

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

We discuss in this chapter how nonsense-mediated mRNA decay (NMD) affects the expression of human genetic diseases resulting from premature termination codons (PTCs) by considering how NMD alters disease phenotype. NMD may exert a beneficial, neutral, or harmful effect, depending on the location of the PTC in the transcript and the properties of the truncated protein. In the case of many PTCs, the resulting truncated protein might be nonfunctional and could be degraded without harmful effects. In these instances, NMD probably does not significantly influence phenotype. In other cases, NMD can prevent expression of potentially dominant negative proteins. Therefore, NMD can sometimes exert a protective effect that benefits heterozygous carriers of PTCs. NMD can also contribute to a disease phenotype when it inhibits expression of partially functional proteins. Therapies that could affect gene expression in such cases are under development and may, in the future, provide avenues for effective clinical treatment of diseases that involve NMD.
“NMD-Neutral” PTCs
In general, whether NMD alters the phenotype of a disease depends on both the affected gene and the location of the disease-causing PTC. PTCs upstream of the so-called “NMD boundary,” which is located 50 to 55 bases 5' of the last exon-exon junction, generally trigger NMD of the affected transcript. In contrast, PTCs 3' of this boundary generally escape detection by the NMD machinery, leading to transcript survival. Perhaps surprisingly, it is likely that NMD often has no discernible effect on disease phenotype, since in the absence of NMD many PTCs would probably lead to expression of inactive peptides rather than dominant-negative proteins. Although it might be expected that many PTCs would fall into this “NMD-neutral” category, experimental data are lacking to demonstrate that particular PTCs lead to an identical phenotype in both the presence and absence of NMD. Such “neutral” PTCs will therefore not be discussed further.
NMD-Mediated Protection of Heterozygotes: The Example of β-Thalassemia
NMD may be expected to be beneficial when it inhibits expression of truncated proteins that could exert deleterious effects. The prototypical genetic condition illustrating the protective effects of NMD is β-thalassemia, which is a disorder of hemoglobin production. Normal hemoglobin, which is necessary for oxygen transport, is a tetramer composed of two α-globin and two β-globin subunits. The common recessive form of β-thalassemia occurs in homozygotes who possess NMD-competent PTCs in both copies of the β-globin gene. The resulting defective β-globin mRNA is degraded by NMD. Free α-globin, which is toxic, is then present in excess and is degraded proteolytically.2 Therefore, the quantity of tetrameric hemoglobin is insufficient, causing severe anemia in affected persons. In comparison, heterozygous carriers of a single NMD-competent PTC generally produce enough β-globin from the normal allele to maintain sufficient amounts of tetrameric hemoglobin, and they are clinically healthy. Rare forms of dominant β-thalassemia, in contrast, are caused by NMD-incompetent PTCs within the last exon of the β-globin gene. These PTCs give rise to a large amount of truncated β-globin that cannot be sufficiently degraded and precipitates in toxic inclusion bodies.3 The remarkable difference between healthy heterozygotes possessing NMD-competent PTCs and anemic heterozygotes possessing NMD-incompetent PTCs indicates that NMD protects many healthy heterozygotes from manifesting clinical disease4 (Fig. 1).
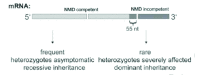
Although β-thalassemia is the only genetic condition so far in which the protective effect of NMD has been thoroughly investigated by experiment, a similar protective role of NMD for heterozygotes with NMD-competent PTCs can be reasonably postulated from similar genotype-phenotype relationships in a number of other diseases (Table 1). These include:
Table 1
Genetic conditions in which NMD can modulate phenotype.
