

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Summary
The techniques that allow spatio-temporal control of gene deletion or gene expression in transgenic and knockout animals have been useful to directly evaluate the roles of the insulin and IGF-1 receptors and proteins in their signaling pathway in islet cells. While a functional role for insulin signaling in islet β-cells is now well established, the precise pathways and proteins that mediate β-cell growth and function during development and adulthood are continuing to be unraveled. This chapter will focus on recent studies derived from using transgenic, knockout and siRNA techniques to directly examine the role of insulin and IGF-I in the regulation of development, growth and function of islet β-cells.
Introduction
Insulin and insulin-like-growth-factor-I (IGF-I) constitute two members of the growth factor family, which play important roles in the regulation of metabolism and growth of virtually all tissues in mammals. The receptors for insulin and IGF-I are expressed ubiquitously and mediate the growth and metabolic effects of the hormones.1-3 Most of the information we currently know regarding insulin/IGF-I signaling pathways is derived from studies underlying the defects in insulin and IGF-I action in type 2 diabetes.2,4,5 Insulin and IGF-1 bind to distinct receptors that in turn transmit signals by phosphorylating insulin receptor substrates (IRS) including the four IRS proteins, Shc, Gab-1, FAK, Cbl, and potentially other substrates. 2,5-10 These insulin receptor substrates play different but crucial roles in cellular processes that are important for the metabolism and growth of tissues including glucose transport and utilization, protein synthesis, cell growth, proliferation and anti-apoptosis. Several reviews provide an update on these signaling networks.2,5,7,8,10 Over the last decade, several laboratories have created global and tissue-specific knockouts of genes that code for protein(s), which are considered potentially important in regulating the effects of insulin and/or IGF-I. The pleiotropic signaling effects of insulin and IGF-I family of growth factors have been studied in great detail in classic insulin sensitive tissues including skeletal muscle, liver and adipose.6,10,11 While IGFs have been studied for their contributions to islet development, a role for insulin during growth of the endocrine cells in the embryonic and post-natal periods is not fully understood. Figure 1 shows a schematic of the pathways and proteins implicated in insulin/IGF-I signaling in islet β-cells.
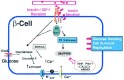
Figure 1
Schematic of insulin/IGF-I signaling pathways in the islet β-cell. Figure adapted with permission form Kulkarni RN. IJBCB 2004; 36:365-371.
Although the presence of functional insulin receptors in β-cells is now undisputable (reviewed in ref. 12), it has been a challenge to study the signaling pathways activated by insulin in β-cells for several reasons. First, the precise localization of receptors on apical and/or basolateral surfaces of different islet cells using immunohistochemistry, has been limited due to lack of a robust anti-insulin receptor antibody. Second, the continuous secretion of insulin by β-cells via the regulated and constitutive pathways allows potential internalization and downregulation of insulin receptors and confounds the effects of added ligand. Thus, several studies have used experimental protocols wherein islets/beta cells are either treated with exogenous insulin.13,14 or with inhibitors of regulated insulin secretion such as somatostatin or diazoxide followed by examining the consequences on insulin secretion or synthesis due to direct or indirect effects. A confounding factor in the latter approach is the inability to completely inhibit insulin secretion since most secretory cells possess regulated and constitutive secretion pathways.15,16 Thus, very small amounts of insulin secreted by the constitutive pathway likely maintain downregulation of insulin receptors in the presence of inhibitors of regulated secretion such as somatostatin and diazoxide, which in turn, can lead to erroneous interpretation of data. Finally, the high degree of homology between insulin and IGF-1 receptors17 and the activation of common signaling proteins precludes accurate assessment of functional end points in response to activation of individual receptors. Thus, insulin, depending on the concentrations used, can also activate the IGF-1 receptors leading to inhibitory effects,18 in contrast to reports of the effects of insulin in single murine19 or human β-cells20,21 demonstrating an increase in intracellular Ca++ flux or alterations in gene expression.22 Recent studies in humans provide further evidence for a role for insulin action in the β-cell in vivo.23,24,24a,24b The development of powerful genetic engineering techniques has circumvented several disadvantages discussed above and allow for disruption of the gene(s) coding for a given protein and enable direct evaluation of the targeted protein(s) in mouse models. This chapter will focus mostly on direct evidence provided by these techniques for a role for insulin and IGF-I during early growth and development of islets and in the maintenance of adult β-cell mass.
Embryonic and Early Post-Natal Development of the Endocrine Pancreas
The pancreas develops from the fusion of two diverticula of primordial gut tissue to form the distinct endocrine and exocrine components observed in adulthood.25 Recent reviews provide insight into the development of the endocrine cells26,27 and the role of numerous transcription factors that are considered essential for the development of the different endocrine cell types.28,29 Several elegant studies provide compelling evidence to disprove previously held dogmas. For example, based on irreversible tagging of progeny through the activity of Cre recombinase it is now accepted that α- and β-cell lineages raise independently from a common precursor expressing the pancreatic homeodomain protein, PDX-1, and not from glucagon-expressing progenitors as was originally suggested.26 Furthermore, direct lineage tracing studies indicate that NGN3+ cells are islet precursors and are distinct from ductal precursors.30 The role(s) of insulin and IGF-I signaling during embryonic development of islet cells has not yet been explored using similar approaches.
Global and Conditional Knockouts of Insulin, IGF-I, IGF-II and Proteins in Their Signaling Pathways
The significance of IGF-I and insulin during early development and growth of islet cells has been a major focus of study for several decades and several important insights have emerged from these experiments.31-35 The fetal pancreas expresses IGF-I, IGF-II, and IGF-binding protein 3 during late gestation.31,36,37 IGF-II levels are higher than IGF-I during fetal development and IGF-II has been localized to islets and duct epithelial cells by immunohistochemistry and in situ hybridization techniques.31,36,38 Together, these studies “indirectly” implicate a role for IGF-I and IGF-II during the post-natal development of the endocrine pancreas.39
The development of genetic engineering techniques over the last decade, to create gain of function or loss of function mutations40,41 has been used successfully to examine the function of specific proteins in different islet cell. Further, the ability to “turn off ” a gene encoding for a particular protein in a time-dependent manner provides a tool to simulate the gradual dysfunction that is usually observed in chronic diseases.41 While adaptation to the creation of a genetic mutation during embryonic life is a natural consequence of this method the information obtained in studies in multiple biological disciplines has been extremely useful to unravel potentially novel and unexpected functions of proteins. These observations are comparable to those made from humans bearing naturally occurring genetic mutations, who of necessity adapt to the mutation, but nevertheless provide important clues to understand the function of the proteins encoded by the gene(s). A partial list of global and conditional knockouts/transgenics of insulin, IGFs and proteins in their signaling pathways is provided in Tables 1 and 2. Unfortunately, many references could not be cited due to space limitations.
Table 1
Global knockouts/transgenics.
Table 2
Tissue-specific knockouts/transgenics.
To “directly” evaluate the role of growth factors, several investigators have utilized homologous recombination in mice. Thus, global knockout of genes coding for the insulin gene in mice leads to growth retardation, and death due to diabetes mellitus with ketoacidosis and liver steatosis.42,43 Interestingly, pancreas examination during the post-natal period revealed large islets and prompted the authors to suggest that insulin is a negative regulator of islet growth.42,43 However, since IGF-II levels are reported to be elevated during the immediate post-natal period,39 it is possible that lack of insulin allows for unopposed action of IGF-II at both insulin and/or IGF-1 receptors to promote islet hyperplasia. Alternatively, the enhanced vascularization in the absence of insulin may lead to an increase in local concentrations of morphogens, derived from the circulation and/or endothelial cells, to promote islet cell growth.44-46 A recent study in which the IGF-I gene was inactivated in islets using the PDX-1 promoter described hyperplastic islets that are resistant to streptozotocin-induced diabetes.47 The increase in the size of islets was disproportionate to the mild hyperglycemia suggesting that IGF-II or insulin acting via insulin and/or IGF-1 receptors enhanced islet growth in the absence of locally produced IGF-I. In this context, it is worth noting that overexpression of IGF-II in β-cells has also been reported to lead to hyperplastic islets48 and intriguingly the mice develop diabetes.49 While the islet growth effects induced by IGF-II could be mediated via the insulin receptor, the creation of a model of IGF-II overexpression in a mouse lacking insulin receptors in β-cells will directly address whether the IGF-II/insulin receptor pathway is indeed critical for growth.
