

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

A number of acute and chronic neurodegenerative conditions are associated by protein misfolding and aggregation of proteins within and outside cells. Misfolded proteins and protein aggregation are controlled by molecular chaperones such as heat shock proteins (HSPs) that are constitutively and inducibly expressed in the nervous system. There is increasing evidence that HSPs could counteract common pathological mechanisms that take place during Alzheimer's disease (AD), Parkinson's disease (PD) and Huntington's disease (HD). This is achieved by HSPs either interfering with the misfolded disease proteins preventing unwanted interactions with other cellular proteins and/or by reducing the risk of formation of toxic oligomeric assemblies of the respective disease proteins such as tau and amyloid-β in AD, α-synuclein in PD and huntingtin in HD. But HSPs are also expected to interfere with detrimental processes that occur during these diseases including oxidative stress and abnormal activation of signaling pathways or act supportive towards degradation systems such as the ubiquitin proteasome- and the autophagic-lysosomal pathway. Specific neuronal structures such as synapses and axons also harbour HSPs that may be misregulated during the disease process. Hence HSPs are expected to be critically involved in the progression of AD, PD and HD making them potential therapeutic targets and the studies discussed in this chapter support this view.
Introduction
The misfolding and progressive polymerisation of otherwise soluble proteins is a common characteristic of a variety of diseases associated with neurodegeneration. These diseases include Alzheimers's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS), the prion disorders, tauopathies (causing frontotemporal dementias such as progressive supranuclear palsy and corticobasal degeneration), other synucleinopathies than PD (multiple system atrophy, dementia with Lewy bodies, progressive autonomic failure, rapid-eye-movement sleep disorders) and the polyglutamine (polyQ) diseases. Neuropathology of these disorders is caused by environmental factors and genetic mutations. In this chapter we will discuss the role of heat shock proteins (HSPs) in some of these disorders with a focus on AD and PD which are the most common neurodegenerative disorders associated with cognitive and motor impairments and Huntington's disease (HD) which is the most common inherited disease caused by an expansion of a polyQ tract in the associated disease gene. The molecular and cellular details vary enormously among these diseases, but the tendency of soluble proteins to develop an altered conformation and aggregate inside and/or outside cells appears to precede early clinical signs in each disease. Protein misfolding and aggregation is associated with profound neuronal dysfunction and death, but the reader should note that currently it is still unclear whether this phenomenon explains the fundamental basis of these diseases or represents a crucial secondary step in the course of the disorder.1
Misfolded proteins are recognized by a set of conserved intracellular proteins known as molecular chaperones that mediate correct folding, assembly and degradation of proteins.2 The heat shock proteins (HSPs) are a family of proteins that can act as molecular chaperones and are classified into six major families: Hsp100, Hsp90, Hsp70, Hsp60, Hsp40 and small HSPs (sHSPs, e.g., αB crystallin and Hsp27). Because HSPs are constitutively expressed and/or inducibly regulated in the nervous system and prevent aggregation of unfolded/misfolded polypeptides, assist refolding and play a role during solubilization of stable protein aggregates, they may be crucial modulators of neurotoxicity in AD, PD and HD. Here we are reviewing recent progress from many laboratories that support this view. After a brief introduction to each disorder we will discuss whether and how HSPs could impact on the common molecular and cellular mechanisms implicated in all three disease pathologies.
Alzheimer's Disease
AD pathology can occur in a sporadic manner (most cases) or is dominantly inherited (ca. 5% of all AD cases).3 Symptoms are progressive and include difficulties in acquisition of new memory, memory loss and depression that typically occur with late onset of the disease. Idiopathic cases of AD appear to be phenocopies of inherited cases, but inherited AD onset occurs earlier in life. AD is the most prevalent human dementia affecting 10% of the population over 65.3 AD has been defined by the occurrence of two types of lesions in brain regions serving memory and cognition: the neurofibrillary tangles (NFTs) and the amyloid plaques. NFTs consist of paired, helically wound or straight protein filaments made of the pathologically altered microtubule (MT)-associated protein tau (Fig. 1A).4 Tau proteins stabilize and promote MT polymerization in neuronal perikarya and processes. NFTs are normally found in the cytoplasm and neuritic processes of affected nerve cells in selectively vulnerable regions of the central nervous system (CNS) in AD.4 Hyperphosphorylation dislodges tau from the MT surface, potentially resulting in compromised axonal integrity and toxic tau peptides. Recent biochemical and animal model studies indicate that phosphorylation of tau's MT-binding domain by MARK (MAP/microtubule affinity regulating kinase) may prime tau for hyperphosphorylation by kinases such as GSK-3 and Cdk5 that in turn trigger the aggregation of tau.5 Filamentous tau tangles are made of hyperphosphorylated and otherwise abnormal tau. The filaments vary between 8-20nm in width and it is the repeat region of tau that forms the core (see Fig. 1). Filaments show a clear cross-β structure, the defining feature of amyloid fibres. A likely mechanism for tau fibrillization is that the protein disengages from MTs and hence the pool of soluble tau increases. Soluble tau may be less degradable and more prone to aggregation, likely due to abnormal phosphorylation events. The transition of the largely unstructured, normal tau into a β-sheet containing species may be facilitated by intracellular interactions with membranes and organelles. Nucleation of tau oligomers and filaments and the formation of mature NFTs likely ocurs via a nonfibrillar step. Amorphous deposits of tau have indeed been detected in human brain and may represent such a “pretangle” stage. Although tau is predominantly a neuronal protein, tau pathologies occur in oligodendrocytes and astrocytes in diverse neurodegenerative disorders.6 For example, the identification of tau mutations in families with frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17) show that mutations reduce binding to MTs and increase NFT formation.6
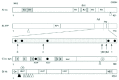
Amyloid plaques are the other major lesion in AD and are extracellular deposits of 8-10 nm wide amyloid fibrils that result from amyloid-β (Αβ) protein polymerisation.1 The prevalent form of amyloid plaques are known as the ‘neuritic plaque’ (NPs). NPs comprise an extracellular core of amyloid fibrils encircled by dystrophic dendrites and axons that can also contain paired helical filaments (PHFs) of tau, activated microglia and reactive astrocytes. 1 NPs are found in the molecular layer of the dentate gyrus of the hippocampus, the amygdala, the association cortices of the frontal, temporal and parietal lobes and also in certain deep brain nuclei projecting to these areas. There are other morphologically distinct forms of Αβ plaques such as “diffuse plaques” found in the limbic and association cortices that are characteristically loose/granular and lacking dystrophic neurites.1 Αβ is a 38-43 residue proteolytic fragment of the amyloid precursor protein (APP). APP is a ubiquitously expressed type 1 membrane glycoprotein that undergoes complex processing and is produced normally throughout life by most mammalian cells. Some of the elaborate processing steps of APP are explained in Figure 1B. The genes implicated in familial AD to date are presenilin-1 and presenilin-2 (PS1/2), APP and the e4 allele of apolipoprotein E (ApoE4).1 Disease causing mutations in APP, PS1/2 and ApoE4 inheritance result in either increased Αβ42 production/oligomerisation or reduced clearance.
