

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Bacterial pathogens against which DNA vaccines are being developed encompass both intracellular and extracellular pathogens as well as vaccines against bacterial toxins. DNA vaccination has an inherent bias towards generating cellular immunity by virtue of the intracellular origin of the antigen, resulting in particular efficacy against intracellular pathogens. However, by manipulating the formulation and delivery, effective antibody responses can also be obtained. The overwhelming majority of publications describe efforts to produce DNA vaccines against tuberculosis, and therefore the first part of this chapter will be dedicated to this organism and other pathogens of the mycobacterial genus like M. leprae and M. avium. In the second part we will give a detailed overview of the endeavors to develop DNA vaccines against other bacterial pathogens.
DNA Vaccines against Mycobacterial Infection
Pathogenic Mycobacteria
Mycobacterium is a bacterial genus containing more than 30 different species including M. tuberculosis and M. bovis causing human and bovine tuberculosis, respectively, and M. leprae the causative agent of leprosy in humans. Other mycobacterial human pathogens comprise M. avium, a pathogen causing opportunistic infections in immuno-compromised individuals like AIDS patients, and M. ulcerans, which causes buruli ulcer, an emerging health problem in developing countries.
The Challenges Presented by Tuberculosis
Human tuberculosis, caused by M. tuberculosis, is a major global human health problem that claims more than 2 millions lives every year.1 The advent of the HIV pandemic has tragically increased the incidence of tuberculosis in developing countries, e.g., sub-Saharan Africa, where AIDS-related tuberculosis is becoming the main cause of mortality in young adults. Indeed, it has been estimated that M. tuberculosis is responsible for the deaths of more youths and adults than any other infectious agent. The WHO has therefore declared tuberculosis a global health emergency.2
BCG (M. bovis Bacille Calmette Guerin), the TB vaccine, is a major component of many national vaccination programs. About 90 million doses are supplied by the World Health Organization (WHO) every year. However, trials performed in a number of countries have revealed that BCG imparts a very heterogeneous degree of protection, ranging from 0 to about 80%.3–5 Although there is usually quite good protection imparted against the severe childhood forms of tuberculosis, such as tuberculous meningitis, there can be poor protection against the infectious adult form.6 Hence a better vaccine is needed, or one that can be given in addition to BCG and results in better protection against both childhood and adult disease. Critically, vaccination must prevent the establishment of the persistent latent state of infection that can follow primary infection and provides a major source of adult disease many years later.
A vaccine against tuberculosis in cattle is also needed. Bovine tuberculosis, caused by the closely related organism M. bovis, can be a significant source of disease in man. For example, around years 1930–1940 about 40% of cows in the UK were infected and 6–7% of total human deaths due to tuberculosis were caused by M. bovis.7 BCG can be effective in cattle, but as in man, the efficacy varies widely and the vaccine response interferes with the hypersensitivity tests deployed in control programs. The strategy of detecting (by skin test hypersensitivity) and slaughtering infected animals is being used to reduce and contain the problem (in conjunction with pasteurization of raw milk) but is not universally applicable especially in the developing world and cannot eliminate the disease in the face of reservoirs of infection in wildlife.8 A recent review of the situation of bovine TB in cattle and wildlife in England and Wales for example has concluded that the development of an effective cattle vaccine against bovine TB has the best prospect of TB control in the National herd.9
The Nature of Protective Immunity Against Tuberculosis
Most constituents of the cellular and humoral immune system have been documented to be involved following infection with M. tuberculosis and several comprehensive review papers have been published recently.10–13 The view that antibody has no role to play in protection may be changing slightly,14,15 but as yet the paradigm of protection in tuberculosis remains cellular immunity. Unfortunately, the disease is also entirely a consequence of the cellular immune response. Accordingly, vaccination is required to enhance the protective and not the harmful aspects of cellular immunity. Numerous studies have shown that DNA vaccination is able to achieve this, although we do not fully understand how, and most remarkably DNA vaccines are even able to exert a therapeutic effect in infected animals.
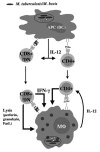
The general principles characterizing protective immune responses are represented graphically in Figure 1. Tubercle bacilli reside and multiply within infected macrophages. Antigen-specific T lymphocytes produce lymphokines including macrophage activating factors such as IFN-γ that enable the macrophages to at least restrict growth of the bacteria and maybe even to kill them. In 90% of people who become infected the organisms are eliminated without ever causing disease, but it has been hard to establish that activated macrophages actually kill the bacteria. However, it may be significant that killing of tubercle bacteria can occur when infected macrophages are themselves killed by the cytotoxic lymphocytes either through induction of apoptosis or by granule-mediated lysis.