Not surprisingly, mice with null mutations of the IGF-I and IGF-II genes show similar but milder defects compared to mice lacking the insulin gene. IGF-I null mice show growth defects similar to IGF-II null mutants17,43,51 and depending on the genetic background, some of the IGF-I knockouts die, while others survive into adulthood.50,51 Mice lacking the IGF-I gene exhibit postnatal lethality, growth retardation, infertility, and defective development of bone and muscle.51,52 Similar findings were reported in a human with homozygous partial deletion of the IGF-I gene.53 Taken together these global knockouts underscore the crucial importance of insulin and IGF-I and their cognate receptors in the overlapping regulatory functions of metabolism and growth in mice and humans.
Insulin and IGF-I mediate their effects via the insulin and IGF-1 receptors respectively. Considering the high degree of homology between the insulin and IGF-1 receptors it is likely that the ligands can also act via their cognate receptors.17 Thus, one would predict either similar phenotypes when either of the receptor is lacking or alternatively one receptor could compensate for the absence of the other receptor in an effort to maintain normal signaling in target tissues. Mice homozygous for a null mutation of the insulin receptor show normal intrauterine growth but die within 48 to 72 h after birth due to severe hyperglycemia and diabetic ketoacidosis.54-56 On the other hand, IGF-1 receptor null mutants show severe growth deficiency and die at birth due to respiratory failure51 and manifest a phenotype similar to the IGF-1 null mutants.50 Although both mutants die early, these studies clearly indicate the mice are born with β-cells. While β-cells in insulin receptor null mutants show degranulation, which likely occurs due to severe hyperglycemia, mice lacking functional IGF-1 receptors show small57 or relatively normal islet/β-cell mass.58 These studies provide evidence that neither receptor is critical for the early development and formation of β-cells. Furthermore, these findings have been confirmed in conditional knockouts of insulin or IGF-1 receptors. Thus, β-cell-specific insulin receptor knockouts (βIRKO)59 or β-cell specific IGF-1 receptor nulls60,61 are both born with a normal complement of islets/β-cells. Considering the overlapping signaling pathways shared by insulin and IGF-1, both these knockouts develop secretory defects characterized by blunted glucose-stimulated insulin secretory responses secondary to poor glucose sensing.22,59-62 Studies using insulin receptor siRNA treatment of β-cells report similar defects in glucose sensing.63 However, one notable difference between the two mutants is the effect on maintenance of β-cell mass in adults. Thus, follow up studies indicate an increased susceptibility of βIRKO mice to develop age-dependent diabetes consequent to a reduced β-cell mass.62,64 In contrast, β-cell mass in 12 month-old βIGFRKO mice is relatively normal60,61 (R.N. Kulkarni unpublished observations). Together, these studies point to similar phenotypes in regard to secretory function of β-cells, when the insulin or IGF-1 receptors are disrupted selectively in β-cells, but suggest a prominent role for insulin signaling in maintenance of adult β-cell mass (see below).
Humans bearing mutations of the insulin receptor (leprechaunism), however, manifest quite a different phenotype, compared to mice lacking insulin receptors, and are characterized by intrauterine growth retardation and only mild hyperglycemia and display large islets.65,66 In these patients, it is unclear whether the insulin receptor is devoid of all signaling capability or whether the mutated protein continues to transmit some signals that allows selective growth pathways to be active.66 Thus, it is possible that the islet hyperplasia in humans with leprechaunism is due to selective activation of proteins critical for mediating growth effects in the β-cells. The lack of reports describing mutations in insulin or IGF-1 receptors that are restricted only to β-cells in humans makes it difficult to directly resolve the issue of whether insulin signaling plays a role in modulating β-cell growth and function in vivo. The creation of mouse models bearing mutations in insulin receptors in β-cells, similar to mutations that occur in leprechaunism in humans, perhaps using a knock-in strategy, is one way to gain insight into this question. Furthermore, recent studies in vivo in humans indicate a potential role for insulin in modulating β-cell function.24a,24b
Gene deletion of proteins downstream to the insulin and IGF-1 receptor, including the IRS proteins, leads to different phenotypes compared to those observed in the receptor mutants. Thus, IRS-1 knockouts exhibit post-natal growth retardation and hyperinsulinemia but a relatively normal lifespan67,68 and interestingly, the IRS-1 null mice show hyperplastic but dysfunctional islets.69-73 While the islet hyperplasia in IRS-1 knockouts may be secondary to upregulation of expression of IRS-2 in β-cells,73 the altered islet function in IRS-1 global mutants has been linked to reduced expression of sarcoplasmic endoplasmic reticulum calcium ATPase (SERCA) proteins.72 In contrast to IRS-1 knockouts, IRS-2 null mice show only mild growth retardation, and depending on the genetic background of the founder mice either manifest a mild phenotype70 or develop β-cell hypoplasia leading to overt diabetes.57 Transgenic mice overexpressing IRS-2 exhibited protection against development of diabetes and transplantation of islets overexpressing IRS-2 promoted glucose tolerance in the recipient mice better than transplantation of islets from wild type mice.74 It must be noted, however that β-cell-specific loss of IRS-2 does not prevent development and growth of β-cells during the embryonic and early post-natal periods.75-77 Notwithstanding the observation that some β-cells “escape” Cre recombination, it is intriguing that lack of IRS-2 in a “majority” of β-cells during the early post-natal period, when Cre expression is maximal and “escape” is minimal, does not lead to a phenotype as severe as that observed in global mutants despite being created on similar genetic backgrounds. This indicates that signals independent of IRS-2 are likely necessary during early growth and development of β-cells. One important role for IRS-2 in β-cell plasticity may be its anti-apoptotic effects linked to cAMP and mediated by the cAMP response binding protein (CREB).78 A reduced expression of IRS-2 has been reported to promote increased apoptosis.57,70,79,80 Several studies to rescue the defects in b-cell growth and function due to loss of IRS-2 have been performed. For example, deficiency of protein tyrosine phosphatase 1b (PTP1B) delayed the onset of diabetes in mice lacking IRS-281 and transgenic expression of PDX-1 in IRS-2 null mice restored β-cell mass and promoted glucose tolerance.82 Recently, IRS-2 null mice crossed with mice haploinsufficient for the 3'-lipid-phosphatase Pten also prevented glucose intolerance and the mice survived without diabetes before succumbing to lymphoproliferative disease.83 Intriguingly, increasing the expression of IRS-4 in β-cell lines that express reduced IRS-2 prevented apoptotic effects.79 These reports indicate that several signaling proteins and transcription factors can restore the defects in β-cell growth and function due to loss of IRS-2.
By contrast to the defects in β-cell function and growth due to loss of function of IRS-1 and IRS-2, IRS-3 null mice develop normally and have normal glucose tolerance.84 IRS-4 deficient mice manifest mild growth defects and glucose intolerance and this is evident only in males because the IRS-4 gene is located on the X chromosome.85 Knockout of the p85 regulatory subunit of PI 3-kinase in mice leads to increased insulin sensitivity and hypoglycemia.86-88 Islets of mice deficient in the p85 sub-unit of PI 3-kinase exhibited reduced insulin content and mass of endoplasmic reticulum while manifesting an increase in insulin secretory response.89 Studies using PI 3-kinase inhibitors report an amplification of glucose-stimulated insulin secretion in islets from lean but not obese mice89-91 and has been suggested to occur at a level distal to mechanisms that alter cytosolic [Ca++].89 While the effects of PI 3-kinase suppression, due to insulin resistance, in classical insulin sensitive tissues leads to a decrease in insulin sensitivity,2 the opposite effect in islet β-cells has been suggested to promote increased insulin secretion to maintain glucose homeostasis.91 These studies, largely based on using PI 3-kinase inhibitors, should be interpreted with caution since wortmannin, at concentrations widely used to inhibit PI 3-kinase, has been reported to exert effects on other proteins in mammalian cells.92
Two independent groups have highlighted the role of Akt/PKB in the regulation of islet mass. Birnbaum and colleagues reported that null mutants for Akt-1 (PKB-β) manifest insulin resistance in muscle and liver and an increased islet mass, while mice lacking the Akt2 enzyme develop insulin resistance and diabetes-like syndrome.93,94 Parallel studies in Permutt's lab reported that constitutive activation of Akt1/PKBalpha increased β-cell size, enhanced islet mass, induced hyperinsulinemia and protected against experimental diabetes.95 Conversely, mice expressing a kinase-dead mutant of Akt manifest defective insulin secretion and increased susceptibility to development of diabetes.96 Together, these experiments highlight an important role for Akt/PKB in the mainenanace of islet mass. Mutants for p70S6 kinase show reduced β-cell size, lower insulin secretion and reduced pancreatic insulin content.97
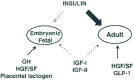
The lack of a defect in the early growth and development of islet/β-cells in mice lacking insulin or IGF-1 receptors indicates that other growth factors are likely important for the development of the insulin-secreting cells (Fig. 2). Indeed, several reports indicate a role for placental lactogen and GH during the early development of the pancreas (reviewed in refs. 98, 99) Overexpression of placental lactogen,100 PThRP,101 and HGF102 individually in β-cells using the rat insulin promoter lead to an increase in β-cell mass and resistance to strepotozotocin-induced β-cell death. Surprisingly, mice with a β-cell-specific knockout of c-met, the receptor mediating the actions of hepatocyte growth factor, developed defects in glucose-stimulated insulin secretion due to reduced glut2 expression but showed little103 or no effects104 on β-cell mass. Further, mice doubly transgenic for parathyroid hormone-related protein and placental lactogen showed defects in β-cell function and growth that was similar in severity to single transgenic mice indicating potential saturation of signaling and common downstream partners for the two hormones105 and also suggest an important role for placental lactogen in β-cell survival. It will be useful to examine potential cross-talk between signaling pathways activated by the receptor tyrosine kinases for c-met, PThRP and placental lactogen and the insulin/IGF-I system to unravel novel interactions underlying the growth and apoptosis of islet cells.