The prevailing view that extracellular Αβ deposition is the primary toxic insult in AD has been challenged because a number of studies suggest that intracellular Αβ accumulation may play an early and relevant pathological role.7 Αβ42 appears to be the prominent form accumulating inside neurons in the brains of AD patients and transgenic mice expressing familial AD mutations.7
The sequence of events underlying the development of AD and the role of the assemblies of tau and/or Αβ is not known. The genetics of AD suggest that Αβ accumulation is the factor that could trigger NFT formation. Based on available evidence it seems that APP molecules at the plasma membrane and in intracellular vesicles are cleaved to liberate the Αβ region (Figs. 1B, 2). A portion of Αβ peptides may then oligomerize, initially intravesicularly, and then be released into the interstitial fluid of the brain resulting in interference with synaptic function.1 Αβ can further polymerize into insoluble fibrils that form plaques that in turn may cause dysfunction of axons and dendrites. Concomitant intraneuronal cytoplasmic kinase activation may then lead to hyperphosphorylation of tau, its dislocation from MTs and aggregation into NFTs resulting in neuritic damage (Fig. 2). The reader should note that not everybody agrees with this view. Activated microglia and reactive astrocytes surrounding the plaques also contribute to neuropathology that may be causally connected to oxidative stress, abnormal activation of signalling pathways, the formation of amyloid ion channels and altered degradation systems (see section Common Mechanisms Leading to Impaired Neuronal Function and Death and the Role of HSPs). It is the diffusable, soluble oligomers of Αβ that are hypothesised to directly compromise neuronal functions first rather than fibrils and plaques.1 Interestingly, recent evidence suggest a connection in AD and PD pathology where tau and α-synuclein inclusions are found in the same cell and a direct link between aggregation of tau and α-synuclein has been demonstrated.6

Parkinson's Disease
Clinical features of PD include tremor, rigidity and bradykinesia (slowness of voluntary movements) and are found in 0.2% of the population.8 The majority of the cases seem to occur sporadically, but in rare cases the disease can also be inherited in an autosomal dominant or recessive fashion. In 1997, a point mutation (A53T) in a gene encoded by α-synuclein was identified in an Italian family and subsequently a second PD-linked dominant mutation in α-synuclein (A30P) was found in a German family.9 Meanwhile other mutations linked to PD in genes encoding parkin, ubiquitin C-terminal hydrolase -L1 (UCH-L1), DJ-1 and PTEN-induced kinase 1 (Pink-1) have been identified.9 As in AD, the infrequent familial forms of PD are expected to be highly informative for the understanding of the pathogenesis of the idiopathic form. The dopaminergic neurons in the substantia nigra (SN) and the noradrenergic neurons in the locus coeruleus frequently exhibit characteristic filamentous inclusion bodies (IBs) in PD called “Lewy bodies” (LBs). There is a depletion of dopamine in the striatum and substantial nerve cell loss in the SN during the disease.9 The main component of LBs is wild type α-synuclein.8 α-synuclein is a ‘natively unfolded’ normally soluble protein which under conditions of oxidative stress (including mitochondrial complex I deficiency) forms amyloid-like filaments. Under physiological conditions in vitro, α-synuclein lacks a well-defined structure and hence is an intrinsically unstructured protein, similar to tau. α-synuclein is a 140 amino acid protein that is predominantly expressed in CNS neurons, where it is concentrated at presynaptic terminals. The function of α-synuclein is not fully characterised but various studies suggest that it is involved in modulating synaptic transmission, regulating the localisation of vesicles in synapses and participating in neuronal plasticity. Human α-synuclein was identified as the precursor protein for the nonamyloid beta component (NAC) of AD plaques. The hydrophobic NAC sequence is important for fibrillization (see Fig. 1C). Like the plaques and tangles in AD, LBs may not initiate the degenerative process as a comparison of neurons in the SN with and without LBs failed to show any obvious differences including apoptotic-like changes. Furthermore, mutations in the parkin gene, linked to PD, that appears to cause degeneration of neurons in the SN, do not result in LB formation (see below). Also the lack of a correlation between the PD causing mutation in α-synuclein and the acceleration of fibril formation suggest that the fibril is not the pathogenic entity. These and other observations have shifted the attention of researchers towards the role of α-synuclein protofibrils which are transient β-sheet containing oligomers. These species have been proposed to form porelike structures that may potentially disrupt ionic and/or metabolic homeostasis.8
Mutations in parkin are the most common cause of autosomal recessive early-onset parkinsonism.9 Parkin mutations have been found in about 50% of familial cases of PD and in 10-20% of cases without a positive family history. Parkin is a protein exhibiting ubiquitin ligase activity.9 The clinical features of parkin-related diseases can be similar to those of late onset sporadic PD (see above). Mutations in parkin result in a loss-of-function of E3 and substrates of parkin abnormally accumulate within dopaminergic neurons. LB formation generally does not occur in parkin-related diseases and there is severe neuronal loss and gliosis in brains of these patients. Several proteins implicated in oxidative stress pathways are down-regulated in parkin deficient mice suggesting that parkin functions are related to oxidative stress control.9 This finding fits with the observation that idiopathic PD may be triggered by chronic exposure to environmental toxins such as rotenone, paraquat or MPTP, all of which increase free radical production and induce LB-like inclusions containing α-synuclein in dopaminergic neurons (Fig. 3). The third gene implicated in PD is UCH-L1 that is an abundant de-ubiquitilating enzyme and hence can be placed in a pathway potentially related to parkin.9 DJ-1 is a cytoplasmic neuronal protein with chaperone activity induced under conditions of oxidative stress and Pink-1 is a mitochondrial protein kinase.9 For the moment it is unclear if these genes are targeting similar cellular and molecular mechanisms implicated in PD. However, there is strong evidence that oxidative stress, mitochondrial dysfunction and impairment of the ubiquitin-proteasome pathway (UPP) is central to the neuropathology of PD (Fig. 3).

Huntington's Disease
HD is a fatal neurodegenerative disorder and together with spinobulbar muscular atrophy (SBMA), dentatorubral pallidoluysian atrophy (DRPLA), and spinocerebellar ataxia (SCA) types, 1, 2, 3, 6, 7, and 17 known as a polyglutamine (polyQ) disease.10 All the polyQ diseases are inherited in a strictly autosomal dominant manner with the exception of SBMA that is X-linked. These diseases are caused by a polyQ expansion mutation. Mutant proteins between diseases do not share any homology except the polyQ tract: this indicates common molecular mechanisms of pathology during polyQ pathology. HD is characterized by abnormal movements including chorea, dementia and emotional disorders and affects ca. one individual out of ten thousand and hence is considered a rare disorder. Age of onset of polyQ diseases correlates inversely with repeat number of the expansion and HD is no exception: longer polyQ repeats lead to earlier age of onset and a more severe pathology.10 The pathologic length of the polyQ repeat varies for each polyQ disease, but is generally ≈ 40 or greater. At the HD locus unaffected individuals typically show 11-34 glutamine residues while in HD patients the polyQ stretch expands to >36 glutamines located in Exon 1 of the HD gene that consists of 67 Exons (see Fig. 1D).