- Dominantly and recessively inherited susceptibility to mycobacterial infections caused by mutations in the IFNGR1 gene.5,6 The recessive form of this condition is often fatal, with patients succumbing to disseminated mycobacterial infections at a young age. This form is caused by PTCs that are probably NMD-competent, since no IFNGR protein was found in a patient with recessive disease. Heterozygous carriers of these PTCs are healthy. The dominant form of the disease arises from PTCs that are predicted to be NMD-incompetent. This form results in production of a truncated IFNGR receptor, as may be expected for a transcript that survives NMD. Heterozygotes manifest increased susceptibility to mycobacterial infection, although such infections are generally not as severe as in the recessive case.
- Brachydactyly type B (see ref. 7), which is a dominant condition involving malformed hands and feet, and Robinow syndrome, which is recessive and characterized by more severe skeletal malformation. Both diseases are caused by mutations in the ROR2 gene. Brachydactyly type B is caused by PTCs that are expected to be NMD-incompetent. Robinow syndrome is characterized by PTCs that should be NMD-competent, and heterozygous carriers are unaffected.
- Dominant and recessive von Willebrand disease,8 which are disorders of blood clotting. PTCs that should be NMD competent lead to severe disease, but only in a pattern of recessive inheritance. In contrast, a mutation that removes the physiological stop codon and results in a new stop codon downstream creates a mutated transcript that should not be an NMD substrate and results in dominant disease.
- Dominant and recessive factor X deficiency,9 which is another disorder of blood clotting. Factor X deficiency is usually transmitted in an autosomal recessive pattern so that heterozygous carriers are healthy. Among the causative mutations, some generate NMD-competent PTCs. A splice site mutation that results in a PTC that should be NMD-incompetent, however, leads to moderate disease in heterozygotes.
A somewhat different example that also demonstrates the protective effect of NMD is provided by the SOX10 gene. In this case, both NMD-competent and NMD-incompetent PTCs give rise to disease. The NMD-competent PTCs appear to cause simple haploinsufficiency, and they result in hereditary neurosensory deafness and intestinal obstruction. However, the NMD-incompetent PTCs results in the production of a dominantly acting truncated protein, which causes a more severe syndrome that includes central and peripheral demyelination.13
Taken in sum, these genetic conditions provide considerable evidence that NMD protects heterozygous carriers of PTCs from expressing dominantly acting deleterious truncated proteins. It is also noteworthy that in many of these conditions, the most severe disease is evident in patients harboring two autosomal recessive mutations. These patients are usually typified by complete or near-complete protein deficiency. Patients harboring dominant mutations, in contrast, usually retain some protein function, which tends to ameliorate the disease phenotype.
NMD in Acquired Genetic Conditions1
Mutations in tumor-suppressor genes are common steps in the development and progression of cancer. As with inherited genetic conditions, NMD appears to provide protection against expression of mutated, truncated tumor-suppressor peptides. For example, NMD has been shown to degrade PTC-containing transcripts arising from the BRCA1 gene.14 PTC-containing mRNAs from the TP53 and Wilms tumor (WT1) loci15-19 are also reduced in abundance compared to wildtype or missense mutated transcripts, presumably due to the action of NMD.
Evidence that NMD protects against dominant truncated forms of these tumor-suppressor proteins derives from experiments in which intronless cDNA constructs encoding unspliced mRNAs that are NMD-incompetent are expressed in cell lines or animals. The resulting C-terminally truncated proteins exert dominant detrimental effects, such as increased chemoresistance, decreased apoptosis, increased tumorigenicity,20-22 interference with transcription-activating ability, and mislocalization of the corresponding cellular tumor suppressor protein23,24 (Table 2). These studies indicate that if abnormal transcripts containing PTCs were not degraded by NMD, clinically recessive tumor-suppressor mutations could instead result in dominant disease due to the synthesis of truncated, dominantly acting oncoproteins. NMD may thus protect heterozygous carriers of PTC-mutated tumor-suppressor genes from developing cancer, at least for as long as the other tumor-suppressor allele remains intact.
Table 2
Effect of NMD on expression of tumor-suppressor genes.