Maintenance of Adult β-Cell Mass
The mechanisms and signaling pathways that maintain adult β-cell mass are currently intense areas of research in type 1 and type 2 diabetes. Several mechanisms have been proposed to influence adult β-cell mass including neogenesis from ductal cells106,107 and apoptosis.108,109 Recent studies using lineage trace analysis provide compelling evidence for β-cell replication as a major pathway for the renewal of adult β-cells in mice.110 Whether a similar mechanism is operative in humans is not known and impossible to prove by lineage trace analysis. Some evidence for DNA duplication is available from two recent studies suggesting de-differentiation and differentiation of human islet precursor cells.111,112 Nevertheless, it is conceivable that all three processes are occurring to maintain an appropriate number of β-cells for glucose homeostasis, and identifying the major pathway that contributes to β-cell regeneration will be key to plan strategies to intervene therapeutically. Therefore, until lineage trace analyses or a similar definitive technique can conclusively prove that neogenesis from duct or periductal cells is also a significant source of β-cells in rodents, or until studies in humans can conclusively demonstrate that β-cell regeneration does not involve mitosis, all efforts must be targeted to understand the basic mechanisms underlying β-cell replication. Some immediate questions include – what are the signals that promote β-cell replication, what are the pathways that mediate the mitotic response and how many times can a single β-cell divide over its life span? Answers to these questions, though not trivial, will likely provide crucial information to focus efforts to develop therapeutic targets to enhance β-cell regeneration.
Previous studies have reported a clear role for cyclin dependent kinase 4 (CDK4) as an essential regulator of β-cell cycle control. Rane and coworkers113 created mice lacking CDK4 and observed the mutants were infertile and developed insulin-deficient diabetes as a consequence of reduced β-cells. Conversely, mice with an activating mutation of CDK4, that inhibited binding to the cell cycle inhibitor P16INK4a, developed pancreatic hyperplasia but did not lead to tumors.113 Recent studies examining the role of cyclin D1 and D2 support the replication hypothesis.114,115 Mice lacking cyclin D2 showed a selective decrease in β-cell expansion while maintaining normal ductal cells suggesting that the cell cycle protein is important for proliferation of β-cells independent of influencing duct cells.
One potential mechanism that can contribute to β-cell expansion, includes the replication process and involves a well-recognized pathway that has been studied in considerable detail in organogenesis and in cancer.116,117 Epithelial-to-mesenchymal transition or EMT occurs in epithelial cells expressing tyrosine kinase receptors and involves disappearance of differentiated junctions, reorganization of cytoskeleton and redistribution of organelles, together transforming epithelial into mesenchymal cells.117-119 After transformation, the mesenchymal cells may eventually regain a fully differentiated epithelial phenotype via a mesenchyme-to-epithelial transition (MET) or reverse EMT.120 A characteristic feature of EMT is repression of epithelial markers including E-cadherin and α- and γ-catenins and induction of mesenchymal markers including vimentin, fibronectin and N-cadherin.117,121 Although the term EMT has been mostly associated either with early development or neoplasia, it is possible that this process is occurring, albeit modified, in normal cells responding to physiological demands that require cell/tissue expansion. Indeed, the E-cadherin/catenin family of proteins can also act as master regulatory and signaling molecules for differentiation, proliferation and apoptosis117,122 indicating that these proteins have the capacity to regulate growth in normal tissues.122 Cell-cell adhesion, as mediated by the cadherin-catenin system, is a prerequisite for normal cell function and for the preservation of tissue integrity in most tissues including islet cells. Both E-cadherin and β-catenin and several other members of the cadherin/catenin family are under the control of growth factors including epidermal growth factor (EGF), hepatocyte growth factor/scatter factor (HGF/SF) and insulin like-growth factors (IGF-I and IGF-II).117,120 Receptors for these growth factors are expressed in β-cells, and proteins in their signaling pathways have been reported to play functional roles in β-cell growth and hormone secretion. Intriguingly, treatment of mouse embryonic stem cells or rat bladder carcinoma cells with IGF-II induces EMT and the withdrawal of IGF-II allows a reversal of the phenotype to an epithelial cell.120 Direct association between insulin/IGF-1 receptors with the E-cadherin-catenin system, forming a multi-element complex, has been recently suggested based on colocalization of IGF-1 receptors with E-cadherin and β-and α-catenins at points of cell contacts.117 The presence of IGF-1 receptors and its substrate proteins insulin receptor substrate-1 and SHC in the same complex with E-cadherin indicates potential cross talk between growth factor and catenin-cadherin signaling pathways.117 Further evidence for a role for cadherin-catenin complex in islet growth is provided by mouse experiments in which dominant-negative expression of E-cadherin on the rat insulin promoter perturbed islet formation without increasing the incidence of tumor formation.117,123 Together, these data provide a basis for a link between growth factor signaling and the potential for EMT in islet/β-cell growth.
Recently, features suggestive of EMT were described to occur in vivo , for the first time to our knowledge, in a mouse model of insulin resistance manifesting robust islet hyperplasia.124 The presence of PCNA+ cells within the islets, which also showed down regulation of E-cadherin, and an increased localization of β-catenin to the nucleus, provided evidence for alterations in adhesion properties and an ability of the cells to replicate (Fig. 3A).124 Furthermore, the lack of close association of replicating cells with lectin (a ductal marker), in multiple pancreas sections, suggests the cells are independent of pancreatic ducts and are likely replicating β-cells that have undergone metaplastic changes. Downregulation of β-catenin, another adhesion protein, in islet cells from a mouse which is haploinsufficient for PDX-1, indicates a potential role for the homeodomain protein in the EMT process (Fig. 3B). In fact, growth factor signaling has been linked to PDX-1-mediated regulation of β-cell growth125 providing additional evidence for a role for PDX-1 in β-cell regeneration. Interestingly, the PCNA+ cells are detectable in some, if not all, of the islets. For example, Figure 3C shows three islets in a pancreas section from a mouse that is doubly heterozygous for insulin receptor and insulin receptor substrate-1. PCNA+ cells are clearly evident within the core of two of the islets but not the third suggesting that the proliferation response occurs in susceptible islets likely in a regulated manner. The mechanisms and signals that allow some cells, but not others, to begin the process of replication in response to stimuli such as insulin resistance require further investigation. Thus, it is possible that EMT or an EMT-like process, may promote β-cell expansion under the appropriate stimulatory conditions. The report that EMT also occurs in human islet cell precursor cells111,112 suggests that this process is a common response across species. Whether this indeed occurs in vivo in humans in early stages of diabetes is an important and timely question to address.

It has been recognized for over a decade that β-cells compensate in order to overcome the ambient hyperinsulinemia.64,126 However, few studies have examined the pathways and proteins underlying the islet hyperplastic process. Since circulating insulin levels are significantly elevated in insulin resistance, an obvious candidate for β-cell proliferation is insulin itself33,127 (Fig. 2). In fact, insulin has been shown to enhance islet β-cell replication in neonatal rat monolayer cultures128 and to increase the regenerative ability of β-cells in transplantation models of fetal rat pancreas.129,130 A role for insulin as a growth factor is also supported by studies in βIRKO mice, which display an age-dependent decrease in β-cell mass59,62 and by reports that treatment of MIN6 β-cells with insulin receptor siRNA leads to altered expression of cell cycle proteins and proliferation.131 Further support for a proliferative role for insulin arises from our recent studies using a transplantation model wherein BrdU+ β-cells show a direct correlation with circulating levels of insulin.73,132 In addition to its nutrient role, glucose has been shown to increase β-cell mass in several models (reviewed in ref. 98).133 Whether the effects of glucose are mediated by the secreted insulin acting in an autocrine manner to promote growth and/or prevent apoptosis in β-cells requires further study in models lacking insulin receptors in β-cells. Similarly, it is possible that the effects of GLP-1 on β-cell proliferation29,134 are mediated, in part, by secreted insulin acting in an autocrine manner.