Huntingtin (htt) is a large protein comprising 3144 amino acids containing a functional nuclear export signal (NES) at its C-terminus. Full-length htt shuttles between the cytoplasm and the nucleus. If the NES is lost due to proteolytic cleavage by caspases (see Fig. 1D) and calpains, smaller fragments are formed that are more toxic and more likely to accumulate in the nucleus giving rise to IBs. Hence htt like APP, tau and synuclein is proteolytically regulated. Htt is associated with various organelles including the endoplasmatic reticulum, Golgi complex, nucleus, synaptic vesicles and mitochondria. Based on the known functions of the many htt interacting proteins it is believed that htt plays a role as a scaffolding protein orchestrating sets of proteins for signalling processes and intracellular transport. Wildtype htt may also have anti-apoptotic properties.
HD typically begins in midlife (but there is juvenile onset) and is characterised by progressive neuronal dysfunction and neuronal loss, and patients die 10 to 15 years after the onset of symptoms. The selective neurodegeneration in the brain occurs most prominently in the striatum and deep layers of the cerebral cortex.10 In advanced stages of the disease other brain regions such as the hippocampus, hypothalamus, cerebellum and amygdala can also be affected. Projection neurons sending their axons to various brain regions are most severely affected. In the striatum the GABAergic medium-sized spiny neurons (MSNs) are most vulnerable. This phenomenon is observed despite ubiquitous expression of the HD gene. PolyQ expansions compromise neuronal function through several deleterious mechanisms which may vary during the long period of HD progression: transcriptional alterations, impairment of degradation systems such as the UPP, abnormal axonal transport and axonal degeneration, bio-energetic defects due to abnormal mitochondrial function and oxidative stress have all been implicated in HD.
The discovery of intraneuronal inclusions that are of a fibrillar nature and contain the mutant proteins in polyQ diseases and transgenic mice with polyQ mutations has provided a common denominator for pathology.11 The polyQ expansion at the N-terminus of htt produces an abnormal conformation inducing a toxic gain-of-function in the mutant protein, a process that is associated with nucleation and subsequent aggregation. It is thought that polyQ fibrillogenesis is an ordered polymerization process similar to amyloid fibrillogenesis in AD and PD. The rate limiting step in the aggregation process is the formation of an oligomeric nucleus which can form after a repeat-length dependent conformational change of the polyQ monomer from a random coil to a parallel, helical β-sheet.12 PolyQ aggregates and IBs are predominantly found in the nucleus and axonal processes, but cytoplasmic aggregates have also been described in the HD brain. Similar to the situation in AD and PD the oligomeric precursors of htt polyQ fibrils are thought to be the most neurotoxic. Htt aggregates have been shown to recruit a number of vital cellular proteins including transcription factors, HSPs and components of the UPP.
Common Mechanisms Leading to Impaired Neuronal Function and Death and the Role of HSPs
Direct Interference of HSPs with Misfolding, Oligomerisation and Fibrillogenesis of Disease Proteins
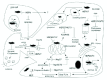
The assembly of misfolded proteins into amyloid fibrils and subsequently into IBs, plaques, NFTs or LBs is a complex process and could be similar among the various proteins that produce these structures. Research into the amyloid aggregation process starting from a partially folded peptide or the native protein to misfolded or unfolded monomeric α-synuclein, Αβ, tau and htt has revealed the production of several, distinct structures such as spherical or annular oligomers and amorphous aggregates and/or fibrils (Fig. 5). HSPs may interfere at various stages during this process by associating with misfolded disease proteins and higher order structures such as oligomers and IBs. This may result in the prevention of a toxic intra-molecular conformational change of the peptide itself or decrease the probability of unwanted homo-and/or heterotypic interactions thereby modifying amyloid assembly. Indeed, various HSPs have been shown to bind in vitro and/or in vivo to mutant htt,13 hyperphosphorylated tau14 and α-synuclein prefibrillar structures15 and modulate amyloid assembly. Consistent with this view, Schaffar and colleagues using fluorescence resonance energy transfer (FRET) showed that HSP40 and HSP70 prevent an intra-molecular conformational change in mutant htt.16 That study also showed that HSPs prevent heterotypic interactions between polyQ expanded htt and transcription factors. Dou and colleagues reported an inverse relationship between aggregated tau and the level of HSP70/90 in transgenic mice and AD brain.17 While increased levels of HSPs promoted tau solubility and binding to MTs, but reduced tau phosphorylation, lowering the levels of HSPs by use of RNA-mediated interference (RNAi) had the opposite effects.17 Several in vitro and in vivo model studies of each disease show that artificial elevation of certain HSPs by over-expression decreases the amount of insoluble disease protein and/or deposits which is associated with a decrease in toxicity in most of the cases (see Table 1). HSPs may reduce aggregation by interfering with the APP secretory pathway (Fig. 2): GRP78, an HSP70 localised in the ER, binds to APP decreasing the secretion of amyloid Αβ40 and Αβ42 in cell culture models.18 Importantly, interactions of HSPs with one disease protein may affect the assembly process of another aggregation prone molecule: α-synuclein binds to tau and increases tau fibrillization and Αβ affects α-synuclein amyloid assembly.6 α-synuclein may also increase htt aggregation and is localised to htt IBs in human brain.19,20

Some HSPs colocalise with intracellular NFTs and Αβ plaques in the extracellular space and in PD many HSPs are sequestered into LBs (see Table 1). Although the colocalisation of HSPs and IBs have not been carefully studied in the HD brain, there are several studies that showed a redistribution of HSP40/HSP70 into cytoplasmic and nuclear inclusions in the related polyQ disorders spinocerebellar ataxias (SCA) 1, 3 and 7.13 It is unclear if such a redistribution reflects an irreversible sequestration and loss of function of HSPs or a failed attempt to refold aggregated proteins or both. A recent study addressed this question using florescence recovery after photobleaching (FRAP) and fluorescence loss in photobleaching (FLIP): the interaction between HSP70 and htt IBs exhibit rapid association and dissociation kinetics and hence HSP70 may actively regulate htt IB formation.21
A study on the assembly behaviour of a fragment of htt showed that similar to α-synuclein and Αβ, mutant htt assembles into spherical, annular and amorphous structures that might be on a pathway to fibril assembly.22 HSP40 and HSP70 attenuated the formation of htt spherical and annular oligomers promoting the accumulation of fibrillar and amorphous aggregates that may be less toxic. This study proposed that HSPs could facilitate the folding of a specific misfolded monomeric conformation that allows on-pathway assembly to occur by monomeric addition to fibril nuclei, while decreasing the likelihood of interactions that promote the formation of off-pathway events during fibril formation (formation of spherical and annular oligomers). This model remains to be tested for other aggregation prone molecules (Fig. 5).
