffics to the cell surface and is then routed through endosomes before entering the insulin responsive compartment.
To distinguish between these possibilities, we recently examined the sorting of GLUT4 immediately following its initial biosynthesis.134 By monitoring the localization of GLUT4 at short time intervals following transient transfection, we have been able to track the GLUT4 protein as it makes its way out of the endoplasmic reticulum and traffics through the Golgi to the insulin-responsive compartment. Although the precise identity of the insulin-responsive compartment remains unknown, it can be operationally defined because GLUT4 becomes insulin-responsive when it is therein sequestered. In contrast, general membrane proteins such as VSV-G and GLUT1 localized to the plasma membrane in as little as 2-3 h post-transfection, whereas GLUT4 was retained within the peri-nuclear region and required 9-12 h to display the full extent of insulin-stimulated translocation.134 Furthermore, there was no apparent requirement for newly synthesized GLUT4 to traffic to the cell surface before acquiring the ability to respond to insulin. These results suggest that newly-synthesized GLUT4 undergoes a time-dependent sorting process that results in its compartmentalization within insulin-responsive vesicular structures.
Golgi-localized, γ-ear-containing, Arf-binding (GGA) proteins are a relatively new family of monomeric clathrin adaptors that function at the TGN to regulate the exit of certain cargo molecules, including the two mannose-6-phosphate receptors and sortilin.135,136 Expression of a dominant-interfering GGA mutant comprising the VHS-GAT domains, completely inhibited the insulin-stimulated translocation of the newly synthesized GLUT4 protein.134 In addition, this mutant had no measurable effect on the insulin-stimulated translocation of endogenous GLUT4. These results suggest that the entry of newly-made GLUT4 into the insulin-responsive compartment is GGA-dependent, whereas insulin-stimulated exit from this compartment is GGA-independent. Furthermore, GGA has recently been shown to be involved in the endocytotic recycling from the plasma membrane back into the insulin-responsive compartment.137 Consistent with these data, Shewan et al (2003) observed that the endocytotic recycling of GLUT4 traffics through the TGN.138 Together, these results are consistent with the involvement of GGA in a specific, GLUT4-selective budding event, most likely at the TGN.
GGA proteins interact with cargo molecules that harbor a specific type of acidic cluster-dileucine motif, such as is present in the mannose-6-phosphate receptors.135 Although GLUT4 contains a region rich in acidic amino acids and a pair of leucines in its C-terminal cytosolic domain, they are not in the correct context for interactions with GGA proteins. Indeed, the GLUT4 protein does not directly bind to GGA. However, GLUT4-containing vesicles do coprecipitate with GGA,137 indicating the likely existence of an as yet unidentified intermediary binding partner. Taken together, the above data suggest that newly synthesized GLUT4 is transported to the Golgi and arrives at the TGN. At the TGN GLUT4 undergoes a specific sorting event that excludes other proteins such as GLUT1 and the TfR, through a GGA-dependent selection process that results in the compartmentalization of GLUT4 within insulin-responsive storage vesicles. This latter sorting step could occur directly from the TGN or through an intermediate endosome compartment. Once in the insulin-responsive compartment, GLUT4 slowly leaks to the cell surface under basal conditions. Following insulin stimulation, exit from this compartment is markedly increased. Although initially identified for newly-synthesized GLUT4, recent work indicates that GLUT4 molecules that have been retrieved from the cell surface may also undergo a GGA-dependent sorting step during their subsequent entry into the insulin-responsive compartment.137 In any event, the ability to functionally isolate the processes responsible for sorting GLUT4 into the insulin-responsive compartment has allowed for a more detailed analysis of the mechanisms and molecules responsible for this critical sorting event. In addition, we now have the means for biochemically isolating and characterizing the GLUT4 storage compartment, an avenue of research currently under vigorous investigation.
Does Insulin Regulate the Intrinsic Transport Activity of GLUT4?
Although insulin clearly induces the translocation of GLUT4 from intracellular storage sites to the cell surface, it remains possible that the intrinsic transport activity of GLUT4 is also regulated by insulin. Indeed, discrepancies between the fold-increase in glucose uptake and the fold-increase in GLUT4 translocation have been reported by several investigators.139 In general, the fold-increase in glucose uptake exceeded the fold-increase in GLUT4 translocation, although results varied considerably. In addition, time course experiments using a GLUT4 reporter construct engineered with an exofacial myc-epitope have shown that GLUT4 translocates to the plasma membrane prior to a measurable increase in glucose uptake. Together, these data raise the possibility that insulin may enhance the intrinsic glucose transport activity of GLUT4 transporters at the plasma membrane.
Over the past several years, evidence implicating the p38 mitogen activated protein kinases (MAPKs) in GLUT4 activation has accumulated. For example, it was shown that inhibitors of p38 MAPK, such as SB203580, prevent glucose uptake without affecting the ability of GLUT4 to translocate to the cell surface in response to insulin.140-142 However, subsequent work showed that the SB203580 compound interacts directly with the endofacial surface of GLUT4 and competitively inhibits glucose transport.143 Nevertheless, recent work has demonstrated that exogenously delivered PI3,4,5P3 can stimulate GLUT4 translocation without causing a concomitant increase in glucose uptake.144 Moreover, it has been reported that a cell-permeable phosphoinositide-binding peptide (PBP10) can also induce the translocation of GLUT4 in the absence of insulin, without an increase in glucose transport. The PBP10 peptide is derived from the N-terminus of gelsolin and binds phosphoinositides at the D3 and D4 positions. Although its mechanism of action with regard to GLUT4 translocation remains unclear, cells pretreated with PBP10 were able to take up glucose when subsequently stimulated with insulin. Thus, insulin's ability to regulate the intrinsic transport activity of GLUT4 remains an open and active area of research. The general acceptance of this hypothesis will probably require the experimental identification of a specific mechanism by which insulin could potentially regulate the intrinsic transport activity of GLUT4.