Other mechanisms that have been reported to contribute to regeneration of β-cells include budding of cells from pancreatic duct tissues,106 transdifferentiation from acinar cells135,136 and the ability of transcription factors to induce hepatocytes to differentiate into insulin-secreting cells.137,138 Figure 4 shows a schematic of current concepts on potential pathways of β-cell regeneration.

Figure 4
A schematic of potential mechanisms contributing to β-cell regeneration. Figure reproduced with permission from Kulkarni RN, Rev Endo Metab Disord 2005; 6(3):199-210.
Growth and Development of Islet α-Cells
Insulin and IGF-1 receptors are also expressed in islet α-cells139,140 and their role in early development and growth of glucagon-producing cells is not fully explored. However, a relative increase in α-cell number is a recognized feature in adult patients with type 2 diabetes.141 Whether potential stimulation of α-cell proliferation is in fact mediated by high circulating levels of insulin in established cases of type 2 diabetes is not clear. Several studies implicate a role for intra-islet insulin to suppress glucagon release as an important factor in poor recovery from hypoglycemia in patients with long-standing type 1 diabetes and in advanced stages of type 2 diabetes on insulin therapy.142,143 Indeed, a report suggests that glucose-stimulated glucagon secretion in αTC6 cells requires expression of insulin receptors in α-cells.144 The signaling proteins and pathways that mediate these effects are not fully defined.
A recent study, in which mice were treated with glucagon receptor inhibitors, reported a significant increase in α-cell hyperplasia providing evidence for therapeutic intervention at the receptor level in modulating the ability of islet cells to grow.145 The potential influence of interactions between β- and α-cells on the modulation of growth of islets during development and for maintenance of mass during adulthood in the face of altering pathophysiological states is worth exploring.
The Liver-Pancreas Connection
The liver and the pancreas are known to share a common developmental pathway146,147 and express several common transcription factors, which are essential for their growth.147 Therefore, it is not surprising that even in the adult organism there is evidence suggestive of communication between the two metabolic tissues. For example, mice with a hepatic glucokinase knock-out manifest impaired insulin secretion in response to glucose suggesting that loss of hepatic glucose sensing impacts on islet function.148 Furthermore, the liver has long been recognized as a source of circulating growth factors including IGF and HGF/SF, both of which are known to especially influence islet growth (reviewed in ref. 64).39,102,149,150 In this context, it is interesting that mice lacking insulin receptors in hepatocytes develop large islets.151 One interpretation of these observations is that in pathophysiological states, the insulin-resistant liver may potentially transmit signals to the islets via secreted growth factors to allow for β-cell compensation.64 Although studies in hepatocytes implicate a role for insulin in regulating the hepatocyte nuclear transcription factor 3β (Foxa2),152 and PDX-1 has been linked to growth factor signaling in the context of β-cell growth,125 further studies to define the proteins linking upstream signals such as insulin with transcription factors in islets/β-cells are worth exploring.153
Future Insights
Genetic engineering techniques have revolutionized the understanding of the role of insulin and IGFs in the development and maintenance of β-cell mass. Understanding the link(s) between insulin/IGF-I/IRS/FoxO1 signaling and PDX-1 with molecules that regulate β-cell cycle control will be crucial for the development of therapeutic strategies aimed at promoting islet cell regeneration.
Acknowledgements
The author thanks Julie Marr and Kezia Frayjo for excellent secretarial assistance. R.N.K. is the recipient of the K08 Clinical Scientist Award (NIH DK 02885) and acknowledges support from NIH R01 DK67536, R01 DK68721, R03 DK66207, the Harvard Stem Cell Institute, the ADA and JDRF Center for Islet Transplantation at Harvard Medical School.
References
- 1.
- Blakesley VA, Butler AA, Koval AP. In: Rosenfeld R, Roberts Jr C, eds. The IGF system. 1999. pp. 143–164.
- 2.
- Cheatham B, Kahn CR. Insulin action and the insulin signaling network. Endocr Rev. 1995;16:117–142. [PubMed: 7781591]
- 3.
- De Meyts P. et al. The insulin-like growth factor-1 receptor. Structure, ligand-binding mechanism and signal transduction. Horm Res. 1994;42:152–169. [PubMed: 7868068]
- 4.
- Bell GI, Polonsky KS. Diabetes mellitus and genetically programmed defects in beta-cell function. Nature. 2001;414:788–791. [PubMed: 11742410]
- 5.
- Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799–806. [PubMed: 11742412]
- 6.
- Kahn CR. Diabetogenes and the cause of type II diabetes (Banting Lecture). Diabetes. 1994:1066–1084. [PubMed: 8039601]
- 7.
- White MF. IRS proteins and the common path to diabetes. Am J Physiol Endocrinol Metab. 2002;283:E413–E422. [PubMed: 12169433]
- 8.
- Accili D. Lilly lecture 2003. The struggle for mastery in insulin action: From triumvirate to republic. Diabetes. 2004;53(7):1633–42. [PubMed: 15220184]
- 9.
- White MF, Yenush L. The IRS-signaling system: A network of docking proteins that mediate insulin and cytokine action. Curr Top Microbiol Immunol. 1998;228:179–208. [PubMed: 9401207]
- 10.
- Virkamaki A, Ueki K, Kahn CR. Protein-protein interaction in insulin signaling and the molecular mechanisms of insulin resistance. J Clin Invest. 1999;103:931–943. [PMC free article: PMC408269] [PubMed: 10194465]
- 11.
- DeFronzo RA. Lilly Lecture 1987. The triumvirate: β-cell, muscle, liver: A collusion responsible for NIDDM. Diabetes. 1988;37:667–687. [PubMed: 3289989]
- 12.
- Kulkarni RN. Receptors for insulin and insulin-like growth factor-1 and insulin receptor substrate-1 mediate pathways that regulate islet function. Biochem Soc Trans. 2002;30:317–322. [PubMed: 12023872]
- 13.
- Leibowitz G. et al. Insulin does not mediate glucose stimulation of proinsulin biosynthesis. Diabetes. 2003;52:998–1003. [PubMed: 12663472]
- 14.
- Wicksteed B, Alarcon C, Briaud I. et al. Glucose-induced translational control of proinsulin biosynthesis is proportional to preproinsulin mRNA levels in islet beta-cells but not regulated via a positive feedback of secreted insulin. J Biol Chem. 2003;278:42080–42090. [PubMed: 12928442]
- 15.
- Irminger JC, Vollenweider FM, Neerman-Arbez M. et al. Human proinsulin conversion in the regulated and the constitutive pathways of transfected AtT20 cells. J Biochem. 1994;269:1756–1762. [PubMed: 7507483]
- 16.
- Halban PA. Proinsulin processing in the regulated and the constitutive secretory pathway. Diabetologia. 1994;37(Suppl 2):S65–S72. [PubMed: 7821742]
- 17.
- Nakae J, Kido Y, Accili D. Distinct and overlapping functions of insulin and IGF-1 receptors. Endo Reviews. 2001;22:818–835. [PubMed: 11739335]
- 18.
- Persaud SJ, AsareAnane H, Jones PM. Insulin receptor activation inhibits insulin secretion from human islets of Langerhans. Growth Regul. 2002;510:225–228. [PubMed: 11801259]
- 19.
- Aspinwall CA, Lakey JRT, Kennedy RT. Insulin-stimulated insulin secretion in single pancreatic beta cells. J Biol Chem. 1998;274:6360–6365. [PubMed: 10037726]
- 20.
- Johnson JD, Misler S. Nicotinic acid-adenine dinucleotide phosphate-sensitive calcium stores initiate insulin signaling in human beta cells. Proc Natl Acad Sci USA. 2002;99:14566–14571. [PMC free article: PMC137923] [PubMed: 12381785]
- 21.
- Luciani DS, Johnson JD. Acute effects of insulin on beta-cells from transplantable human islets. Mol Cell Endocrinol. 2005;241:88–98. [PubMed: 16099589]
- 22.
- Leibiger B, Leibiger IB, Moede T. et al. Selective insulin signaling through A and B insulin receptors regulates transcription of insulin and glucokinase genes in pancreatic beta cells. Mol Cell. 2001;7:559–570. [PubMed: 11463381]
- 23.
- Bouche C, Kulkarni RN, Kahn CR. et al. Exogenous insulin enhances glucose-stimulated insulin secretion in healthy humans. Diabetes. 2005
- 24.