Prevention of Oxidative Stress by Heat Shock Proteins
The cause of neuronal dysfunction and death in each disease is multi-factorial, but evidence is emerging that the pathological depositions of abnormal proteins is associated with alterations in redox state homeostasis and mitochondrial dysfunction.23 PD is characterized by increased oxidative damages, altered activities of antioxidant defence enzymes and decreased mitochondrial complex I activity (Fig. 3). Increased oxidative damage and altered mitochondrial activity (probably because of decreased complex IV activity) has also been reported in AD.23 In the HD brain, oxidative damage has been detected together with a decreased mitochondrial complex II and IV activity24 and in HD mouse models free radical damage has been observed.25 Similar observations were made in a cellular model of HD.26
Evidence for the presence of oxidative stress can also be provided by the finding that (i) the pathological protein deposits can be recognized by antibodies that are specific to protein side-chains modified either directly by reactive oxygen or nitrogen species, or by products of lipid peroxidation or glycoxidation and (ii) the brain regions that are the most affected often contain deposits of redox-active transition metals, such as iron and copper. Moreover, membrane-associated oxidative stress (MAOS) which is a metal-catalyzed oxidative disruption of membrane protein and lipid signalling has been suggested to occur in the pathogenesis of AD, PD and HD.27 The origin of the oxidative stress that is concomitant to the presence of aggregated neuronal proteins is not clear. In this respect, cellular models based on the expression of mutant htt have been useful to show that the oxidative events are probably not cell type specific since they can still be detected if the abnormal protein is expressed in cells other than neurones.26 Moreover, the degree of aggregation of mutant htt, which is a direct consequence of the number of polyQ expansions, appears to be a key factor generating oxidative stress.26 In vitro assays have been performed in which aggregated α-synuclein and β-amyloid are incubated in the presence of redox-active transition metals.28 These assays revealed that iron and copper catalyze the formation of reactive oxygen species (ROS) by the aggregating proteins probably through Fenton-Haber-Weiss types of reaction. This suggests that an aggregating protein in contact with redox-active transition metals is capable of ROS generation. Htt is an iron-regulated protein and similar results could be found. Moreover, using a cellular model of HD26, we recently noticed that the transient expression of mutant htt increased the intracellular level of iron, a phenomenon that correlated with the number of polyQ repeats (W. Firdaus, A. Wyttenbach, B. Currie, A.P. Arrigo, unpublished). It is not known if specific properties of some neuronal proteins are required for production of ROS in the presence of metals or if this phenomenon is more general and can be observed in proteins that are prone to aggregation.
It is well known that HSPs can protect cells against the damage induced by oxidative stress (Figs. 2-4). HSP mediated resistance against oxidative stress has been recently shown in an HD cellular model.26 HSP mediated protection of cells against oxidative stress appears to occur via the molecular chaperone activity of these proteins. For example, in addition of being able to take care of the elimination of oxidized proteins, HSPs can protect specific enzymes involved in the detoxification of reactive oxygen and are involved in the reduction of oxidized glutathione.29 It is also notable that the forced expression of Hsp70/Hsp40 and/or Hsp27 attenuated the intracellular increase in iron found in cells transiently expressing mutant htt polypeptides (W. Firdaus, A. Wyttenbach, B. Currie, A.P. Arrigo, unpublished). This finding is consistent with our observation that HSP27 plays a role in intracellular iron homeostasis.29
What is the importance of oxidative stress in neurodegenerative diseases? Delineation of the profile of oxidative damage in each disease must be determined as it will provide clues to how the specific neuronal populations are affected by the individual disease conditions. In this respect, studying single markers of oxidative damage outside the context of oxidative balance is probably not sufficient. Concerning anti-oxidants, in the case of a cellular model of HD it is clear that they abolish some of the intracellular cytotoxicity of the aggregates26 and various antioxidants are known to protect against Aβ toxicity in cell models. However, aggregates are still present hence suggesting that anti-oxidants will have only a transient beneficial effect. In vivo, testing of anti-oxidants is more problematic, however, because of the proximal role that transition metals appear to play in the pathology of neurodegenerative diseases.
Maintenance of Signalling Homeostasis: Can Heat Shock Proteins Suppress the Activation of Detrimental Signalling Pathways?
Genomic and proteomic screens on either diseased human tissue or cell and animal models accelerate the identification of signalling pathways that are impaired and/or abnormally activated in each disease. Many candidate pathways have not yet been functionally validated in appropriate disease models and although various pathways are modulated in a disease-specific manner, there are several signalling modules that are similarly activated in AD, PD and HD. Apoptotic pathways that typically involve caspase activation have been reported to be engaged in all three diseases. Evidence in PD suggests that a p53-glyceraldehyde-3 phosphate dehydrogenase (GAPDH) and/or FAS receptor-FADD-caspase 8 pathway converge onto Bax30, a key neuronal death molecule that regulates the neuronal mitochondrial death pathway. Αβ production in both FAD and sporadic AD has been linked to the intrinsic, mitochondrial apoptotic pathway involving caspase-2, caspase-9 and Bax and the extrinsic receptor mediated apoptotic pathway (caspase-8) both converging onto the effector caspase-3.31 Evidence for engagement of the mitochondrial pathway, caspase-1, -3, -8 and 9 activation and hence apoptosis in HD comes from studies on human brain tissue and many in vitro and in vivo models of the disease, but some evidence suggests that neuronal death in HD is not apoptotic and does not depend on Bax.32 HSPs are known to interact with and suppress the activation of several molecules/arms of the apoptotic cascades (Figs. 2-4).33 For example, HSP27, HSP70 and HSP90 all interfere with the formation of a functional apoptosome and are able to suppress caspase-9 activation (Fig. 4). HSP27 binds to cytochrome c and caspase-3 thereby negatively regulating cell death.33 Therefore, HSPs are potentially important suppressors of death signalling pathways (Figs. 2-4). However, it is unlikely that only apoptotic pathways regulate neuronal death in these diseases. It will be crucial to elucidate the role of death pathways that involve the autophagic/lysosomal system and pathways linked to axonal degeneration upon which HSPs may act.
ER-stress sometimes associated with the unfolded protein response (UPR) has been associated with FAD and sporadic AD, PD and HD.34 Common downstream effectors such as the ER stress kinases IRE and PERK are activated in these situations likely due to an overload of misfolded proteins. Here up-regulation of ER-resident chaperones of the HSP70 family such as GRP78/BiP normally occurs. There is indeed evidence of involvement of GRP78 in HD and AD.18,35 A consequence of ER-stress in HD is the activation of Ask1, an upstream kinase that regulates pro-apoptotic JNK and p38MAPK activity (see below). Ask has also been implicated in Αβ induced neuronal death and since Ask1 is negatively regulated by HSP70 it could inhibit ER-stress activated kinases.
Stress activated mitogen-activated protein kinase (MAPKs) pathways appear to be modulated in AD, PD and HD and represent attractive therapeutic targets.36 While there is strong evidence for over-activation of the p38 MAPK pathway in early stages of AD36 the situation in PD and HD is less clear. Involvement of a JNK-dependent death pathway has been suggested in AD, PD and HD.13,36 HSP70 inhibits JNK activity by either a direct interaction or by negatively regulating a JNK phosphatase.13 It should also be noted that tau, APP, htt and α-synuclein have multiple phosphorylation sites on which many of the above mentioned stress kinases could act on when activated and hence they could modulate the toxic aggregation process. Therefore HSPs have the potential to indirectly impact on the assembly behaviour of each polypeptide by modulating their respective kinase activities. Most significantly, the physical state of oligomers may decide which and to what extent MAPK pathways are engaged37 If specific HSPs regulate distinct populations of aggregation intermediates they could also impact on the balance of death/survival MAPK pathways. As molecular chaperones, HSPs may further impact in as yet unidentified ways on the various disease specific pathways implicated in AD (wnt and cdk5/p35 pathway or protein kinase C and IGF-1/insulin receptor pathway), PD (phosphatidyl inositol lipid signaling pathways) and HD (cAMP signalling cascades, mTOR signalling).