Conclusions and Future Directions
The past several years have seen many advances on two separate investigative fronts: Insulin signaling and GLUT4 vesicle trafficking, two distinct fields united by the common theme of subcellular compartmentalization. At some point the information carried by the insulin receptor signaling cascade must be translated into the language of vesicular trafficking. How this is accomplished remains unclear, however the recent discovery of AS160 as a downstream substrate of PKB provides a potential mechanism. AS160 contains a Rab GAP domain, and Rab proteins are thought to function at several steps of the vesicular transport process. However, the AS160 target Rab remains unidentified and this is an area that clearly is of great interest. In addition to AS160, the recent identification of Exo70 as a binding partner for activated TC10 provides another link between signaling and trafficking processes. In this case Exo70 appears to act at a relatively late stage in the translocation process, during the docking/ fusion step with the plasma membrane. Similarly, the identification of Synip as a downstream substrate of PKB also provides a link between insulin signaling and GLUT4 trafficking, again occurring during the docking/fusion step of vesicular transport. Thus, although several important connections have been made between signaling processes and trafficking events, it still remains unclear how the precise mobilization of GLUT4 storage compartments in response to insulin is accomplished.
GLUT4 traffics through several internal compartments during its exocytsosis and subsequent retrieval from the cell surface. Although GGA appears to play a role in selecting GLUT4 molecules at the TGN for delivery to the insulin responsive compartment, there almost certainly are analogous sorting processes occurring in endosomes such that GLUT4 is efficiently retrieved back to its storage compartment. Indeed, following its internalization at the cell surface, GLUT4 is routed through a series of endosome compartments before returning to the insulin-responsive compartment. Although the precise trafficking itinerary remains unclear, for example whether GLUT4 is routed back through the TGN before entering its storage compartment, it nevertheless seem apparent that many sorting decisions are made in the process of correctly routing GLUT4 back to its storage depot. Some of these sorting decisions likely employ general factors, such as AP2 at the cell surface. However, other sorting events may be specifically selective for GLUT4, and deciphering these cargo-specific steps represents an important area for current and future work.
References
- 1.
- Jhun BH, Rampal AL, Liu H. et al. Effects of insulin on steady state kinetics of GLUT4 subcellular distribution in rat adipocytes. J Biol Chem. 1992;268:17710–5. [PubMed: 1517217]
- 2.
- Satoh S, Nishimura H, Clark AE. et al. Use of bismannose photolabel to elucidate insulin-regulated GLUT4 subcellular trafficking kinetics in rat adipose cells. Evidence that exocytosis is a critical site of hormone action. J Biol Chem. 1993;268:17820–9. [PubMed: 8349666]
- 3.
- Yang J, Holman GD. Comparison of GLUT4 and GLUT1 subcellular trafficking in basal and insulin-stimulated 3T3-L1 cells. J Biol Chem. 1993;268:4600–3. [PubMed: 8444835]
- 4.
- Bryant NJ, Govers R, James DE. Regulated transport of the glucose transporter GLUT4. Nat Rev Mol Cell Biol. 2002;3:267–77. [PubMed: 11994746]
- 5.
- Rea S, James DE. Moving GLUT4: The biogenesis and trafficking of GLUT4 storage vesicles. Diabetes. 1997;46:1667–77. [PubMed: 9356011]
- 6.
- Watson RT, Kanzaki K, Pessin JE. Regulated membrane trafficking of the insulin-responsive glucose transporter 4 in adipocytes. Endocr Rev. 2004;25:177–204. [PubMed: 15082519]
- 7.
- Thurmond DC, Pessin JE. Molecular machinery involved in the insulin-regulated fusion of GLUT4-containing vesicles with the plasma membrane. Mol Membr Biol. 2001;18:237–45. [PubMed: 11780752]
- 8.
- Kanzaki M, Pessin JE. Insulin signaling: GLUT4 vesicles exit via the exocyst. Curr Biol. 2003;13:R574–R6. [PubMed: 12867054]
- 9.
- Saltiel AR. New perspectives into the molecular pathogenesis and treatment of type 2 diabetes. Cell. 2001;104:517–29. [PubMed: 11239409]
- 10.
- Czech MP. The nature and regulation of the insulin receptor: structure and function. Annu Rev Physiol. 1985;47:357–81. [PubMed: 2986534]
- 11.
- Frattali AL, Pessin JE. Relationship between a subunit ligand occupancy and b subunit autophosphorylation in insulin/insulin-like growth factor-1 hybrid receptors. J Biol Chem. 1993;268:7393–400. [PubMed: 8463272]
- 12.
- Lee J, O'Hare T, Pilch PF. et al. Insulin receptor autophosphorylation occurs asymmetrically. J Biol Chem. 1993;268:4092–8. [PubMed: 8440700]
- 13.
- Pessin JE, Saltiel AR. Signaling pathways in insulin action: Molecular targets of insulin resistance. J Clin Invest. 2000;106:165–9. [PMC free article: PMC314316] [PubMed: 10903329]
- 14.
- White MF, Yenush L. The IRS-signaling system: A network of docking proteins that mediate insulin and cytokine action. Curr Top Microbiol Immunol. 1998;228:179–208. [PubMed: 9401207]
- 15.
- Toker A, Newton AC. Akt/protein kinase B is regulated by autophosphorylation at the hypothetical PDK-2 site. J Biol Chem. 2000;275:8271–4. [PubMed: 10722653]
- 16.
- Mossessova E, Gulbis JM, Goldberg J. Structure of the guanine nucleotide exchange factor Sec7 domain of human ARNO and analysis of the interaction with Arf GTPase. Cell. 1998;92:415–23. [PubMed: 9476900]
- 17.