- Rasouli N, Hale T, Kahn SE. et al. Effects of short term experimental insulin resistance and family history of diabetes on pancreatic {beta}-cell function in nondiabetic individuals. J Clin Endocrinol Metab. 2005;90(10):5825–33. [PubMed: 16091496]
- 24a.
- Gunton JE, Kulkarni RN, Yim S. et al. Loss of ARNT/HIF1beta mediates altered gene expression and pancreatic-islet dysfunction in human type 2 diabetes. Cell. 2005;122(3):337–49. [PubMed: 16096055]
- 24b.
- Hribal ML, Perego L, Lovari S. et al. Chronic hyperglycemia impairs insulin secretion by affecting insulin receptor expression splicing and signaling in RIN beta cell line and human islets of Langerhans. FASEB J. 2003;17(10):1340–2. [PubMed: 12738810]
- 25.
- Pictet R, Rutter WJ. In: Steiner DF, Freinkel M, eds. Handbook of Physiology. Washington, DC: American Physiological Society. 1972:25–66.
- 26.
- Herrera PL. Defining the cell lineages of the islets of Langerhans using transgenic mice. Int J Dev Biol. 2002;46:97–103. [PubMed: 11902693]
- 27.
- Murtaugh LC, Melton DA. Genes, signals, and lineages in pancreas development. Annu Rev Cell Dev Biol. 2003;19:71–89. [PubMed: 14570564]
- 28.
- Sander M, German MS. The beta cell transcription factors and development of the pancreas. J Mol Med. 1997;75:327–340. [PubMed: 9181474]
- 29.
- Habener JF, Kemp DM, Thomas MK. Minireview: Transcriptional regulation in pancreatic development. Endocrinology. 2005;146:1025–1034. [PubMed: 15604203]
- 30.
- Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129:2447–2457. [PubMed: 11973276]
- 31.
- Fowden AL, Hill DJ. Intra-uterine programming of the endocrine pancreas. Br Med Bull. 2001:123–142. [PubMed: 11809622]
- 32.
- Dupont J, Holzenberger M. Biology of insulin-like growth factors in development. Birth Defects Res C Embryo Today. 2003;69:257–271. [PubMed: 14745968]
- 33.
- Hill DJ, Milner RD. Insulin as a growth factor. Pediatr Res. 1985;19:879–886. [PubMed: 2413420]
- 34.
- Milner RD, Hill DJ. Fetal growth control: The role of insulin and related peptides. Clin Endocrinol (Oxf.). 1984;21:415–433. [PubMed: 6096045]
- 35.
- Hill DJ. Development of the endocrine pancreas. Rev Endocr Metab Disord. 2005;6:229–238. [PubMed: 16151627]
- 36.
- Hogg J, Han VKM, Clemmons DR. et al. Interactions of glucose, insulin-like growth factors (IGFs) and IGF binding proteins in the regulation of DNA synthesis by isolated fetal rat islets of langerhans. J Endocrinol. 1993:401–412. [PubMed: 7506287]
- 37.
- Fehmann HC, Jehle P, Goke B. IGF-I and IGF-II: Expression and function in the endocrine pancreas. Exp Clin Endocrinol Diabetes. 1995;103(Suppl 2):37–41. [PubMed: 8839252]
- 38.
- Hill DJ, Hogg J. Growth factor control of pancreatic B cell hyperplasia. Baillieres. Clin Endocrinol Metab. 1991;5:689–698. [PubMed: 1755812]
- 39.
- Hill DJ, Petrik J, Arany E. Growth factors and the regulation of fetal growth. Diabetes Care. 1998;(Suppl 2):B60–B69. [PubMed: 9704229]
- 40.
- Orban PC, Chui D, Marth JD. Tissue- and site-specific DNA recombination in transgenic mice. Proc Natl Acad Sci USA. 1992;89:6861–6865. [PMC free article: PMC49604] [PubMed: 1495975]
- 41.
- Ryding AD, Sharp MG, Mullins JJ. Conditional transgenic technologies. J Endocrinol. 2001;171:1–14. [PubMed: 11572785]
- 42.
- Duvillie B. et al. Increased islet cell proliferation, decreased apoptosis, and greater vascularization leading to beta-cell hyperplasia in mutant mice lacking insulin. Endocrinology. 2002;143:1530–1537. [PubMed: 11897712]
- 43.
- Duvillie B. et al. Phenotypic alterations in insulin-deficient mutant mice. Proc Natl Acad Sci USA. 1997;94:5137–5140. [PMC free article: PMC24644] [PubMed: 9144203]
- 44.
- Lammert E, Cleaver O, Melton D. Induction of pancreatic differentiation by siganls from blood vessels. Science. 2001:1–10. [PubMed: 11577200]
- 45.
- Lammert E, Cleaver O, Melton D. Role of endothelial cells in early pancreas and liver development. Mech Dev. 2003;120:59–64. [PubMed: 12490296]
- 46.
- Cleaver O, Melton DA. Endothelial signaling during development. Nat Med. 2003;9:661–668. [PubMed: 12778164]
- 47.
- Lu Y. et al. Pancreatic-specific inactivation of IGF-I gene causes enlarged pancreatic islets and significant resistance to diabetes. Diabetes. 2004;53:3131–3141. [PubMed: 15561943]
- 48.
- Petrik J. et al. Overexpression of insulin-like growth factor-II in transgenic mice is associated with pancreatic islet cell hyperplasia. Endocrinology. 1999;140:2353–2363. [PubMed: 10218989]
- 49.
- Devedjian JC. et al. Transgenic mice overexpressing insulin-like growth factor-II in B cells develop type 2 diabetes. J Clin Invest. 2000;105:731–740. [PMC free article: PMC377454] [PubMed: 10727441]
- 50.
- Efstratiadis A. Genetics of mouse growth. Int J Dev Biol. 1998;955-976 [PubMed: 9853827]
- 51.
- Liu JP, Baker J, Perkins JA. et al. Mice carrying null mutations of the genes encoding insulin-like growth factor I (lgf-1) and type 1 IGF receptor (lgf1r). Cell. 1993;75:59–72. [PubMed: 8402901]
- 52.
- Powell-Braxton L. et al. IGF-I is required for normal embryonic growth in mice. Genes Dev. 1993;7:2609–2617. [PubMed: 8276243]
- 53.
- Woods KA, Camacho-Hubner C, Savage MO. et al. Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N Engl J Med. 1996;335:1363–1367. [PubMed: 8857020]
- 54.
- Accili D. et al. Early neonatal death in mice homozygous for a null allele of the insulin receptor gene. Nat Genet. 1996;12:106–109. [PubMed: 8528241]
- 55.
- Joshi RL. et al. Targeted disruption of the insulin receptor gene in the mouse results in neonatal lethality. EMBO J. 1996;15:1542–1547. [PMC free article: PMC450062] [PubMed: 8612577]
- 56.
- Joshi RL. et al. Targeted disruption of the insulin receptor gene in the mouse results in neonatal lethality. EMBO J. 1996;15:1542–1547. [PMC free article: PMC450062] [PubMed: 8612577]
- 57.
- Withers DJ. et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature. 1998;391:900–904. [PubMed: 9495343]
- 58.
- Kido Y, Nakae J, Xuan S. et al. Beta cell development in mice lacking insulin and type I IGF receptors. Diabetes. 2000;49(Suppl 1)
- 59.
- Kulkarni RN. et al. Tissue-specific knockout of the insulin receptor in pancreatic β cells creates an insulin secretory defect similar to that in Type 2 diabetes. Cell. 1999;96:329–339. [PubMed: 10025399]
- 60.
- Kulkarni RN. et al. beta-cell-specific deletion of the Igf1 receptor leads to hyperinsulinemia and glucose intolerance but does not alter beta-cell mass. Nat Genet. 2002;31:111–115. [PubMed: 11923875]
- 61.
- Xuan S. et al. Defective insulin secretion in pancreatic beta cells lacking type 1 IGF receptor. J Clin Invest. 2002;110:1011–1019. [PMC free article: PMC151144] [PubMed: 12370279]
- 62.
- Otani K. et al. Reduced beta-cell mass and altered glucose sensing impair insulin-secretory function in betaIRKO mice. Am J Physiol Endocrinol Metab. 2004;286:E41–E49. [PubMed: 14519599]
- 63.
- Da Silva X, Qian Q, Cullen PJ. et al. Distinct roles for insulin and insulin-like growth factor-1 receptors in pancreatic beta-cell glucose sensing revealed by RNA silencing. Biochem J. 2004;377:149–158. [PMC free article: PMC1223855] [PubMed: 14563207]
- 64.
- Kulkarni RN, Kahn CR. Molecular basis of pancreas development and function. In: Habener JF, Hussain M, eds. New York City: Kluwer Academic Publishers. 2001:299–323.
- 65.