The Recruitment Hypothesis: Heat Shock Proteins as Molecular Players
Misfolding and the subsequent assembly into oligomers and higher order structures of tau, Aβ, htt and α-synuclein may result in abnormal binding and recruitment of cellular factors. The recruitment of vital proteins to deposits and their precursors may result in inactivation and/or loss of function of the molecules, abnormal intracellular localisation and abnormal activation of signaling complexes. In HD (and other polyQ diseases) it is well documented that proteins as diverse as transcription factors (e.g., CREB-binding protein/CBP, SP1, p53), HSPs and components of the UPP are recruited into aggregates.13 Recruitment of proteasome subunits to NFTs in dystrophic neurites and LBs is also observed in AD and PD.38 However, it should be noted that UPP proteins (and HSPs) are only found in a subset of protein deposits in some neurons. Donaldson and colleagues have also reported that proteins containing ubiquitin-binding motifs are recruited into polyQ aggregates.39 Hence the depletion of normal ubiquitin-binding proteins caused by sequestration into aggregates may be a common event contributing to neurodegenerative diseases. The p62/sequestosome is found in tau assemblies in AD and LBs in PD supporting this hypothesis. Caspases are a further group of proteins that are recruited to aggregates: Caspase-8 is recruited to htt IBs and APP/Aβ multimers with subsequent activation probably due to the close proximity of several pro-caspase-8 molecules.13 Hence there is evidence that recruitment of cellular proteins in these diseases may play a role in neuropathology.
The recruitment/sequestration model is particularly attractive in the case of polyQ diseases such as HD where aggregates often reside in the nucleus and coaggregate with transcription factors. Aggregates formed by mutant htt also sequester normal htt that may contribute to a ‘secondary’ loss-of-function. Furthermore, Wetzel and colleagues proposed a structural, molecular mechanism that endows the recruitment process with some degree of specificity by reporting an inverse relationship between the size and recruitment activity of different forms of aggregates.40 This is in line with the idea that smaller aggregates than IBs are a more toxic species. Here, HSPs could interact with misfolded proteins and aggregated species reducing the probability of abnormal associations with other cellular proteins. Indeed, Schaffar and colleagues showed that mutant htt fragments structurally destabilise the transcription factor TATA-binding protein (TBP), but HSP40/70 chaperones inhibited its deactivation by interfering with the conformational change in mutant htt (Fig. 4).16 Whether HSPs are able to prevent yet other events as the ones described above remains to be tested.
Heat Shock Proteins and the Formation of Ion Channels
Over ten years ago “the channel hypothesis” of amyloid toxicity was proposed. This postulated that the toxic aggregated species form nonspecific porelike channels in the membranes of cells exposed to aggregating proteins.8 Porelike activity leading to an imbalance of ion homeostasis has been suggested to occur in AD, PD and HD. α-synuclein protofibrils exist as individual spheres, elongated chains, or rings. In contrast to the monomer or fibril, α-synuclein protofibrils bind and permeabilize lipid vesicles and maybe other membranes, perhaps via a porelike mechanism similar to beta-sheet-rich, membrane-permeabilising toxins.8 The A53T and A30P mutants of α-synuclein promote the formation of such “amyloid pores”.8 Annular, porelike protofibrils may also be responsible for Αβ permeabilizing or channel-like activity in AD: a ring-like structure has been found in in vitro aggregation assays of the arctic variant (E22G) of Αβ40.41 As mentioned above, htt fragments also produce annular structures and could possess pore-activity (Fig. 5). Alternatively, Monoi and colleagues proposed that polyQ sequences may form a novel structure called “μ-helix” where L-amino acid polypeptide chains form cylindrical pores with a 3.7 Ådiameter, large enough to accommodate passage of small ions and water.42 Significantly, such channels form in artificial planar lipid bilayer membranes with 40-glutamine residues (the pathological threshold for many polyQ diseases including HD), but not those with 29 residues.
Not only α-synuclein (see above), but also Aβ and htt bind to lipids and interactions of these hydrophobic misfolded proteins (including prion peptides) with membranes may not only cause direct damage, but also affect their aggregation behaviour frequently increasing the aggregation potential.43,44 HSPs prevent misfolding and the exposure of hydrophobic regions and therefore may suppress the interactions of the peptides with cellular membranes. This could reduce the ability of lipids to further destabilise proteins and reduce unwanted protein-membrane interactions. HSPs could prevent channel formation since they seem to reduce the formation of annular, perhaps pore-forming species in favour of amorphous structures and fibrils as shown for the htt fragment (Fig. 5).
HSP Communication with Protein Degradation Systems and Processing of Disease Proteins
Under conditions of stress and toxic insults degradation pathways such as the UPP and the lysosomal system could be impaired leading to accumulation of misfolded/damaged proteins. While the controlled proteolysis of substrates by the UPP is crucial in controlling the levels of short-lived proteins, the lysosomal pathway contributes to total rates of protein degradation and organelle turnover. The lysosome is the catabolic factory in eukaryotic cells to which proteins can be transported following several different pathways including endocytosis, micro- and macroautophagy and chaperone-mediated autophagy. Evidence for a causal relationship between UPP impairment and neurodegeneration comes from studies on PD where mutations in Parkin and UCHL1 are linked to familial PD. Although LBs are associated with UPP components and both monomeric and aggregated α-synuclein selectively binds to a subunit of the 19S cap 45 suggesting a link to dysfunctional proteasomal degradation it is currently unclear whether α-synuclein is a substrate for the proteasome. Wildtype α-synuclein appears to be selectively translocated into lysosomes for degradation by chaperone-mediated autophagy and the A53T and A30P mutants may act as uptake blockers, inhibiting both their own degradation and that of other substrates.46 UPP impairment has also been reported in a model of sporadic PD where systemic complex I inhibition with rotenone induces dopaminergic degeneration of the nigrostriatal pathway and formation of LBs (Fig. 3).
There are several studies that reported a reduction in proteasome- and ubiquitinationactivity in patients with AD. Tau PHFs reduce proteasome activity in AD brain and Αβ inhibits the proteasome in neuronal cells.38,47 Furthermore, presenilins and APP appear to be actively degraded by the proteasome under normal conditions and a subtle inhibition of proteasome activity could lead to an accumulation of PS and APP and their fragments and hence enhanced Αβ production. Activation of the neuronal lysosomal system and pathways converging to the lysosome, namely endocytosis and autophagy, is also a feature of brain pathology in AD and several studies support the view that progressive alterations of lysosomal function observed during aging and AD may impact on neuropathology.48
Studies on HD cellular models show both UPP abnormalities and interference with proteasome inhibitors modulate htt aggregation.13,49,50 However, in vivo studies do not confirm these results so far. Cell-type specific analysis is needed and the ageing process needs to be taken into consideration too. Eukaryotic proteasomes cannot cleave within polyQ stretches and hence it is unknown how long perfect polyQ sequences are disposed of.51 Autophagic degradation is an alternative route for disposal that has been implicated in HD.52
HSPs facilitate the ubiquitination and degradation of many proteins. Misfolded proteins such as mutant htt may be maintained in a degradation-competent state by HSPs and their cochaperones directing them more efficiently to degradation machineries. Indeed, the cochaperone CHIP (C-terminus of Hsp70 interacting protein) associates with expanded htt and its overexpression suppresses htt aggregation in an Hsc70 dependent manner.53 Because HSPs appear to promote the formation of fibrils and amorphous aggregates (Fig. 5) they may facilitate aggresome formation and subsequent degradation via macroautophagy (Figs. 3, 4). HSPs are also required for lysosomal protein degradation. Hsc70 forms a chaperone complex at the lysosomal membrane that is needed for unfolding the substrate proteins to efficiently translocate and degrade them in the lumen during chaperone-mediated autophagy. Therefore this process may critically regulate a-synuclein degradation (see above). HSP27 associates with the proteasome 54 and our results suggest that HSP27 partially restores proteasome activities impaired by mutant htt (W. Firdaus, A. Wyttenbach, P. Arrigo, unpublished observation). HSPs may also improve decreased proteasome activity in AD and PD. However, further research must determine whether HSPs beneficially interact with various signalling components that regulate autophagy or prevent deleterious processes arising from protein aggregation in each disease such as oxidative stress and hence prevent secondary damage to the UPP. Finally, since tau, APP and htt are caspase substrates (Fig. 1) and HSPs are known to prevent caspase activation, HSPs could suppress proteolysis of these proteins and diminish their aggregation potential.