NMD may also affect expression of certain tumor-suppressor genes by modulating the quantity of splice variants produced by these genes, although this hypothesis remains somewhat speculative. For example, the TP53 gene produces a splice variant that contains a PTC at low levels in normal tissues.25 In contrast, a much larger amount of this splice variant —accounting for approximately half of all TP53 transcripts—was found in a leukemic cell line.26 Although a causal relationship has not yet been established, large amounts of the splice variant could potentially contribute to leukemogenesis, which would be prevented under normal conditions by NMD-induced degradation of the variant.
Transcripts from the WT1 gene also undergo alternative splicing to produce two major splice forms. The more abundant splice form (+KTS) encodes three additional amino acids at the 3' end of the penultimate exon, while the less abundant splice form (-KTS) lacks these residues. Interestingly, a mutation found in acute myelogenous leukaemia,27 Wilms tumor28 and Frasier syndrome (male pseudohermaphroditism and progressive glomerulopathy)29 is also located within the penultimate exon of the WT1 gene. This mutation introduces an NMD-competent PTC, but only for the +KTS form. In contrast, for the -KTS form, the PTC is within the terminal 50 bases of the penultimate exon. Therefore, -KTS transcripts probably escape NMD and are translated to produce a truncated protein (Fig. 2). NMD could thus limit expression of the primary transcript. This is important because, in general, Frasier syndrome is attributable to mutations that selectively reduce the abundance of the +KTS isoform.30,31 Therefore, Frasier syndrome in individuals carrying this particular PTC mutation could be caused by NMD-induced degradation of the +KTS isoform. In addition, the truncated form of the minor splice variant might act in a dominant fashion to promote tumorigenesis.

Medical Therapies for PTC-Related Disease
In contrast to its role in preventing dominant disease, NMD can also eliminate mRNAs that would otherwise result in the production of partly or fully functional truncated protein, thereby contributing to the protein deficiency that is the hallmark of many recessive genetic conditions. In such instances, interventions to prevent degradation of transcripts containing PTCs may be therapeutically useful. Drug therapy with these aims is the subject of Chapter 10 in this book and therefore will be discussed only briefly here.
At this time, the therapeutic approach that is closest to clinical applicability is the use of aminoglycoside antibiotics that allow read-through of nonsense codons. These drugs bind to the decoding center of the ribosome32 and decrease the accuracy requirements for codon-anticodon pairing, thereby resulting in incorporation of an amino acid into the polypeptide chain instead of chain termination. Thus, full-length, albeit missense-mutated, proteins are synthesized. Aminoglycosides have been used to treat Duchenne muscular dystrophy, Hurler syndrome, X-linked nephrogenic diabetes insipidus, ataxia-telangiectsia, and cystinosis, resulting in some functional improvement in cell lines33-39 and animal models.36,40 Some trials of aminoglycoside therapy also have been carried out in humans with PTC-associated diseases. Promising results have been obtained in a controlled clinical trial for cystic fibrosis, in which full-length CFTR protein was detected in nasal epithelial cells of two treated individuals.41 In contrast, clinical studies of individuals with muscular dystrophy have not shown functional improvement.42,43 Overall, although early results indicate that aminoglycoside treatment may have potential applicability, a therapeutic benefit has yet to be demonstrated. The effect of prolonged treatment with aminoglycosides is also a concern. First, there is the problem of toxicity. Second, there is the issue of whether the general, long-term suppression of PTCs, which is accompanied by the suppression of physiologic termination codons and the potential for pseudogene transcript translation, will result in the build-up of abnormal proteins that could trigger other cellular problems.