- Elders MJ. et al. Endocrine-metabolic relationships in patients with leprechaunism. J Natl Med Assoc. 1982;74:1195–1210. [PMC free article: PMC2561419] [PubMed: 7154104]
- 66.
- Taylor SI. Lilly Lecture: Molecular mechanisms of insulin resistance-Lessons from patients with mutations in the insulin receptor gene. Diabetes. 1992;41:1473–1490. [PubMed: 1327927]
- 67.
- Araki E. et al. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature. 1994;372:186–190. [PubMed: 7526222]
- 68.
- Tamemoto H. et al. Insulin resistance and growth retardation in mice lacking insulin receptor substrate-1. Nature. 1994;372:182–186. [PubMed: 7969452]
- 69.
- Kulkarni RN. et al. Altered function of insulin receptor substrate-1-deficient mouse islets and cultured beta-cell lines. J Clin Invest. 1999;104:R69–R75. [PMC free article: PMC409887] [PubMed: 10606633]
- 70.
- Kubota N. et al. Disruption of insulin receptor substrate 2 causes type 2 diabetes because of liver insulin resistance and lack of compensatory β-cell hyperplasia. Diabetes. 2000;49:1880–1889. [PubMed: 11078455]
- 71.
- Aspinwall CA. et al. Roles of insulin receptor substrate-1, phosphatidylinositol 3-kinase, and release of intracellular Ca2+ stores in insulin-stimulated insulin secretion in beta -cells. J Biol Chem. 2000;275:22331–22338. [PubMed: 10764813]
- 72.
- Kulkarni RN, Roper M, Dahlgren GM. et al. Insulin secretory defect in IRS-1 null mice is linked with reduced calcium signaling and altered expression of SERCA-2b and-3. Diabetes. 2004;53:1517–1525. [PubMed: 15161756]
- 73.
- Hennige AM. et al. Alterations in growth and apoptosis of insulin receptor substrate-1 deficient beta-cells. Am J Physiol Endocrinol Metab. 2005;289:E337–E346. [PubMed: 15827066]
- 74.
- Hennige AM. et al. Upregulation of insulin receptor substrate-2 in pancreatic beta cells prevents diabetes. J Clin Invest. 2003;112:1521–1532. [PMC free article: PMC259126] [PubMed: 14617753]
- 75.
- Lin X. et al. Dysregulation of insulin receptor substrate 2 in beta cells and brain causes obesity and diabetes. J Clin Invest. 2004;114:908–916. [PMC free article: PMC518668] [PubMed: 15467829]
- 76.
- Kubota N. et al. Insulin receptor substrate 2 plays a crucial role in beta cells and the hypothalamus. J Clin Invest. 2004;114:917–927. [PMC free article: PMC518663] [PubMed: 15467830]
- 77.
- Choudhury AI. et al. The role of insulin receptor substrate 2 in hypothalamic and beta-cell function. J Clin Invest. 2005;115:940–950. [PMC free article: PMC1069106] [PubMed: 15841180]
- 78.
- Jhala US. et al. cAMP promotes pancreatic beta cell survival via CREB mediated induction of IRS2. Genes Dev. 2003;17:1575–1580. [PMC free article: PMC196130] [PubMed: 12842910]
- 79.
- Lingohr MK. et al. Decreasing IRS-2 expression in pancreatic beta-cells (INS-1) promotes apoptosis, which can be compensated for by introduction of IRS-4 expression. Mol Cell Endocrinol. 2003;209:17–31. [PubMed: 14604813]
- 80.
- Briaud I. et al. Insulin receptor substrate-2 proteasomal degradation mediated by a mammalian target of rapamycin (mTOR)-induced negative feedback downregulates protein kinase B-mediated signaling pathway in beta-cells. J Biol Chem. 2005;280:2282–2293. [PubMed: 15537654]
- 81.
- Kushner JA. et al. Islet-sparing effects of protein tyrosine phosphatase-1b deficiency delays onset of diabetes in IRS2 knockout mice. Diabetes. 2004;53:61–66. [PubMed: 14693698]
- 82.
- Kushner JA. et al. Pdx1 restores beta cell function in Irs2 knockout mice. J Clin Invest. 2002;109:1193–1201. [PMC free article: PMC150960] [PubMed: 11994408]
- 83.
- Kushner JA. et al. Pten regulation of islet growth and glucose homeostasis. J Biol Chem. 2005 [PubMed: 16170201]
- 84.
- Liu SC, Wang Q, Lienhard GE. et al. Insulin receptor substrate 3 is not essential for growth or glucose homeostasis. J Biol Chem. 1999;274:18093–18099. [PubMed: 10364263]
- 85.
- Fantin VR, Wang GE, Lienhard GE. et al. Mice lacking insulin receptor substrate 4 exhibit mild defects in growth, reproduction, and glucose homostasis. Am J Physiol. 2000;278:E127–133. [PubMed: 10644546]
- 86.
- Ueki K. et al. Increased insulin sensitivity in mice lacking p85beta subunit of phosphoinositide 3-kinase. Proc Natl Acad Sci USA. 2002;99:419–424. [PMC free article: PMC117575] [PubMed: 11752399]
- 87.
- Terauchi Y. et al. Increased insulin sensitivity and hypoglycaemia in mice lacking the p85 alpha subunit of phosphoinositide 3-kinase. Nat Genet. 1999;21:230–235. [PubMed: 9988280]
- 88.
- Mauvais-Jarvis F. et al. Reduced expression of the murine p85a subunit of phosphoinositide 3-kinase improves insulin signaling and ameliorates diabetes. J Clin Invest. 2002;109:141–149. [PMC free article: PMC150818] [PubMed: 11781359]
- 89.
- Eto K. et al. Phosphatidylinositol 3-kinase suppresses glucose-stimulated insulin secretion by affecting post-cytosolic [Ca(2+)] elevation signals. Diabetes. 2002;51:87–97. [PubMed: 11756327]
- 90.
- Zawalich WS, Tesz GJ, Zawalich KC. Inhibitors of phosphatidylinositol 3-kinase amplify insulin release from islets of lean but not obese mice. J Endocrinol. 2002;174:247–258. [PubMed: 12176663]
- 91.
- Zawalich WS, Zawalich KC. A link between insulin resistance and hyperinsulinemia: Inhibitors of phosphatidylinositol 3-kinase augment flucose-induced insulin secretion from islets of lean, but not obese, rats. Endocrinology. 2000;141:3287–3295. [PubMed: 10965900]
- 92.
- Liu Y. et al. Wortmannin, a widely used phosphoinositide 3-kinase inhibitor, also potently inhibits mammalian polo-like kinase. Chem Biol. 2005;12:99–107. [PubMed: 15664519]
- 93.
- Cho H. et al. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKBβ). Science. 2001;292:1728–1731. [PubMed: 11387480]
- 94.
- Tuttle RL. et al. Regulation of pancreatic beta-cell growth and survival by the serine/threonine protein kinase Akt1/PKBalpha. Nat Med. 2001;7:1133–1137. [PubMed: 11590437]
- 95.
- Bernal-Mizrachi E, Wen W, Stahlhut S. et al. Islet beta cell expression of constitutively active Akt1/PKB alpha induces striking hypertrophy, hyperplasia, and hyperinsulinemia. J Clin Invest. 2001;108:1631–1638. [PMC free article: PMC200992] [PubMed: 11733558]
- 96.
- Bernal-Mizrachi E. et al. Defective insulin secretion and increased susceptibility to experimental diabetes are induced by reduced Akt activity in pancreatic islet beta cells. J Clin Invest. 2004;114:928–936. [PMC free article: PMC518659] [PubMed: 15467831]
- 97.
- Pende M. et al. Hypoinsulinaemia,glucose intolerance and diminished beta-cell size in S6K1-deficient mice. Nature. 2000;408:994–997. [PubMed: 11140689]
- 98.
- Bonner-Weir S, Smith FE. Islet cell growth and the growth factors involved. TEM. 1994;5:60–64. [PubMed: 18407189]
- 99.
- Bonner-Weir S. Regulation of pancreatic β-cell mass in vivo. Recent Prog Horm Res. 1994;49:91–104. [PubMed: 8146438]
- 100.
- Vasavada RC. et al. Targeted expression of placental lactogen in the beta cells of transgenic mice results in beta cell proliferation, islet mass augmentation, and hypoglycemia. J Biol Chem. 2000;275:15399–15406. [PubMed: 10809775]
- 101.
- Vasavada RC. et al. Overexpression of parathyroid hormone-related protein in the pancreatic islets of transgenic mice causes islet hyperplasia, hyperinsulinemia, and hypoglycemia. J Biol Chem. 1996;271:1200–1208. [PubMed: 8557651]
- 102.