Early Neuropathology, Synaptic Function and Axonal Transport
Early symptoms in AD correlate with dysfunctional cholinergic and glutamatergic synapses and alterations in synaptic efficacy prior to frank neurodegeneration may be caused by soluble, oligomeric assemblies of Αβ.1 Early synaptic dysfunction and/or loss prior to overt cell death may also occur in PD and HD. Both wildtype htt and α-synuclein localise to and are abundant at the synapse and a role in synaptic homeostasis has been suggested for both proteins. In HD mouse models, mutant htt inhibits the uptake of glutamate in synaptic vesicles and a reduction in long term potentiation and depression and other abnormalities in the hippocampus have been reported.55 Together with the finding that there is a loss of specific synaptic proteins and impairment of endo- and exocytosis in HD55, the current results suggest an early deficit in the machinery that regulates neurotransmitter release. In PD, mutations in α-synuclein alter its association with vesicular membranes, disrupting normal recycling of vesicles, which in turn can decrease the capacity of vesicular storage and increase cytosolic levels of dopamine and its toxic metabolites.8 Additionally, since both mutations in Parkin and UCH-L1 are linked to PD it is conceivable that a general impairment of the UPP in the synaptic compartment contributes to PD.
Could HSPs modulate synaptic pathology? Hsp40/70 members and sHSPs are present at the synapse and are increased after hyperthermia in animals.56 Mild metabolic stress increases the resistance of synaptic terminals to Αβ dysfunction through increased expression of Hsp70 and GRP78.57 Furthermore, the synaptic compartment contains a specific trimeric chaperone machinery consisting of cysteine string protein (CSP), small glutamine-rich tetratricopeptide repeat (TPR) containing proteins (SGTs) and HSC70 and it appears that these chaperones significantly maintain synaptic integrity58 and that alterations of these molecules could induce pathology. Indeed, SGT proteins redistribute to inclusion bodies in HD mice and SGT expression levels are significantly decreased.59 It remains to be seen if these chaperones are impaired in AD and PD.
Results obtained from in vitro and in vivo models of HD show that fast axonal transport is impaired due to a polyQ expansion.60 Additionally, htt specifically enhances vesicular transport of BDNF along microtubules and a loss of this function may contribute to pathogenesis.61 PD α-synuclein mutations show defective axonal transport in cultured neurons62 and mutations in APP and tau disrupt axonal transport in cultured neurons and in animal models.63 HSPs are expected to modulate axonal transport. Initial evidence for this idea comes from a study showing that elevated expression of Hsc70 in Drosophila improved axonal transport impaired by mutant htt.60 A role for HSPs during abnormal axonal/dendritic transport in AD and PD has to be established. In this respect it is interesting to note that HSPs can be synthesised in axons after injury.64 Finally, it should not be forgotten that glial cells critically impact on synapse and axon function and glial HSPs are highly inducible.
Heat Shock Proteins and Therapy
Animal studies support the view that modulating the levels of HSPs could prove benefical for the treatment of AD, PD and HD. However, an accurate balance (stoechiometry) of HSPs will be required to maximize their benefical roles. Excessive up-regulation of HSPs leads to unwanted effects such as cancer. A promising first target may be the heat shock transcription factor 1 (HSF1) which regulates HSP expression in concert with HSP90 that negatively regulates HSF-1. Drugs such as geldanamycin and radicicol that block the interaction of HSP90 with HSF-1, promoting HSF1 activation and synthesis of HSPs have been found to indeed induce HSPs and confer neuroprotection in in vitro and in vivo models of AD, PD and HD.59,65,66 Treatments using drugs that up-regulate HSPs in combination with compounds that act as chemical chaperones increasing the stability of native proteins in vitro (e.g., glycerol, trehalose, dimethyl sulphoxide) await testing and may well turn out to be a valid approach to combat not only AD, PD and HD, but also other conformational disorders such as ALS and prion disease.
Conclusions
HSPs provide a first line of defence against misfolded proteins and hence could act on early steps in misfolding disease pathology. Early synaptic and axonal abnormalities in AD, PD and HD may therefore be partially reversed by HSPs. Evidence is emerging that HSPs interact with the most toxic oligomeric precursors of protein deposits and perhaps also with misfolded conformations of monomers. This points to the ability of HSPs to prevent aberrant protein interactions due to altered conformations of disease proteins with other cellular components. But other activities of HSPs including the regulation of protein degradation, the control of signalling pathways and the redox state of the cell will be equally important to consider. Since the mechanisms by which HSPs improve neuroprotection are poorly understood, a lot more research is needed to get a glimpse of the full therapeutic potential of HSPs during neurodegeneration.
Acknowledgements
Andreas Wyttenbach is grateful to the Medical Research Council (MRC, UK) and the HighQ Foundation for financial support. André Patrick Arrigo is supported by ARC grant 4602 and the Region Rhone-Alpes. We apologize to the authors of many studies that are relevant to this chapter that could not be cited due to space limitations.
References
- 1.
- Selkoe DJ. Cell biology of protein misfolding: The examples of Alzheimer's and Parkinson's diseases. Nat Cell Biol. 2004;6(11):1054–61. [PubMed: 15516999]
- 2.
- Hartle FU. Molecular chaperones in cellular protein folding. Nature. 1996;381:571–579. [PubMed: 8637592]
- 3.
- Walsh DM, Selkoe DJ. Oligomers on the brain: The emerging role of soluble protein aggregates in neurodegeneration. Protein and Pept Lett. 2004;11(3):1–16. [PubMed: 15182223]
- 4.
- Goedert M. Tau protein and neurodegeneration. Semin Cell Dev Biol. 2004;15(1):45–49. [PubMed: 15036206]
- 5.
- Drewes G. MARKing tau for tangles and toxicity. Trends Biochem Sci. 2004;29(10):548. [PubMed: 15450610]
- 6.
- Lee MY, Giasson BI, Trojanowski JQ. More than just two peas in a pod: Common amyloidogenic properties of tau and alpha-synuclein in neurodegenerative diseases. Trends Neurosci. 2004;27(3):129–133. [PubMed: 15036877]
- 7.