- Cheatham B, Vlahos CJ, Cheatham L. et al. Phosphatidylinositol 3-kinase activation is required for insulin stimulation of pp70 S6 kinase, DNA synthesis, and glucose transporter translocation. Mol Cell Biol. 1994;14:4902–11. [PMC free article: PMC358862] [PubMed: 8007986]
- 18.
- Okada T, Kawano Y, Sakakibara R. et al. Essential role of phophatidylinositol 3-kinase in insulin-induced glucose transport and antilypolysis in rat adipocytes. Studies with a selective inhibitor wortmannin. J Biol Chem. 1994;269:3568–73. [PubMed: 8106400]
- 19.
- Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–8. [PubMed: 9593664]
- 20.
- Nakashima N, Sharma PM, Imamura T. et al. The tumor suppressor PTEN negatively regulates insulin signaling in 3T3-L1 adipocytes. J Biol Chem. 2000;275:12889–95. [PubMed: 10777587]
- 21.
- Frank S, Upender S, Hansen SH. et al. ARNO is a guanine nucleotide exchange factor for ADP-ribosylation factor 6. J Biol Chem. 1998;273:23–7. [PubMed: 9417041]
- 22.
- Chavrier P, Goud B. The role of Arf and Rab GTPases in membrane transport. Curr Opin Cell Biol. 1999;11:466–75. [PubMed: 10449335]
- 23.
- D'Souza-Schorey C, van Donselaar E, Hsu VW. et al. Arf6 targets recycling vesicles to the plasma membrane: Insights from an ultrastructural investigation. J Cell Biol. 1998;140:603–16. [PMC free article: PMC2140168] [PubMed: 9456320]
- 24.
- Vitale N, Chasserot-Golaz S, Bailly Y. et al. Calcium-regulated exocytosis of dense-core vesicles requires the activation of ADP-ribosylation factor (Arf)6 by arf nucleotide binding site opener at the plasma membrane. J Cell Biol. 2002;159:79–89. [PMC free article: PMC2173505] [PubMed: 12379803]
- 25.
- Bose A, Cherniack AD, Langille SE. et al. G(alpha)11 signaling through Arf6 regulates F-actin mobilization and GLUT4 glucose transporter translocation to the plasma membrane. Mol Cell Biol. 2001;21:5262–75. [PMC free article: PMC87250] [PubMed: 11438680]
- 26.
- Jackson CL, Casanova JE. Turning on arf: The Sec7 family of guanine-nucleotide-exchange factors. Trends Cell Biol. 2000;10:60–7. [PubMed: 10652516]
- 27.
- Emoto M, Langille SE, Czech MP. A role for kinesin in insulin-stimulated GLUT4 glucose transporter translocation in 3T3-L1 adipocytes. J Biol Chem. 2001;276:10677–82. [PubMed: 11145966]
- 28.
- Kristiansen S, Richter EA. GLUT4-containing vesicles are released from membranes by phospholipase D cleavage of a GPI anchor. Am J Physiol Endocrinol Metab. 2002;283:E374–E82. [PubMed: 12110545]
- 29.
- Huang P, Altshuller YM, Hou JC. et al. Insulin-stimulated plasma membrane fusion of GLUT4 glucose transporter-containing vesicles is regulated by phospholipase D1. Mol Biol Cell. 2005;16(6):2614–23. [PMC free article: PMC1142410] [PubMed: 15772157]
- 30.
- Liscovitch M, Czarny M, Fiucci G. et al. Phospholipase d: Molecular and cell biology of a novel gene family. Biochem J. 2000;345:401–15. [PMC free article: PMC1220771] [PubMed: 10642495]
- 31.
- Li HS, Shome K, Rojas R. et al. The guanine nucleotide exchange factor ARNO mediates the activation of Arf and phospholipase D by insulin. BMC Cell Biol. 2003;4:13. [PMC free article: PMC212319] [PubMed: 12969509]
- 32.
- Stokoe D, Stephens LR, Copeland T. et al. Dual role of phosphatidylinositol-3,4,5-trisphosphate in the activation of protein kinase B. Science. 1997;277:567–70. [PubMed: 9228007]
- 33.
- Kohn AD, Summers SA, Birnbaum MJ. et al. Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J Biol Chem. 1996;271:31372–8. [PubMed: 8940145]
- 34.
- Cong LN, Chen H, Li Y. et al. Physiological role of Akt in insulin-stimulated translocation of GLUT4 in transfected rat adipose cells. Mol Endocrinol. 1997;11:1881–90. [PubMed: 9415393]
- 35.
- Hajduch E, Alessi DR, Hemmings BA. et al. Constitutive activation of protein kinase B alpha by membrane targeting promotes glucose and system a amino acid transport, protein synthesis, and inactivation of glycogen synthase kinase 3 in L6 muscle cells. Diabetes. 1998;47:1006–13. [PubMed: 9648821]
- 36.
- Kohn AD, Barthel A, Kovacina KS. et al. Construction and characterization of a conditionally active version of the Serine/Threonine kinase Akt. J Biol Chem. 1998;273:11937–43. [PubMed: 9565622]
- 37.
- Wang Q, Somwar R, Bilan PJ. et al. Protein kinase B/Akt participates in GLUT4 translocation by insulin in L6 myoblasts. Mol Cell Biol. 1999;19:4008–18. [PMC free article: PMC104360] [PubMed: 10330141]
- 38.
- Heller-Harrison RA, Morin M, Guilherme A. et al. Insulin-mediated targeting of phosphatidylinositol 3-kinase to GLUT4-containing vesicles. J Biol Chem. 1996;271:10200–4. [PubMed: 8626583]
- 39.
- Kupriyanova TA, Kandror KV. Akt-2 binds to GLUT4-containing vesicles and phosphorylates their component proteins in response to insulin. J Biol Chem. 1999;274:1458–64. [PubMed: 9880520]
- 40.