Other approaches to modulating PTC-induced transcript degradation are also under investigation. One potential approach is to use antisense oligoribonucleotides to redirect splicing, thereby avoiding the production of PTCs in the first place. Initially described as a method for correcting the in vitro aberrant splicing of a disease-associated beta globin gene,44 this strategy employs antisense 2'-O-methylribonucleotides (2OMeAO) that hybridize to splice sites or branch point junctions of aberrantly spliced pre-mRNA, thereby restoring normal splicing in a significant fraction of molecules. This approach was modified for use with a disease-associated dystrophin transcript.45 In this case, the targeted PTC was located within an exon coding for a dispensable protein region. Antisense 2OMeAO that targeted splice sites flanking the PTC promoted in-frame skipping of the affected exon, effectively removing the PTC. Treatment of mdx mice with these antisense oligonucleotides resulted in low-level expression of shortened but functional dystrophin. In a further step toward the clinic, the efficiency of oligonucleotide delivery to tissues has been enhanced by the use of vehicles such as block copolymer.46 Before trials of 2OMeAO are feasible in humans, however, a systemic delivery method needs to be developed. As with all forms of gene therapy, the issues of transfection efficiency, potential immune responses, and side effects must be addressed. Unfortunately, this sort of treatment would be feasible only for mutations in which manipulation of splicing maintains in-frame translation, and—if exon skipping is the result—that does not remove essential protein regions or result in protein mis-folding. Therefore, this treatment will probably be limited to specific cases rather than provide a general therapy for PTC-associated diseases.
A further potential approach—although currently very far from realization—involves modulating NMD per se, rather than modulating recognition of PTCs. Down-regulating the central NMD protein Upf1 using RNA interference has been shown to inhibit NMD in cultured cells, and it might constitute a starting point for therapeutic developments. Additionally, some evidence exists to suggest that different individuals with identical genetic mutations may exhibit distinct phenotypic severities due to differences in the efficiency of NMD.47 Thus, identifying factors that regulate the efficiency of NMD could permit development of therapies that fine-tune NMD, potentially allowing more targeted interventions in patients with PTC-associated diseases.
Conclusions
NMD plays an important role in modulating the manifestation of hereditary and acquired genetic diseases, and a deepening understanding of NMD will augment the establishment of genotype-phenotype relationships in a number of conditions. The contribution of NMD to genetic diseases may be beneficial, neutral or harmful, depending on the specific PTC, its location, and the type of mutation that generates the PTC. Therefore, the potential role of NMD in disease must be appreciated when the functional effect of a mutation is considered. Therapies that target PTC-containing transcripts are under development, and continued research in this direction should help lead to viable strategies to treat PTC-associated diseases.
References
- 1.
- Holbrook JA, Neu-Yilik G, Hentze MW. et al. Nonsense-mediated decay approaches the clinic. Nat Genet. 2004;36(8):801–808. [PubMed: 15284851]
- 2.
- Hall GW, Thein S. Nonsense codon mutations in the terminal exon of the beta-globin gene are not associated with a reduction in beta-mRNA accumulation: A mechanism for the phenotype of dominant beta-thalassemia. Blood. 1994;83(8):2031–2037. [PubMed: 8161774]
- 3.
- Thein SL, Hesketh C, Taylor P. et al. Molecular basis for dominantly inherited inclusion body beta-thalassemia. Proc Natl Acad Sci USA. 1990;87(10):3924–3928. [PMC free article: PMC54016] [PubMed: 1971109]
- 4.
- Kugler W, Enssle J, Hentze MW. et al. Nuclear degradation of nonsense mutated beta-globin mRNA: A post-transcriptional mechanism to protect heterozygotes from severe clinical manifestations of beta-thalassemia? Nucleic Acids Res. 1995;23(3):413–418. [PMC free article: PMC306691] [PubMed: 7885837]
- 5.
- Jouanguy E, Altare F, Lamhamedi S. et al. Interferon-gamma-receptor deficiency in an infant with fatal bacille Calmette-Guerin infection. N Engl J Med. 1996;335(26):1956–1961. [PubMed: 8960475]
- 6.