- Magrane J, Smith RC, Walsh K. et al. Heat shock protein 70 participates in the neuroprotective response to intracellularly expressed beta-amyloid in neurons. J Neurosci. 2004;24(7):1700–1706. [PMC free article: PMC6730449] [PubMed: 14973234]
- 8.
- Rochet JC, Outeiro TF, Conway KA. et al. Interactions among alpha-synuclein, dopamine, and biomembranes. J Mol Neurosci. 2004;23:23–33. [PubMed: 15126689]
- 9.
- Healy DG, Abou-Sleiman PM, Wood NW. PINK, PANK, or PARK? A clinicians's guide to familial parkinsonism. The Lancet. 2004;3:652–662. [PubMed: 15488458]
- 10.
- Zoghbi HY, Orr HT. Glutamine repeats and neurodegeneration. Ann Rev Neurosci. 2000;23:217–247. [PubMed: 10845064]
- 11.
- Davies SW, Turmaine M, Cozens BA. et al. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90(3):537–548. [PubMed: 9267033]
- 12.
- Perutz MF. Glutamine repeats and neurodegenerative diseases: Molecular aspects. Trends Biochem Sci. 1999;24(2):58–63. [PubMed: 10098399]
- 13.
- Wyttenbach A. Role of heat shock proteins during polyglutamine neurodegeneration: Mechanisms and hypothesis. J Mol Neurosci. 2004;23(1-2):69–96. [PubMed: 15126694]
- 14.
- Shimura H, Miura-Shimura Y, Kosik K. Binding of tau to heat shock protein 27 leads to decreased concentration of hyperphosphorylated tau and enhanced cell survival. J Biol Chem. 2004;279(17):17957–17962. [PubMed: 14963027]
- 15.
- Dedmon MM, Christodoulou J, Wilson MR. et al. Heat shock protein 70 inhibits alpha-synuclein fibril formation via preferential binding to prefibrillar species. J Biol Chem. 2005;280(15):14733–14740. [PubMed: 15671022]
- 16.
- Schaffar G, Breuer P, Boteva R. et al. Cellular toxicity of polyglutamine expansion proteins: Mechanism of transcription factor deactivation. Mol Cell. 2004;15(1):95–105. [PubMed: 15225551]
- 17.
- Dou F, Netzer WJ, Tanemura K. et al. Chaperones increase association of tau protein with microtubules. Proc Natl Acad Sci USA. 2003;100(2):721–726. [PMC free article: PMC141063] [PubMed: 12522269]
- 18.
- Yang Y, Turner RS, Gaut JR. The chaperone BiP/GRP78 binds to amyloid precursor protein and decreases Abeta40 and Abeta42 secretion. J Biol Chem. 1998;273:25552–25555. [PubMed: 9748217]
- 19.
- Furlong RA, Narain Y, Rankin J. et al. Alpha-synuclein overexpression promotes aggregation of mutant huntingtin. Biochem J. 2000;346(3):577–581. [PMC free article: PMC1220887] [PubMed: 10698681]
- 20.
- Charles V, Mezey E, Reddy PH. et al. Alpha-synuclein immunoreactivity of huntingtin polyglutamine aggregates in striatum and cortex of Huntington's disease patients and transgenic mouse models. Neurosci Lett. 2000;289(1):29–32. [PubMed: 10899401]
- 21.
- Kim S, Nollen EA, Kitagawa K. et al. Polyglutamine protein aggregates are dynamic. Nat Cell Biol. 2002;4(10):826–831. [PubMed: 12360295]
- 22.
- Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci. 2005;6:11–22. [PubMed: 15611723]
- 23.
- dersen JK. idative stress in neurodegeneration: Cause or consequence? Nat Rev Neurosci 2004; 10:S18-25. [PubMed: 15298006]
- 24.
- Browne SE, Ferrante RJ, Beal MF. Oxidative stress in ntington's disease. Brain Pathol. 1999;147-163 [PMC free article: PMC8098260] [PubMed: 9989457]
- 25.
- Tabrizi SJ, Workman J, Hart PE. et al. Mitochondrial dysfunction and free radical damage in the Huntington R6/2 transgenic mouse. Ann Neurol. 2000;47(1):–86. [PubMed: 10632104]
- 26.
- Wyttenbach A, Sauvageot O, Carmichael J. et al. Heat shock protein 27 prevents cellular polyglutamine toxicity and suppresses the increase of reactive oxygen species caused by huntingtin. Hum Mol Genet. 2002;11(9):1137–1151. [PubMed: 11978772]
- 27.
- Mattson MP. Metal-catalyzed disruption of membrane protein and lipid signaling in the pathogenesis of neurodegenerative disorders. Ann NY Acad Sci. 2004;1012:37–50. [PubMed: 15105254]
- 28.
- Tabner BJ, Turnbull S, El-Agnaf OM. et al. Formation of hydrogen peroxide and hydroxyl radicals from A(beta) and alpha-synuclein as a possible mechanism of cell death in Alzheimer's disease and Parkinson's disease. Free Radic Biol Med. 2002;32(11):1076–1083. [PubMed: 12031892]
- 29.
- Arrigo AP, Virot S, Chaufour S. et al. Hsp27 consolidates intracellular redox homeostasis by upholding glutathione in its reduced form and by decreasing iron intracellular levels. Antioxid Redox Sigal. 2005;7(3-4):414–422. [PubMed: 15706088]
- 30.
- Tatton W, Chalmers-Redman R, Brown D. et al. Apoptosis in Parkinson's Disease: Signals for Neruonal Degradation. Ann Neurol. 2003;53(3):61–72. [PubMed: 12666099]
- 31.
- Takuma K, Yan SS, Stern DM. et al. Mitochondrial dysfunction, endoplasmic reticulum stress, and apoptosis in Alzheimer's disease. J Pharmacol Sci. 2005;97(3):312–316. [PubMed: 15750290]
- 32.
- Hickey MA, Chesselet MF. Apoptosis in Huntington's disease. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27(2):255–265. [PubMed: 12657365]
- 33.
- Beere HM. Stressed to death: Regulation of apoptotic signaling pathways by the heat shock proteins. Sci STKE. 2001;93:RE1. [PubMed: 11752668]
- 34.
- Kadowaki H, Nishitoh H, Ichijo H. Survival and apoptosis in ER stress: The role of protein kinases. J Chem Neuroanat. 2004;28(1-2):93–100. [PubMed: 15363494]
- 35.
- Kouroku Y, Fujita E, Jimbo A. et al. Polyglutamine aggregates stimulate ER stress signals and caspase-12 activation. Hum Mol Genet. 2002;11(13):1505–1515. [PubMed: 12045204]
- 36.
- Harper SJ, Wilkie N. MAPKs: New targets for neurodegeneration. Expert Opin Ther Targets. 2003;7(2):187–200. [PubMed: 12667097]
- 37.
- Bell KA, O'Riordan KJ, Sweatt JD. et al. MAPK recruitment by beta-amyloid in organotypic hippocampal slice cultures depends on physical state and exposure time. J Neurochem. 2004;91:349–361. [PubMed: 15447668]
- 38.
- Ross CA, Pickart CM. The ubiquitin-proteasome pathway in Parkinson's disease and other neurodegenerative diseases. Trends Cell Biol. 2004;14(12):703–711. [PubMed: 15564047]
- 39.