- Calera MR, Martinez C, Liu H. et al. Insulin increases the association of Akt-2 with GLUT4-containing vesicles. J Biol Chem. 1998;273:7201–4. [PubMed: 9516411]
- 41.
- Katome T, Obata T, Matsushima R. et al. Use of RNA interference-mediated gene silencing and adenoviral overexpression to elucidate the roles of Akt/protein kinase B isoforms in insulin actions. J Biol Chem. 2003;278:28312–23. [PubMed: 12734182]
- 42.
- Jiang ZY, Zhou QL, Coleman KA. et al. Insulin signaling through Akt/protein kinase B analyzed by small interfering RNA-mediated gene silencing. Proc Natl Acad Sci USA. 2003;100:7569–74. [PMC free article: PMC164627] [PubMed: 12808134]
- 43.
- Bae SS, Cho H, Mu J. et al. Isoform-specific regulation of insulin-dependent glucose uptake by Akt/protein kinase B. J Biol Chem. 2003;278:49530–6. [PubMed: 14522993]
- 44.
- Cho H, Mu J, Kim JK. et al. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta). Science. 2001;292:1728–31. [PubMed: 11387480]
- 45.
- Cho H, Thorvaldsen JL, Chu Q. et al. Akt1/PKB alpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem. 2001;276:38349–52. [PubMed: 11533044]
- 46.
- Verdu J, Buratovich MA, Wilder EL. et al. Cell-autonomous regulation of cell and organ growth in drosophila by Akt/PKB. Nat Cell Biol. 1999;1:500–6. [PubMed: 10587646]
- 47.
- Kane S, Sano H, Liu SC. et al. A method to identify serine kinase substrates. Akt phosphorylates a novel adipocyte protein with a Rab GTPase-activating protein (GAP) domain. J Biol Chem. 2002;277:22115–8. [PubMed: 11994271]
- 48.
- Sano H, Kane S, Sano E. et al. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem. 2003;278:14599–602. [PubMed: 12637568]
- 49.
- Colicelli J. Human Ras superfamily proteins and related GTPases. Sci STKE. 2004;250:RE13. [PMC free article: PMC2828947] [PubMed: 15367757]
- 50.
- Pfeffer SR. Structural clues to Rab GTPase functional diversity. J Biol Chem. 2005;280(16):15485–8. [PubMed: 15746102]
- 51.
- Maxfield FR, McGraw TE. Endocytic recycling. Nat Rev Mol Cell Biol. 2004;2:121–32. [PubMed: 15040445]
- 52.
- Zeigerer A, McBayer MK, McGraw TE. Insulin stimulation of GLUT4 exocytosis, but not its inhibition of endocytosis, is dependent on RabGAP as160. Mol Biol Cell. 2004;10:4406–15. [PMC free article: PMC519136] [PubMed: 15254270]
- 53.
- Yamada E, Okada S, Saito T. et al. Akt2 phosphorylates synip to regulate docking and fusion of GLUT4-containing vesicles. J Cell Biol. 2005;168:921–8. [PMC free article: PMC2171785] [PubMed: 15753124]
- 54.
- Min J, Okada S, Coker K. et al. Synip: A novel insulin-regulated syntaxin 4 binding protein mediating GLUT4 translocation in adipocytes. Mol Cell. 1999;3:751–60. [PubMed: 10394363]
- 55.
- Standaert ML, Galloway L, Karnam P. et al. Protein kinase C-zeta as a downstream effector of phosphatidylinositol 3-kinase during insulin stimulation in rat adipocytes. Potential role in glucose transport. J Biol Chem. 1997;272:30075–82. [PubMed: 9374484]
- 56.
- Kotani K, Ogawa W, Matsumoto M. et al. Requirement of atypical protein kinase C lambda for insulin stimulation of glucose uptake but not for Akt activation in 3T3-L1 adipocytes. Mol Cell Biol. 1998;18:6971–82. [PMC free article: PMC109280] [PubMed: 9819385]
- 57.
- Bandyopadhyay G, Standaert ML, Kikkawa U. et al. Effects of transiently expressed atypical (zeta, lambda), conventional (alpha, beta) and novel (delta, epsilon) protein kinase C isoforms on insulin- stimulated translocation of epitope-tagged GLUT4 glucose transporters in rat adipocytes: Specific interchangeable effects of protein kinases C-zeta and C-lambda. Biochem J. 1999;337:461–70. [PMC free article: PMC1219997] [PubMed: 9895289]
- 58.
- Standaert ML, Bandyopadhyay G, Perez L. et al. Insulin activates protein kinases C-zeta and C-lambda by an autophosphorylation-dependent mechanism and stimulates their translocation to GLUT4 vesicles and other membrane fractions in rat adipocytes. J Biol Chem. 1999;274:25308–16. [PubMed: 10464256]
- 59.
- Bandyopadhyay G, Standaert ML, Sajan MP. et al. Dependence of insulin-stimulated glucose transporter 4 translocation on 3-phosphoinositide-dependent protein kinase-1 and its target Threonine-410 in the activation loop of protein kinase C-zeta. Mol Endocrinol. 1999;13:1766–72. [PubMed: 10517677]
- 60.
- Farese RV. Function and dysfunction of aPKC isoforms for glucose transport in insulin-sensitive and insulin-resistant states. Am J Physiol Endocrinol Metab. 2002;283:E1–E11. [PubMed: 12067836]
- 61.
- Imamura T, Huang J, Usui I. et al. Insulin-induced GLUT4 translocation involves protein kinase C-lambda-mediated functional coupling between Rab4 and the motor protein kinesin. Mol Cell Biol. 2003;23:4892–900. [PMC free article: PMC162221] [PubMed: 12832475]
- 62.
- Semiz S, Park JG, Nicoloro SM. et al. Conventional kinesin KIF5B mediates insulin-stimulated GLUT4 movements on microtubules. EMBO J. 2003;22:2387–9239. [PMC free article: PMC155995] [PubMed: 12743033]
- 63.