- Jouanguy E, Lamhamedi-Cherradi S, Lammas D. et al. A human IFNGR1 small deletion hotspot associated with dominant susceptibility to mycobacterial infection. Nat Genet. 1999;21(4):370–378. [PubMed: 10192386]
- 7.
- Schwabe GC, Tinschert S, Buschow C. et al. Distinct mutations in the receptor tyrosine kinase gene ROR2 cause brachydactyly type B. Am J Hum Genet. 2000;67(4):822–831. [PMC free article: PMC1287887] [PubMed: 10986040]
- 8.
- Schneppenheim R, Budde U, Obser T. et al. Expression and characterization of von Willebrand factor dimerization defects in different types of von Willebrand disease. Blood. 2001;97(7):2059–2066. [PubMed: 11264172]
- 9.
- Millar DS, Elliston L, Deex P. et al. Molecular analysis of the genotype-phenotype relationship in factor X deficiency. Hum Genet. 2000;106(2):249–257. [PubMed: 10746568]
- 10.
- Rivolta C, Berson EL, Dryja TP. Dominant Leber congenital amaurosis, cone-rod degeneration, and retinitis pigmentosa caused by mutant versions of the transcription factor CRX. Hum Mutat. 2001;18(6):488–498. [PubMed: 11748842]
- 11.
- Rosenfeld PJ, Cowley GS, McGee TL. et al. A null mutation in the rhodopsin gene causes rod photoreceptor dysfunction and autosomal recessive retinitis pigmentosa. Nat Genet. 1992;1(3):209–213. [PubMed: 1303237]
- 12.
- Sung CH, Davenport CM, Hennessey JC. et al. Rhodopsin mutations in autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci USA. 1991;88(15):6481–6485. [PMC free article: PMC52109] [PubMed: 1862076]
- 13.
- Inoue K, Khajavi M, Ohyama T. et al. Molecular mechanism for distinct neurological phenotypes conveyed by allelic truncating mutations. Nat Genet. 2004 [PubMed: 15004559]
- 14.
- Perrin-Vidoz L, Sinilnikova OM, Stoppa-Lyonnet D. et al. The nonsense-mediated mRNA decay pathway triggers degradation of most BRCA1 mRNAs bearing premature termination codons|hHum Mol Genet 200211(23):2805–2814. [PubMed: 12393792]
- 15.
- Kawasaki T, Tomita Y, Watanabe R. et al. mRNA and protein expression of p53 mutations in human bladder cancer cell lines. Cancer Lett. 1994;82(1):113–121. [PubMed: 8033064]
- 16.
- Williams C, Norberg T, Ahmadian A. et al. Assessment of sequence-based p53 gene analysis in human breast cancer: Messenger RNA in comparison with genomic DNA targets. Clin Chem. 1998;44(3):455–462. [PubMed: 9510848]
- 17.
- Magnusson KP, Sandstrom M, Stahlberg M. et al. p53 splice acceptor site mutation and increased HsRAD51 protein expression in Bloom's syndrome GM1492 fibroblasts. Gene. 2000;246(1-2):247–254. [PubMed: 10767546]
- 18.
- Usuda J, Inomata M, Fukumoto H. et al. Restoration of p53 gene function in 12-O-tetradecanoylphorbor 13-acetate-resistant human leukemia K562/TPA cells. Int J Oncol. 2003;22(1):81–86. [PubMed: 12469188]
- 19.
- King-Underwood L, Pritchard-Jones K. Wilms' tumor (WT1) gene mutations occur mainly in acute myeloid leukemia and may confer drug resistance. Blood. 1998;91(8):2961–2968. [PubMed: 9531607]
- 20.
- Fan S, Yuan R, Ma YX. et al. Mutant BRCA1 genes antagonize phenotype of wild-type BRCA1. Oncogene. 2001;20(57):8215–8235. [PubMed: 11781837]
- 21.