- Donaldson KM, Li W, Ching KA. et al. Ubiquitin-mediated sequestration of normal cellular proteins into polyglutamine aggregates. Proc Natl Acad Sci USA. 2003;100(15):8892–8897. [PMC free article: PMC166409] [PubMed: 12857950]
- 40.
- Chen S, Berthelier V, Yang W. et al. Polyglutamine aggregation behavior in vitro supports a recruitment mechanism of cytotoxicity. J Mol Biol. 2001;311:173–182. [PubMed: 11469866]
- 41.
- Lashuel HA, Hartley D, Petre BM. et al. Mixtures of wild-type and a pathogenic (E22G) form of Ab40 in vitro accumulate protofibrils, including amyloid pores. J Mol Biol. 2003;332:795–808. [PubMed: 12972252]
- 42.
- Monoi H, Futaki S, Kugimiya S. et al. Poly-L-glutamine forms cation channels: Relevance to the pathogenesis of the polyglutamine diseases. Biophys J. 2000;78(6):2892–2899. [PMC free article: PMC1300875] [PubMed: 10827970]
- 43.
- Kourie JI, Henry CL. Ion channel formation and membrane-linked pathologies of misfolded hydrophobic proteins: The role of dangerous unchaperoned molecules. Clin Exp Pharmacol Physiol. 2002;29:741–753. [PubMed: 12165037]
- 44.
- Kegel KB, Sapp E, Yoder JL. et al. Huntingtin associates with membranes by directly binding phospholipids Washington, DC: Society for Neuroscience 200444517 (Abstract)
- 45.
- Snyder H, Mensah K, Theisler C. et al. Aggregated and monomeric alpha-synuclein bind to the S6' proteasomal protein and inhibit proteasomal function. J Biol Chem. 2003;278(14):11753–9. [PubMed: 12551928]
- 46.
- Cuervo AM, Stefanis L, Fredenburg R. et al. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305(5688):1292–5. [PubMed: 15333840]
- 47.
- Keck S, Nitsch R, Grune T. et al. Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer's disease. J Neurochem. 2003;85(1):115–122. [PubMed: 12641733]
- 48.
- Nixon RA, Cataldo AM, Mathews PM. The endosomal-lysosomal system of neurons in Alzheimer's disease pathogenesis: A review. Neurochem Res. 2000;25(9-10):1161–72. [PubMed: 11059790]
- 49.
- Wyttenbach A, Carmichael J, Swartz J. et al. Effects of heat shock, heat shock protein 40 (HDJ-2), and proteasome inhibition on protein aggregation in cellular models of Huntington's disease. Proc Natl Acad Sci USA. 2000;97(6):2898–2903. [PMC free article: PMC16027] [PubMed: 10717003]
- 50.
- Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2000;292(5521):1552–2. [PubMed: 11375494]
- 51.
- Venkatraman P, Wetzel R, Tanaka M. et al. Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Mol Cell. 2004;14(1):95–104. [PubMed: 15068806]
- 52.
- Ravikumar B, Vacher C, Berger Z. et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36(6):585–595. [PubMed: 15146184]
- 53.
- Jana NR, Dikshit P, Goswami A. et al. Cochaperone CHIP associates with expanded polyglutamine protein and promotes their degradation by proteasomes. J Biol Chem. 2005;280(12):11635–40. [PubMed: 15664989]
- 54.
- Parcellier A, Schmitt E, Gurbuxani S. et al. HSP27 is a ubiquitin-binding protein involved in I-kappaBalpha proteasomal degradation. Mol Cell Biol. 2003;23(16):5790–5802. [PMC free article: PMC166315] [PubMed: 12897149]
- 55.
- Li JY, Plomann M, PB. Huntington's disease: A synaptopathy? Trends Mol Med. 2003;9(10):414–420. [PubMed: 14557053]
- 56.
- Bechtold DA, Rush SJ, Brown IR. Localisation of the heat-shock protein Hsp70 to the synapse following hyperthermic stress in the brain. J Neurochem. 2000;74:641–646. [PubMed: 10646515]
- 57.
- Guo ZH, Mattson MP. In Vivo 2-Deoxyglucose Administration Preserves Glucose and Glutamate Transport and Mitochondrial Function in Cortical Synaptic Terminals after Exposure to Amyloid. Exp Neurol. 2000;166:173–179. [PubMed: 11031093]
- 58.
- Tobaben S, Thakur P, Fernandez-Chacon R. et al. A trimeric protein complex functions as a synaptic chaperone machine. Neuron. 2001;31:987–999. [PubMed: 11580898]
- 59.
- Hay DG, Sathasivam K, Tobaben S. et al. Progressive decrease in chaperone protein levels in a mouse model of Huntington's disease and induction of stress proteins as a therapeutic approach. Hum Mol Genet. 2004;13(13):1389–405. [PubMed: 15115766]
- 60.
- Gunawardena S, LSG. Polyglutamine diseases and transport problems: Deadly traffic jams on neuronal highways. Arch Neurol. 2005;62(1):46–51. [PubMed: 15642849]
- 61.
- Gauthier LR, Charrin BC, Borrell-Pages M. et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118(1):127–138. [PubMed: 15242649]
- 62.
- Saha AR, Hill J, Utton MA. et al. Parkinson's disease alpha-synuclein mutations exhibit defective axonal transport in cultured neurons. J Cell Sci. 2004;117(7):1017–24. [PubMed: 14996933]
- 63.
- Terwel D, Dewachter I, Van Leuven F. Axonal transport, tau protein, and neurodegeneration in Alzheimer's disease. Neuromoleculer Med. 2002;2(2):151–165. [PubMed: 12428809]
- 64.
- Willis D, Li KW, Zheng JQ. et al. Differential transport and local translation of cytoskeletal, injury-response, and neurodegeneration protein mRNAs in axons. J Neurosci. 2005;25(4):778–791. [PMC free article: PMC6725618] [PubMed: 15673657]
- 65.
- Auluck PK, Meulener MC, Bonini NM. Mechanisms of suppression of alpha-synuclein neurotoxicity by Geldanamycin in Drosophila. J Biol Chem. 2005;280(4):2873–8. [PubMed: 15556931]
- 66.
- Klettner A. The induction of heat shock proteins as a potential strategy to treat neurodegenerative disorders. Drug News Perspect. 2004;17(5):299–306. [PubMed: 15334179]
- The Role of Heat Shock Proteins during Neurodegeneration in Alzheimer's, Parkins...The Role of Heat Shock Proteins during Neurodegeneration in Alzheimer's, Parkinson's and Huntington's Disease - Madame Curie Bioscience Database
- Abl Family Kinases in Mammalian Development - Madame Curie Bioscience DatabaseAbl Family Kinases in Mammalian Development - Madame Curie Bioscience Database
- Cell-Cell Channels and Their Implications for Cell Theory - Madame Curie Bioscie...Cell-Cell Channels and Their Implications for Cell Theory - Madame Curie Bioscience Database
- Antibiotic Resistance in Bacteria Caused by Modified Nucleosides in 23S Ribosoma...Antibiotic Resistance in Bacteria Caused by Modified Nucleosides in 23S Ribosomal RNA - Madame Curie Bioscience Database
- Proteolytic Degradation of Αβ by Neprilysin and Other Peptidases - Madame Curie ...Proteolytic Degradation of Αβ by Neprilysin and Other Peptidases - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...