- Isakoff SJ, Taha C, Rose E. et al. The inability of phosphatidylinositol 3-kinase activation to stimulate GLUT4 translocation indicates additional signaling pathways are required for insulin-stimulated glucose uptake. Proc Natl Acad Sci USA. 1995;92:10247–51. [PMC free article: PMC40773] [PubMed: 7479761]
- 64.
- Guilherme A, Czech MP. Stimulation of IRS-1-associated phosphatidylinositol 3-kinase and Akt/ protein kinase B but not glucose transport by beta1-integrin signaling in rat adipocytes. J Biol Chem. 1998;273:33119–22. [PubMed: 9837876]
- 65.
- Jiang T, Sweeney G, Rudolf MT. et al. Membrane-permeant esters of phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:11017–24. [PubMed: 9556583]
- 66.
- Somwar R, Koterski S, Sweeney G. et al. A dominant-negative p38 MAPK mutant and novel selective inhibitors of p38 MAPK reduce insulin-stimulated glucose uptake in 3T3-L1 adipocytes without affecting GLUT4 translocation. J Biol Chem. 2002;277:50386–95. [PubMed: 12393894]
- 67.
- Konrad D, Bilan PJ, Nawaz Z. et al. Need for GLUT4 activation to reach maximum effect of insulin-mediated glucose uptake in brown adipocytes isolated from GLUT4myc-expressing mice. Diabetes. 2002;51:2719–26. [PubMed: 12196464]
- 68.
- Ribon V, Saltiel AR. Insulin stimulates tyrosine phosphorylation of the proto-oncogene product of c-Cbl in 3T3-L1 adipocytes. Biochem J. 1997;324:839–45. [PMC free article: PMC1218500] [PubMed: 9210408]
- 69.
- Ribon V, Printen JA, Hoffman NG. et al. A novel, multifuntional c-Cbl binding protein in insulin receptor signaling in 3T3-L1 adipocytes. Mol Cell Biol. 1998;18:872–9. [PMC free article: PMC108798] [PubMed: 9447983]
- 70.
- Ribon V, Herrera R, Kay BK. et al. A role for CAP, a novel, multifunctional Src homology 3 domain-containing protein in formation of actin stress fibers and focal adhesions. J Biol Chem. 1998;273:4073–80. [PubMed: 9461600]
- 71.
- Ahmed Z, Smith BJ, Pillay TS. The APS adapter protein couples the insulin receptor to the phosphorylation of c-Cbl and facilitates ligand-stimulated ubiquitination of the insulin receptor. FEBS Lett. 2000;475:31–4. [PubMed: 10854852]
- 72.
- Moodie SA, Alleman-Sposeto J, Gustafson TA. Identification of the APS protein as a novel insulin receptor substrate. J Biol Chem. 1999;274:11186–93. [PubMed: 10196204]
- 73.
- Hu J, Liu J, Ghirlando R. et al. Structural basis for recruitment of the adaptor protein APS to the activated insulin receptor. Mol Cell. 2003;12:1379–89. [PubMed: 14690593]
- 74.
- Liu J, Kimura A, Baumann CA. et al. Aps facilitates c-Cbl tyrosine phosphorylation and GLUT4 translocation in response to insulin in 3T3-L1 adipocytes. Mol Cell Biol. 2002;22:3599–609. [PMC free article: PMC133825] [PubMed: 11997497]
- 75.
- Ahmed Z, Smith BJ, Kotani K. et al. APS, an adapter protein with a PH and SH2 domain, is a substrate for the insulin receptor kinase. Biochem J. 1999;341:665–8. [PMC free article: PMC1220404] [PubMed: 10417330]
- 76.
- Ribon V, Johnson JH, Camp HS. et al. Thiazolidinediones and insulin resistance: Peroxisome proliferatoractivated receptor gamma activation stimulates expression of the CAP gene. Proc Natl Acad Sci USA. 1998;95:14751–6. [PMC free article: PMC24521] [PubMed: 9843961]
- 77.
- Baumann CA, Chokshi N, Saltiel AR. et al. Cloning and characterization of a functional peroxisome proliferator activator receptor-gamma-responsive element in the promoter of the CAP gene. J Biol Chem. 2000;75:9131–5. [PubMed: 10734046]
- 78.
- Baumann CA, Ribon V, Kanzaki M. et al. CAP defines a second signalling pathway required for insulin-stimulated glucose transport. Nature. 2000;407:202–7. [PubMed: 11001060]
- 79.
- Ahn MY, Katsanakis KD, Bheda F. et al. Primary and essential role of the adaptor protein APS for recruitment of both c-Cbl and its associated protein CAP in insulin signaling. J Biol Chem. 2004;279:21526–32. [PubMed: 15031295]
- 80.
- Alcazar O, Ho RC, Fujii N. et al. cDNA cloning and functional characterization of a novel splice variant of c-Cbl-associated protein from mouse skeletal muscle. Biochem Biophys Res Commun. 2004;317:285–93. [PubMed: 15047181]
- 81.
- Mitra P, Zheng X, Czech MP. RNAi-based analysis of CAP, Cbl, and CrkII function in the regulation of GLUT4 by insulin. J Biol Chem. 2004;279:37431–5. [PubMed: 15258163]
- 82.
- Mastick CC, Saltiel AR. Insulin-stimulated tyrosine phosphorylation of caveolin is specific for the differentiated adipocyte phenotype in 3T3-L1 cells. J Biol Chem. 1997;272:20706–14. [PubMed: 9252391]
- 83.
- Gustavsson J, Parpal S, Karlsson M. et al. Localization of the insulin receptor in caveolae of adipocyte plasma membrane. FASEB J. 1999;13:1961–71. [PubMed: 10544179]
- 84.