- Sylvain V, Lafarge S, Bignon YJ. Dominant-negative activity of a Brca1 truncation mutant: Effects on proliferation, tumorigenicity in vivo, and chemosensitivity in a mouse ovarian cancer cell line. Int J Oncol. 2002;20(4):845–853. [PubMed: 11894135]
- 22.
- Cardinali M, Kratochvil FJ, Ensley JF. et al. Functional characterization in vivo of mutant p53 molecules derived from squamous cell carcinomas of the head and neck. Mol Carcinog. 1997;18(2):78–88. [PubMed: 9049183]
- 23.
- Reddy JC, Morris JC, Wang J. et al. WT1-mediated transcriptional activation is inhibited by dominant negative mutant proteins. J Biol Chem. 1995;270(18):10878–10884. [PubMed: 7738027]
- 24.
- Englert C, Vidal M, Maheswaran S. et al. Truncated WT1 mutants alter the subnuclear localization of the wild-type protein. Proc Natl Acad Sci USA. 1995;92(26):11960–11964. [PMC free article: PMC40275] [PubMed: 8618823]
- 25.
- Flaman JM, Waridel F, Estreicher A. et al. The human tumour suppressor gene p53 is alternatively spliced in normal cells. Oncogene. 1996;12(4):813–818. [PubMed: 8632903]
- 26.
- Chow VT, Quek HH, Tock EP. Alternative splicing of the p53 tumor suppressor gene in the Molt-4 T-lymphoblastic leukemia cell line. Cancer Lett. 1993;73(2-3):141–148. [PubMed: 8221626]
- 27.
- King-Underwood L, Renshaw J, Pritchard-Jones K. Mutations in the Wilms' tumor gene WT1 in leukemias. Blood. 1996;87(6):2171–2179. [PubMed: 8630376]
- 28.
- Little MH, Prosser J, Condie A. et al. Zinc finger point mutations within the WT1 gene in Wilms tumor patients. Proc Natl Acad Sci USA. 1992;89(11):4791–4795. [PMC free article: PMC49173] [PubMed: 1317572]
- 29.
- Kohsaka T, Tagawa M, Takekoshi Y. et al. Exon 9 mutations in the WT1 gene, without influencing KTS splice isoforms, are also responsible for Frasier syndrome. Hum Mutat. 1999;14(6):466–470. [PubMed: 10571943]
- 30.
- Barbaux S, Niaudet P, Gubler MC. et al. Donor splice-site mutations in WT1 are responsible for Frasier syndrome. Nat Genet. 1997;17(4):467–470. [PubMed: 9398852]
- 31.
- Klamt B, Koziell A, Poulat F. et al. Frasier syndrome is caused by defective alternative splicing of WT1 leading to an altered ratio of WT1 +/-KTS splice isoforms. Hum Mol Genet. 1998;7(4):709–714. [PubMed: 9499425]
- 32.
- Eustice DC, Wilhelm JM. Fidelity of the eukaryotic codon-anticodon interaction: Interference by aminoglycoside antibiotics. Biochemistry. 1984;23(7):1462–1467. [PubMed: 6722101]
- 33.
- Zsembery A, Jessner W, Sitter G. et al. Correction of CFTR malfunction and stimulation of Ca-activated Cl channels restore HCO3- Secretion in cystic fibrosis bile ductular cells. Hepatology. 2002;35(1):95–104. [PubMed: 11786964]
- 34.
- Bedwell DM, Kaenjak A, Benos DJ. et al. Suppression of a CFTR premature stop mutation in a bronchial epithelial cell line. Nat Med. 1997;3(11):1280–1284. [PubMed: 9359706]
- 35.
- Keeling KM, Brooks DA, Hopwood JJ. et al. Gentamicin-mediated suppression of Hurler syndrome stop mutations restores a low level of alpha-L-iduronidase activity and reduces lysosomal glycosaminoglycan accumulation. Hum Mol Genet. 2001;10(3):291–299. [PubMed: 11159948]
- 36.