- Garcia-Ocana A. et al. Hepatocyte growth factor overexpression in the islet of transgenic mice increases beta cell proliferation, enhances islet mass, and induces mild hypoglycemia. J Biol Chem. 2000;275:1226–1232. [PubMed: 10625667]
- 103.
- Dai C, Huh CG, Thorgeirsson SS. et al. Beta-cell-specific ablation of the hepatocyte growth factor receptor results in reduced islet size, impaired insulin secretion, and glucose intolerance. Am J Pathol. 2005;167:429–436. [PMC free article: PMC1603568] [PubMed: 16049329]
- 104.
- Roccisana J. et al. Targeted inactivation of hepatocyte growth factor receptor c-met in beta-cells leads to defective insulin secretion and GLUT-2 downregulation without alteration of beta-cell mass. Diabetes. 2005;54:2090–2102. [PubMed: 15983210]
- 105.
- Fujinaka Y, Sipula D, Garcia-Ocana A. et al. Characterization of mice doubly transgenic for parathyroid hormone-related protein and murine placental lactogen: A novel role for placental lactogen in pancreatic beta-cell survival. Diabetes. 2004;53:3120–3130. [PubMed: 15561942]
- 106.
- Bonner-Weir S. et al. In vitro cultivation of human islets from expanded ductal tissue. Proc Natl Acad Sci USA. 2000;97:7999–8004. [PMC free article: PMC16659] [PubMed: 10884429]
- 107.
- Suarez-Pinzon WL, Lakey JR, Brand SJ. et al. Combination therapy with epidermal growth factor and gastrin induces neogenesis of human islet {beta}-cells from pancreatic duct cells and an increase in functional {beta}-cell mass*. J Clin Endocrinol Metab. 2005 [PubMed: 15769977]
- 108.
- Yoon KH. et al. Selective beta-Cell loss and alpha-cell expansion in patients with type 2 diabetes mellitus in korea. J Clin Endocrinol Metab. 2003;88:2300–2308. [PubMed: 12727989]
- 109.
- Butler AE. et al. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–110. [PubMed: 12502499]
- 110.
- Dor Y, Brown J, Martinez OI. et al. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004;429:41–46. [PubMed: 15129273]
- 111.
- Gershengorn MC. et al. Epithelial-to-mesenchymal transition generates proliferative human islet precursor cells. Science. 2004;306:2261–2264. [PubMed: 15564314]
- 112.
- Lechner A, Nolan AL, Blacken RA. et al. Redifferentiation of insulin-secreting cells after in vitro expansion of adult human pancreatic islet tissue. Biochem Biophys Res Commun. 2005;327:581–588. [PubMed: 15629153]
- 113.
- Rane SG. et al. Loss of cyclin-dependent kinase (Cdk4) expression causes insulin-deficient diabetes and cdk4 activation results in β-islet cell hyperplasia. Nat Genet. 1999;22:44–52. [PubMed: 10319860]
- 114.
- Georgia S, Bhushan A. Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. J Clin Invest. 2004;114:963–968. [PMC free article: PMC518666] [PubMed: 15467835]
- 115.
- Kushner JA, Ciemerych MA, Sicinska E. et al. Cyclins D2 and D1 are essential for postnatal pancreatic beta-cell growth. Mol Cell Biol. 2005;25:3752–3762. [PMC free article: PMC1084308] [PubMed: 15831479]
- 116.
- Thiery JP. Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2003;15:740–746. [PubMed: 14644200]
- 117.
- Potter E, Bergwitz C, Brabant G. The cadherin-catenin system: Implications for growth and differentiation of endocrine tissues. Endocr Rev. 1999;20:207–239. [PubMed: 10204118]
- 118.
- Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. [PubMed: 12189386]
- 119.
- Savagner P. Leaving the neighborhood: Molecular mechanisms involved during epithelial-mesenchymal transition. BioEssays. 2001;23:912–923. [PubMed: 11598958]
- 120.
- Morali OG. et al. IGF-II induces rapid beta-catenin relocation to the nucleus during epithelium to mesenchyme transition. Oncogene. 2001;20:4942–4950. [PubMed: 11526479]
- 121.
- Kang Y, Massague J. Epithelial-mesenchymal transitions: Twist in development and metastasis. Cell. 2004;118:277–279. [PubMed: 15294153]
- 122.
- El Bahrawy MA, Pignatelli M. E-cadherin and catenins: Molecules with versatile roles in normal and neoplastic epithelial cell biology. Microsc Res Tech. 1998;43:224–232. [PubMed: 9840800]
- 123.
- Dahl U, Sjodin A, Semb H. Cadherins regulate aggregation of pancreatic beta-cells in vivo. Development. 1996;122:2895–2902. [PubMed: 8787762]
- 124.
- Kulkarni RN. et al. PDX-1 haploinsufficiency limits the compensatory islet hyperplasia that occurs in response to insulin resistance. J Clin Invest. 2004;114:828–836. [PMC free article: PMC516265] [PubMed: 15372107]
- 125.
- Kitamura T. et al. The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic beta cell growth. J Clin Invest. 2002;110:1839–1847. [PMC free article: PMC151657] [PubMed: 12488434]
- 126.
- Bonner-Weir S, Scaglia L, Montana E. et al. Diabetes 1994. In: Baba S, Kaneko T, eds. Excerta Medica International Congress. 1995:179–228.
- 127.
- Accili D. A kinase in the life of the beta cell. J Clin Invest. 2001;108:1575–1576. [PMC free article: PMC200994] [PubMed: 11733550]
- 128.
- Rabinovitch A, Quigley C, Russell T. et al. Insulin and multiplication stimulating activity (and insulin-like growth factor) stimulate islet beta-cell replication in neonatal rat pancreatic monolayer cultures. Diabetes. 1982:160–164. [PubMed: 6759233]
- 129.
- McEvoy RC, Schmitt RV, Hegre OD. Syngeneic transplantation of fetal rat pancreas. I. Effect of insulin treatment of the reversal of alloxan diabetes. Diabetes. 1978;27:982–987. [PubMed: 359388]
- 130.
- Movassat J, Saulnier C, Portha B. Insulin administration enhances growth of the beta-cell mass in streptozotocin-treated newborn rats. Diabetes. 1997;46:1445–1452. [PubMed: 9287045]
- 131.
- Ohsugi M. et al. Reduced expression of the insulin receptor in mouse insulinoma (MIN6) cells reveals multiple roles of insulin signaling in gene expression, proliferation, insulin content, and secretion. J Biol Chem. 2005;280:4992–5003. [PubMed: 15546857]
- 132.
- Flier SN, Kulkarni RN, Kahn CR. Evidence for a circulating islet cell growth factor in insulin- resistant states. Proc Natl Acad Sci USA. 2001;98:7475–7480. [PMC free article: PMC34693] [PubMed: 11404474]
- 133.
- Bonner-Weir S, Deery D, Leahy JL. et al. Compensatory growth of pancreatic β-cells in adult rats after short-term glucose infusion. Diabetes. 1989;38:49–53. [PubMed: 2642434]
- 134.
- Stoffers DA. The development of beta-cell mass: Recent progress and potential role of GLP-1. Horm Metab Res. 2004;36:811–821. [PubMed: 15655713]
- 135.
- Song KH. et al. In vitro transdifferentiation of adult pancreatic acinar cells into insulin-expressing cells. Biochem Biophys Res Commun. 2004;316:1094–1100. [PubMed: 15044097]
- 136.
- Paris M, Tourrel-Cuzin C, Plachot C. et al. Review: Pancreatic beta-cell neogenesis revisited. Exp Diabesity Res. 2004;5:111–121. [PMC free article: PMC2496878] [PubMed: 15203882]
- 137.
- Ferber S. et al. Pancreatic and duodenal homeobox gene 1 induces expression of insulin genes in liver and ameliorates streptozotocin-induced hyperglycemia. Nat Med. 2000;6:568–572. [PubMed: 10802714]
- 138.
- Kojima H. et al. NeuroD-betacellulin gene therapy induces islet neogenesis in the liver and reverses diabetes in mice. Nat Med. 2003;9:596–603. [PubMed: 12704384]
- 139.
- Kaneko K. et al. Insulin inhibits glucagon secretion by the activation of PI3-kinase in In-R1-G9 cells. Diabetes Res Clin Pract. 1999;44:83–92. [PubMed: 10414926]
- 140.
- Van Schravendijk CF, Foriers A, Van den Brande JL. et al. Evidence for the presence of type I insulin-like growth factor receptors on rat pancreatic A and B cells. Endocrinology. 1987;121:1784–1788. [PubMed: 2959469]
- 141.
- Unger RH. Glucagon physiology and pathophysiology in the light of new advances. Diabetologia. 1985;28:574–578. [PubMed: 3902546]
- 142.