- Parpal S, Karlsson M, Thorn H. et al. Cholesterol depletion disrupts caveolae and insulin receptor signaling for metabolic control via insulin receptor substrate-1, but not for mitogen-activated protein kinase control. J Biol Chem. 2001;276:9670–8. [PubMed: 11121405]
- 85.
- Kimura A, Baumann CA, Chiang SH. et al. The Sorbin homology domain: A motif for the targeting of proteins to lipid rafts. Proc Natl Acad Sci USA. 2001;98:9098–103. [PMC free article: PMC55379] [PubMed: 11481476]
- 86.
- Chiang S-H, Baumann CA, Kanzaki M. et al. Insulin-stimulated GLUT4 translocation requires the CAP-dependent activation of the small GTP binding protein TC10. Nature. 2001;410:944–8. [PubMed: 11309621]
- 87.
- Watson RT, Shigematsu S, Chiang SH. et al. Lipid raft microdomain compartmentalization of TC10 is required for insulin signaling and GLUT4 translocation. J Cell Biol. 2001;154:829–40. [PMC free article: PMC2196453] [PubMed: 11502760]
- 88.
- Neudauer CL, Joberty G, Tatsis N, Macara IG. et al. Distinct cellular effects and interactions of the Rho-family GTPase TC10. Curr Biol. 1998;8:1151–60. [PubMed: 9799731]
- 89.
- Murphy GA, Solski PA, Jillian SA. et al. Cellular functions of TC10, a Rho family GTPase: Regulation of morphology, signal transduction and cell growth. Oncogene. 1999;18:3831–45. [PubMed: 10445846]
- 90.
- Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–35. [PubMed: 12478284]
- 91.
- Kanzaki M, Watson RT, Hou JC. et al. Small GTP-binding protein TC10 differentially regulates two distinct populations of filamentous actin in 3T3L1 adipocytes. Mol Biol Cell. 2002;13:2334–46. [PMC free article: PMC117317] [PubMed: 12134073]
- 92.
- Joberty G, Perlungher RR, Macara IG. The Borgs, a new family of Cdc42 and TC10 GTPase-interacting proteins. Mol Cell Biol. 1999;19:6585–97. [PMC free article: PMC84628] [PubMed: 10490598]
- 93.
- Joberty G, Petersen C, Gao L. et al. The cell-polarity protein Par6 links Par3 and atypical protein kinase C to Cdc42 . Nat Cell Biol. 2000;2:531–9. [PubMed: 10934474]
- 94.
- Kanzaki M, Furukawa M, Raab W. et al. Phosphatidylinositol-4, 5-bisphosphate (PI4,5P2) regulates adipocyte actin dynamics and GLUT4 vesicle recycling. J Biol Chem. 2004;279:30622–33. [PubMed: 15123724]
- 95.
- Neudauer CL, Joberty G, Macara IG. PIST: A novel PDZ/coiled-coil domain binding partner for the Rho-family GTPase TC10. Biochem Biophys Res Commun. 2001;280:541–7. [PubMed: 11162552]
- 96.
- Chang L, Adams RD, Saltiel AR. The TC10-interacting protein CIP4/2 is required for insulin-stimulated GLUT4 translocation in 3T3L1 adipocytes. Proc Natl Acad Sci USA. 2002;99:12835–40. [PMC free article: PMC130546] [PubMed: 12242347]
- 97.
- Inoue M, Chang L, Hwang J. et al. The exocyst complex is required for targeting of GLUT4 to the plasma membrane by insulin. Nature. 2003;422:629–33. [PubMed: 12687004]
- 98.
- Scherer PE, Lisanti MP, Baldini G. et al. Induction of caveolin during adipogenesis and association of GLUT4 with caveolin-rich vesicles. J Cell Biol. 1994;127:1233–43. [PMC free article: PMC2120260] [PubMed: 7962086]
- 99.
- Kanzaki M, Pessin JE. Caveolin-associated filamentous actin (Cav-actin) defines a novel F-actin structure in adipocytes. J Biol Chem. 2002;277:25867–9. [PubMed: 12039946]
- 100.
- Watson RT, Furukawa M, Chiang SH. et al. The exocytotic trafficking of TC10 occurs through both classical and nonclassical secretory transport pathways in 3T3L1 adipocytes. Mol Cell Biol. 2003;23:961–74. [PMC free article: PMC140699] [PubMed: 12529401]
- 101.
- Kanzaki M, Pessin JE. Insulin-stimulated GLUT4 translocation in adipocytes is dependent upon cortical actin remodeling. J Biol Chem. 2001;276(45):42436–44. [PubMed: 11546823]
- 102.
- Rothberg KG, Heuser JE, Donzell WC. et al. Caveolin, a protein component of caveolae membrane coats. Cell. 1992;68:673–82. [PubMed: 1739974]
- 103.
- Tsakiridis T, Vranic M, Klip A. Disassembly of the actin network inhibits insulin-dependent stimulation of glucose transport and prevents recruitment of glucose transporters to the plasma membrane. J Biol Chem. 1994;269:29934–42. [PubMed: 7961991]
- 104.
- Omata W, Shibata H, Li L. et al. Actin filaments play a critical role in insulin-induced exocytotic recruitment but not in endocytosis of GLUT4 in isolated rat adipocytes. Biochem J. 2000;346 Pt 2:321–8. [PMC free article: PMC1220856] [PubMed: 10677349]
- 105.
- Eyster CA, Duggins QS, Olson AL. Expression of a constitutively active Akt/PKB signals GLUT4 translocation in the absence of an intact actin cytoskeleton. J Biol Chem. 2005;280(18):17978–85. [PubMed: 15738003]
- 106.
- Kanzaki M, Watson RT, Khan A. et al. Insulin stimulates actin comet tails on intracellular GLUT4-containing compartments in differentiated 3t3l1 adipocytes. J Biol Chem. 2001;276(52):49331–6. [PubMed: 11606595]
- 107.