- Barton-Davis ER, Cordier L, Shoturma DI. et al. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J Clin Invest. 1999;104(4):375–381. [PMC free article: PMC481050] [PubMed: 10449429]
- 37.
- Sangkuhl K, Schulz A, Rompler H. et al. Aminoglycoside-mediated rescue of a disease-causing nonsense mutation in the V2 vasopressin receptor gene in vitro and in vivo. Hum Mol Genet. 2004 [PubMed: 14998935]
- 38.
- Lai CH, Chun HH, Nahas SA. et al. Correction of ATM gene function by aminoglycoside-induced read-through of premature termination codons Proc Natl Acad Sci USA 2004101(44):15676–15681. (Epub 12004 Oct 15621) [PMC free article: PMC524838] [PubMed: 15498871]
- 39.
- Helip-Wooley A, Park MA, Lemons RM. et al. Expression of CTNS alleles: Subcellular localization and aminoglycoside correction in vitro. Mol Genet Metab. 2002;75(2):128–133. [PubMed: 11855931]
- 40.
- Du M, Jones JR, Lanier J. et al. Aminoglycoside suppression of a premature stop mutation in a Cftr-/- mouse carrying a human CFTR-G542X transgene. J Mol Med. 2002;80(9):595–604. [PubMed: 12226741]
- 41.
- Wilschanski M, Yahav Y, Yaacov Y. et al. Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N Engl J Med. 2003;349(15):1433–1441. [PubMed: 14534336]
- 42.
- Wagner KR, Hamed S, Hadley DW. et al. Gentamicin treatment of Duchenne and Becker muscular dystrophy due to nonsense mutations. Ann Neurol. 2001;49(6):706–711. [PubMed: 11409421]
- 43.
- Politano L, Nigro G, Nigro V. et al. Gentamicin administration in Duchenne patients with premature stop codon. Preliminary results. Acta Myol. 2003;22(1):15–21. [PubMed: 12966700]
- 44.
- Dominski Z, Kole R. Restoration of correct splicing in thalassemic pre-mRNA by antisense oligonucleotides. Proc Natl Acad Sci USA. 1993;90(18):8673–8677. [PMC free article: PMC47420] [PubMed: 8378346]
- 45.
- Mann CJ, Honeyman K, Cheng AJ. et al. Antisense-induced exon skipping and synthesis of dystrophin in the mdx mouse. Proc Natl Acad Sci USA. 2001;98(1):42–47. [PMC free article: PMC14541] [PubMed: 11120883]
- 46.
- Lu QL, Mann CJ, Lou F. et al. Functional amounts of dystrophin produced by skipping the mutated exon in the mdx dystrophic mouse. Nat Med. 2003;9(8):1009–1014. [PubMed: 12847521]
- 47.
- Kerr TP, Sewry CA, Robb SA. et al. Long mutant dystrophins and variable phenotypes: Evasion of nonsense-mediated decay? Hum Genet. 2001;109(4):402–407. [PubMed: 11702221]
- NMD and Human Disease - Madame Curie Bioscience DatabaseNMD and Human Disease - Madame Curie Bioscience Database
- Telomeres: Guardians of Genomic Integrity or Double Agents of Evolution? - Madam...Telomeres: Guardians of Genomic Integrity or Double Agents of Evolution? - Madame Curie Bioscience Database
- Preprotein Translocation through the Sec Translocon in Bacteria - Madame Curie B...Preprotein Translocation through the Sec Translocon in Bacteria - Madame Curie Bioscience Database
- Functional Evidence of Pain Control by the Immune System - Madame Curie Bioscien...Functional Evidence of Pain Control by the Immune System - Madame Curie Bioscience Database
- Future Perspectives of Interstitial and Perfusional Hyperthermia - Madame Curie ...Future Perspectives of Interstitial and Perfusional Hyperthermia - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...