- Harvel PJ, Veith RC, Dunning BE. et al. Role for autonomic nervous system to increase pancreatic glucagon secretion durning marked insulin-induced hypoglycemia in dogs. Diabetes. 1991;40:1107–1114. [PubMed: 1682196]
- 143.
- Cryer PE. Banting Lecture. Hypoglycemia: The limiting factor in the management of IDDM. Diabetes. 1994;43:1378–1389. [PubMed: 7926315]
- 144.
- Diao J, Asghar Z, Chan CB. et al. Glucose-regulated glucagon secretion requires insulin receptor expression in pancreatic alpha-cells. J Biol Chem. 2005;280:33487–33496. [PubMed: 16027126]
- 145.
- Sloop KW. et al. Hepatic and glucagon-like peptide-1-mediated reversal of diabetes by glucagon receptor antisense oligonucleotide inhibitors. J Clin Invest. 2004;113:1571–1581. [PMC free article: PMC419489] [PubMed: 15173883]
- 146.
- Duncan SA, Navas MA, Dufort D. et al. Regulation of a transcription factor network required for differentiation and metabolism. Science. 1998;281:692–695. [PubMed: 9685261]
- 147.
- Odom DT. et al. Control of pancreas and liver gene expression by HNF transcription factors. Science. 2004;303:1378–1381. [PMC free article: PMC3012624] [PubMed: 14988562]
- 148.
- Postic C. et al. Dual roles for glucokinase in flucose homeostasis as determined by liver and pancreatic β cell-specific gene knock-outs using cre recombinase. J Biol Chem. 1998:1–11. [PubMed: 9867845]
- 149.
- Scharf JG, Ramadori G, Braulke T. et al. Cellular localization and hormonal regulation of biosynthesis of insulin-like growth factor binding proteins and of the acid-labile subunit within rat liver. Prog Growth Factor Res. 1995;6:175–180. [PubMed: 8817659]
- 150.
- Funakoshi H, Nakamura T. Hepatocyte growth factor: From diagnosis to clinical applications. Clin Chim Acta. 2003;327:1–23. [PubMed: 12482615]
- 151.
- Michael MD. et al. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol Cell. 2000;6:87–97. [PubMed: 10949030]
- 152.
- Wolfrum C, Besser D, Luca E. et al. Insulin regulates the activity of forkhead transcription factor Hnf-3beta/Foxa-2 by Akt-mediated phosphorylation and nuclear/cytosolic localization. Proc Natl Acad Sci USA. 2003;100:11624–11629. [PMC free article: PMC208808] [PubMed: 14500912]
- 153.
- Kulkarni RN, Kahn CR. Molecular biology. HNFs—linking the liver and pancreatic islets in diabetes. Science. 2004;303:1311–1312. [PubMed: 14988544]
- 154.
- Duvillie B. et al. Phenotypic alterations in insulin-deficient mutant mice. Proc Natl Acad Sci USA. 1997;94:5137–5140. [PMC free article: PMC24644] [PubMed: 9144203]
- 155.
- Liu JP, Baker J, Perkins AS. et al. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell. 1993;75:59–72. [PubMed: 8402901]
- 156.
- Accili D. et al. Early neonatal death in mice homozygous for a null allele of the insulin receptor gene. Nat Genet. 1996;12:106–109. [PubMed: 8528241]
- 157.
- Aspinwall CA. et al. Roles of insulin receptor substrate-1, phosphatidylinositol 3-kinase, and release of intracellular Ca2+ stores in insulin-stimulated insulin secretion in beta -cells. J Biol Chem. 2000;275:22331–22338. [PubMed: 10764813]
- 158.
- Chen WS. et al. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev. 2001;15:2203–2208. [PMC free article: PMC312770] [PubMed: 11544177]
- 159.
- Cho H, Thorvaldsen JL, Chu Q. et al. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem. 2001;276:38349–38352. [PubMed: 11533044]
- 160.
- Kitamura T. et al. Preserved pancreatic beta-cell development and function in mice lacking the insulin receptor-related receptor. Mol Cell Biol. 2001;21:5624–5630. [PMC free article: PMC87283] [PubMed: 11463843]
- 161.
- Yakar S. et al. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc Natl Acad Sci USA. 1999;96:7324–7329. [PMC free article: PMC22084] [PubMed: 10377413]
- 162.
- Liu JL, Yakar S, LeRoith D. Mice deficient in liver production of insulin-like growth factor I display sexual dimorphism in growth hormone-stimulated postnatal growth. Endocrinology. 2000;141:4436–4441. [PubMed: 11108252]
- 163.
- Yakar S. et al. Liver-specific igf-1 gene deletion leads to muscle insulin insensitivity. Diabetes. 2001;50:1110–1118. [PubMed: 11334415]
- 164.
- Devedjian JC. et al. Transgenic mice overexpressing insulin-like growth factor-II in beta cells develop type 2 diabetes. J Clin Invest. 2000;105:731–740. [PMC free article: PMC377454] [PubMed: 10727441]
- 165.
- Moller DE. et al. Transgenic mice with muscle-specific insulin resistance develop increased adiposity, impaired glucose tolerance, and dyslipidemia. Endocrinology. 1996;137:2397–2405. [PubMed: 8641192]
- 166.
- Bruning JC. et al. A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol Cell. 1998;2:559–569. [PubMed: 9844629]
- 167.
- Mauvais-Jarvis F. et al. A model to explore the interaction between muscle insulin resistance and beta-cell dysfunction in the development of type 2 diabetes. Diabetes. 2000;49:2126–2134. [PubMed: 11118016]
- 168.
- Bruning JC. et al. Role of brain insulin receptor in control of body weight and reproduction. Science. 2000;289:2122–2125. [PubMed: 11000114]
- 169.
- Bluher M. et al. Adipose tissue selective insulin receptor knockout protects against obesity and obesity-related glucose intolerance. Dev Cell. 2002;3:25–38. [PubMed: 12110165]
- 170.
- Lauro D, Kido Y, Hayashi H. et al. Transgenic knock-out mice with a targeted impairment of insulin action in skeletal muscle and adipose tissue. Diabetes. 1998;47(Supp1):A45. [PubMed: 9806552]
- 171.
- Choudhury AI. et al. The role of insulin receptor substrate 2 in hypothalamic and beta cell function. J Clin Invest. 2005;115:940–950. [PMC free article: PMC1069106] [PubMed: 15841180]
- 172.
- Fernandez AM. et al. Functional inactivation of the IGF-I and insulin receptors in skeletal muscle causes type 2 diabetes. Genes Dev. 2001;15:1926–1934. [PMC free article: PMC312754] [PubMed: 11485987]
- 173.
- Chen D, Mauvais-Jarvis F, Bluher M. et al. p50alpha/p55alpha phosphoinositide 3-kinase knockout mice exhibit enhanced insulin sensitivity. Mol Cell Biol. 2004;24:324–329. [PMC free article: PMC303335] [PubMed: 14673165]
- 174.
- Kulkarni RN. The islet beta-cell. Int J Biochem Cell Biol. 2004;36(3):365–71. [PubMed: 14687913]
- 175.
- Kulkarni RN. New insights into the roles of Insulin/IGF-I in the development and maintenance of beta-cell mass. Rev Endocr Metab Disord. 2005;6(3):199–210. [PubMed: 16151624]
- Summary
- Introduction
- Embryonic and Early Post-Natal Development of the Endocrine Pancreas
- Global and Conditional Knockouts of Insulin, IGF-I, IGF-II and Proteins in Their Signaling Pathways
- Maintenance of Adult β-Cell Mass
- Growth and Development of Islet α-Cells
- The Liver-Pancreas Connection
- Future Insights
- Acknowledgements
- References
- Insulin Action in the Islet β-Cell - Madame Curie Bioscience DatabaseInsulin Action in the Islet β-Cell - Madame Curie Bioscience Database
- Chemokine Binding Proteins Encoded by Pathogens - Madame Curie Bioscience Databa...Chemokine Binding Proteins Encoded by Pathogens - Madame Curie Bioscience Database
- Cytokines, Receptors and Signalling Pathways Involved in Macrophage and Dendriti...Cytokines, Receptors and Signalling Pathways Involved in Macrophage and Dendritic Cell Development - Madame Curie Bioscience Database
- Physiological Roles and Mechanisms of Signaling by TRAF2 and TRAF5 - Madame Curi...Physiological Roles and Mechanisms of Signaling by TRAF2 and TRAF5 - Madame Curie Bioscience Database
- Sympathetic Nervous System Regulation of Metastasis - Madame Curie Bioscience Da...Sympathetic Nervous System Regulation of Metastasis - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...