- Jiang ZY, Chawla A, Bose A. et al. A phosphatidylinositol 3-kinase-independent insulin signaling pathway to N-WASP/Arp2/3/F-actin required for GLUT4 glucose transporter recycling. J Biol Chem. 2002;277:509–15. [PubMed: 11694514]
- 108.
- Lipschutz JH, Mostov KE. Exocytosis: The many masters of the exocyst. Curr Biol. 2002;12:R212–4. [PubMed: 11909549]
- 109.
- Rothman JE, Warren G. Implications of the SNARE hypothesis for intracellular membrane topology and dynamics. Curr Biol. 1994;4:220–33. [PubMed: 7922327]
- 110.
- Pessin JE, Thurmond DC, Elmendorf JS. et al. Molecular basis of insulin-stimulated GLUT4 vesicle trafficking. Location! Location! Location! J Biol Chem. 1999;274:2593–6. [PubMed: 9915783]
- 111.
- Chamberlain LH, Gould GW. The vesicle- and target-SNARE proteins that mediate GLUT4 vesicle fusion are localized in detergent-insoluble lipid rafts present on distinct intracellular membranes. J Biol Chem. 2002;277:49750–4. [PubMed: 12376543]
- 112.
- Thurmond DC, Ceresa BP, Okada S. et al. Regulation of insulin-stimulated GLUT4 translocation by Munc18c in 3T3L1 adipocytes. J Biol Chem. 1998;273:33876–83. [PubMed: 9837979]
- 113.
- Widberg CH, Bryant NJ, Girotti M. et al. Tomosyn interacts with the t-SNAREs syntaxin4 and SNAP23 and plays a role in insulin-stimulated GLUT4 translocation. J Biol Chem. 2003;278:35093–101. [PubMed: 12832401]
- 114.
- Chiang S-H, Hwang J, Legendre M. et al. TCGAP, a multidomain Rho GTPase-activating protein involved in insulin-stimulated glucose transport. EMBO J. 2003;22:2679–91. [PMC free article: PMC156759] [PubMed: 12773384]
- 115.
- Chou MM, Hou W, Johnson J. et al. Regulation of protein kinase C zeta by PI 3-kinase and PDK-1. Curr Biol. 1998;8:1069–77. [PubMed: 9768361]
- 116.
- Le Good JA, Ziegler WH, Parekh DB. et al. Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science. 1998;281:2042–5. [PubMed: 9748166]
- 117.
- Lin D, Edwards AS, Fawcett JP. et al. A mammalian Par-3-Par-6 complex implicated in Cdc4 2/ Rac1 and aPKC signalling and cell polarity . Nat Cell Biol. 2000;2:540–7. [PubMed: 10934475]
- 118.
- Etemad-Moghadam B, Guo S, Kemphues KJ. Asymmetrically distributed Par-3 protein contributes to cell polarity and spindle alignment in early C. elegans embryos. Cell. 1995;83:743–52. [PubMed: 8521491]
- 119.
- Ponting CP, Ito T, Moscat J. et al. OPR, PC and AID: All in the PB1 family. Trends Biochem Sci. 2002;27:10. [PubMed: 11796218]
- 120.
- Izumi Y, Hirose T, Tamai Y. et al. An atypical PKC directly associates and colocalizes at the epithelial tight junction with ASIP, a mammalian homologue of caenorhabditis elegans polarity protein Par-3. J Cell Biol. 1998;143:95–106. [PMC free article: PMC2132825] [PubMed: 9763423]
- 121.
- Noda Y, Takeya R, Ohno S. et al. Human homologues of the caenorhabditis elegans cell polarity protein Par6 as an adaptor that links the small GTPases Rac and Cdc42 to atypical protein kinase C. Genes Cells. 2001;6:107–19. [PubMed: 11260256]
- 122.
- Nishikawa K, Toker A, Johannes FJ. et al. Determination of the specific substrate sequence motifs of protein kinase C isozymes. J Biol Chem. 1997;272:952–60. [PubMed: 8995387]
- 123.
- Chieregatti E, Meldolesi J. Regulated exocytosis: New organelles for non-secretory purposes. Nat Rev Mol Cell Biol. 2005;6:181–7. [PubMed: 15688003]
- 124.
- Bogan JS, Hendon N, McKee AE. et al. Functional cloning of TUG as a regulator of GLUT4 glucose transporter trafficking. Nature. 2003;425:727–33. [PubMed: 14562105]
- 125.
- Karylowski O, Zeigerer A, Cohen A. et al. GLUT4 is retained by an intracellular cycle of vesicle formation and fusion with endosomes. Mol Biol Cell. 2004;15:870–82. [PMC free article: PMC329400] [PubMed: 14595108]
- 126.
- Zeigerer A, Lampson MA, Karylowski O. et al. GLUT4 retention in adipocytes requires two intracellular insulin-regulated transport steps. Mol Biol Cell. 2002;13:2421–35. [PMC free article: PMC117324] [PubMed: 12134080]
- 127.
- Sargeant RJ, Paquet MR. Effect of insulin on the rates of synthesis and degradation of GLUT1 and GLUT4 glucose transporters in 3T3-L1 adipocytes. Biochem J. 1993;290:913–9. [PMC free article: PMC1132367] [PubMed: 8457217]
- 128.
- Holman GD, Sandoval IV. Moving the insulin-regulated glucose transporter GLUT4 into and out of storage. Trends Cell Biol. 2001;11:173–9. [PubMed: 11306298]
- 129.
- Palacios S, Lalioti V, Martinez-Arca S. et al. Recycling of the insulin-sensitive glucose transporter GLUT4. Access of surface internalized GLUT4 molecules to the perinuclear storage compartment is mediated by the Phe5-Gln6-Gln7-Ile8 motif. J Biol Chem. 2001;276:3371–83. [PubMed: 11031262]
- 130.
- Foster LJ, Li D, Randhawa VK. et al. Insulin accelerates inter-endosomal GLUT4 traffic via phosphatidylinositol 3-kinase and protein kinase B. J Biol Chem. 2001;276:44212–21. [PubMed: 11560920]
- 131.
- Lampson MA, Racz A, Cushman SW. et al. Demonstration of insulin-responsive trafficking of GLUT4 and vpTR in fibroblasts. J Cell Sci. 2000;113:4065–76. [PubMed: 11058093]
- 132.
- Shigematsu S, Khan AH, Kanzaki M. et al. Intracellular insulin-responsive glucose transporter (GLUT4) distribution but not insulin-stimulated GLUT4 exocytosis and recycling are microtubule dependent. Mol Endocrinol. 2002;16:1060–8. [PubMed: 11981040]
- 133.
- Gleeson PA, Lock JG, Luke MR. et al. Domains of the tgn: Coats, tethers and G proteins. Traffic. 2004;5:315–26. [PubMed: 15086781]
- 134.
- Watson RT, Khan AH, Furukawa M. et al. Entry of newly synthesized GLUT4 into the insulin-responsive storage compartment is GGA-dependent. EMBO J. 2004;23:2059–70. [PMC free article: PMC424358] [PubMed: 15116067]
- 135.
- Bonifacino JS. The GGA proteins: Adaptors on the move. Nat Rev Mol Cell Biol. 2004;5:23–32. [PubMed: 14708007]
- 136.
- Ghosh P, Kornfeld S. The GGA proteins: Key players in protein sorting at the trans-Golgi network. Eur J Cell Biol. 2004;83:257–62. [PubMed: 15511083]
- 137.
- Li LV, Kandror KV. GGA adaptors mediate insulin-responsive trafficking of GLUT4 in 3T3-L1 adipocytes. Mol Endocrinol. 2005;19:2145–53. [PubMed: 15774496]
- 138.
- Shewan AM, Van Dam EM, Martin S. et al. GLUT4 recycles via a trans-Golgi network (TGN) subdomain enriched in syntaxins 6 and 16 but not TGN38: Involvement of an acidic targeting motif. Mol Biol Cell. 2003;14:973–86. [PMC free article: PMC151573] [PubMed: 12631717]
- 139.
- Furtado LM, Somwar R, Sweeney G. et al. Activation of the glucose transporter GLUT4 by insulin. Biochem Cell Biol. 2002;80:569–78. [PubMed: 12440698]
- 140.
- Sweeney G, Somwar R, Ramlal T. et al. An inhibitor of p38 mitogen-activated protein kinase prevents insulin-stimulated glucose transport but not glucose transporter translocation in 3T3-L1 adipocytes and L6 myotubes. J Biol Chem. 1999;274:10071–8. [PubMed: 10187787]
- 141.
- Somwar R, Kim DY, Sweeney G. et al. GLUT4 translocation precedes the stimulation of glucose uptake by insulin in muscle cells: Potential activation of GLUT4 via p38 mitogen-activated protein kinase. Biochem J. 2001;359:639–49. [PMC free article: PMC1222186] [PubMed: 11672439]
- 142.
- Bazuine M, Carlotti F, Rabelink MJ. et al. The p38 mitogen-activated protein kinase inhibitor SB203580 reduces glucose turnover by the glucose transporter-4 of 3T3-L1 adipocytes in the insulin- stimulated state. Endocrinology. 2005;146:1818–24. [PubMed: 15665038]
- 143.
- Ribe D, Yang J, Patel S, Koumanov F, Cushman SW, Holman GD. et al. Endofacial competitive inhibition of glucose transporter-4 intrinsic activity by the mitogen-activated protein kinase inhibitor SB203580. Endocrinology. 2005;146:1713–7. [PubMed: 15661859]
- 144.
- Sweeney G, Garg RR, Ceddia RB. et al. Intracellular delivery of phosphatidylinositol (3,4,5)-trisphosphate causes incorporation of glucose transporter 4 into the plasma membrane of muscle and fat cells without increasing glucose uptake. J Biol Chem. 2004;279:32233–42. [PubMed: 15166230]
- Introduction
- The Insulin Receptor and Its Immediate Downstream Substrate Proteins
- The PI 3-Kinase Is Necessary for Insulin-Stimulated GLUT4 Translocation
- Is There a Second Signaling Pathway Required for Insulin-Stimulated Glucose Uptake?
- The APS-CAP-Cbl Pathway Is Compartmentalized Within Plasma Membrane Microdomains
- TC10 Generates Spatially Compartmentalized Signals That Contribute to the Specificity of Insulin Action
- Downstream Targets of TC10
- Sorting GLUT4 In to and Out of the Insulin-Responsive Storage Compartment
- Does Insulin Regulate the Intrinsic Transport Activity of GLUT4?
- Conclusions and Future Directions
- References
- Subcellular Compartmentalization of Insulin Signaling Processes and GLUT4 Traffi...Subcellular Compartmentalization of Insulin Signaling Processes and GLUT4 Trafficking Events - Madame Curie Bioscience Database
- Immunity and Autoimmunity Induced by Polyomaviruses: Clinical, Experimental and ...Immunity and Autoimmunity Induced by Polyomaviruses: Clinical, Experimental and Theoretical Aspects - Madame Curie Bioscience Database
- Methylenetetrahydrofolate Reductase Polymorphisms: Pharmacogenetic Effects - Mad...Methylenetetrahydrofolate Reductase Polymorphisms: Pharmacogenetic Effects - Madame Curie Bioscience Database
- NMD and Human Disease - Madame Curie Bioscience DatabaseNMD and Human Disease - Madame Curie Bioscience Database
- Telomeres: Guardians of Genomic Integrity or Double Agents of Evolution? - Madam...Telomeres: Guardians of Genomic Integrity or Double Agents of Evolution? - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...