TB Vaccines for Prophylactic Vaccination and Identification of Protective Antigens
The era of DNA vaccination against mycobacterial diseases began in 1994 when Silva and Lowrie demonstrated protection against M. tuberculosis after vaccination of mice with the macrophage cell line J774 that had been stably transfected with the heat-shock protein HSP65 from M. leprae.16 This provided proof of principle that a mycobacterial antigen expressed by a eukaryotic expression vector could induce protective immunity and was quickly followed by the successful use of a naked DNA vaccine expressing the same antigen.17 Similar protection could be obtained with DNA vaccines expressing either the secreted fibronectin binding protein antigen 85A (Ag85A)18 of M. tuberculosis or the M. leprae HSP65.19 This was followed soon after by a report indicating that another secreted TB antigen, the phosphate transporter PstS-1 (38 kDa antigen) also induced protective immunity.20 Since then a large number of antigens have been expressed as DNA vaccines and tested in mouse, guinea pig, or cattle models of tuberculosis. Antigens, which in at least one of these animal models have been proven protective, are listed in Table 1. The overwhelming majority of antigens are secreted antigens. This is consistent with (and in part a consequence of) the long-held belief that such antigens will be the earliest targets seen by the immune system upon infection and therefore likely to be protective.21 Nevertheless, a number of other antigens that are probably not secreted also impart protection when given as DNA vaccines. Examples are the heat-shock protein HSP65, or the cytoplasmic enzyme KatG (Table 1, see refs. 19,22, 23). Although immune responses to the antigens listed in Table 1 conferred protection against M. tuberculosis to some degree, only very rarely did the protection surpass that observed after BCG vaccination (Table 1). We will now briefly discuss the results obtained so far for several of the more intimately studied antigens, namely HSP65, Ag85A, ESAT-6, and phosphate transporters.
Table 1
Mycobacterial antigens protecting against tuberculosis when applied as prophylactic vaccine.
HSP65
As mentioned above, vaccination of mice with M. leprae-derived HSP65 imparted a good degree of protection when vaccinated mice were challenged with M. tuberculosis.19,24 In initial studies, several inbred strains of mice could be protected and subsequently good protection was also observed in outbred mice. The immune responses described following vaccination included strong IFN-γ production by both CD4 and CD8 T cells as well as the induction of cytotoxic T cells. We have recently found that cattle vaccinated with M. leprae HSP65 generated strong IFN-γ and T cell proliferative responses, but did not respond subsequently to a tuberculin skin test positively (Vordermeier, and Hewinson, unpublished data). This is a significant observation because it suggests that vaccination strategies can be developed which would allow the continuation of tuberculin-based test and slaughter control strategies of bovine tuberculosis alongside vaccination. If this were also shown to be the case for humans, it would allow, by tuberculin skin testing, the identification of those vaccinated but infected individuals that required therapy. In subsequent studies cattle were not protected when vaccinated with DNA expressing a cocktail of antigens containing M. leprae HSP65 and the M. tuberculosis HSP70 and Apa proteins (Vordermeier, Hewinson and Buddle, unpublished results, see Table 1). However, this cocktail significantly improved the efficacy of BCG when applied in a prime-boost protocol (see below). In contrast to the results obtained after vaccination with M. leprae HSP65, the group of Orme-vaccinated mice and guinea pigs with DNA expressing the M. tuberculosis homologue of this protein and were unable to demonstrate protection.25 However, the vaccine was designed to give enhanced secretion of the antigen by including a secretion signal from tissue plasminogen activator. This approach has been reported to enhance the antibody response23,26 and may have contributed to the severe lung damage seen in guinea pigs that was characterized by necrotizing bronchointerstitial pneumonia and bronchiolitis.25 The authors voiced grave safety concerns against vaccination with heat-shock proteins that have close homologues in eukaryotic organisms as T cells recognizing cross-reactive determinants could give rise to autoimmune responses. Interestingly, naked DNA vaccination with M. leprae HSP65, rather than exacerbating adjuvant arthritis, an experimental autoimmune disease, protected rats against disease development.27 The striking differences observed between M. leprae and M. tuberculosis HSP65 DNA vaccines await adequate explanation.
Ag85A
Antigen 85A is part of a complex of secreted antigens (consisting of Ag85A, Ag85B, Ag85C) that constitute a major fraction of the secreted proteins in culture filtrates. These fibronectin-binding proteins are also mycolyl transferases, which play an important role in the synthesis of mycobacterial cell wall components. Huygen and colleagues demonstrated that DNA vaccination with Ag85A-induced substantial cellular and humoral immune responses (CTL, IFN-γ, etc.) and conferred a significant degree of protection to mice against aerosol challenge with virulent M. tuberculosis.18 This group also provided evidence that the inclusion of the secretion signal peptide from tissue plasminogen activator (tPA) resulted not only in stronger immune responses but also in sustained protection when the resting period between vaccination and challenge was increased from 30 to 90 days. The nonmodified form of Ag85A was not protective after 90 days.28 DNA vaccination with Ag85B has also been shown to be protective in mice, whereas vaccination with Ag85C is not.29 Although Ag85A DNA vaccinated mice could control the numbers of viable bacilli in lungs and spleens for prolonged periods (up to 10 weeks postinfection), this protection waned rapidly and bacterial loads 12 weeks following infection were indistinguishable from those of vector-vaccinated control animals.30
Ag85A has also been tested in a guinea pig aerosol challenge model of tuberculosis. DNA vaccination did not lead to a reduction of bacterial burdens in the lungs of infected guinea pigs but did reduce dissemination to the spleen and resulted in prolonged survival. The mean survival time for DNA vaccinated guinea pigs was still shorter than for BCG vaccinated animals.31 Surviving animals had extensive granulomatous pneumonia in which a high percentage of lymphocytes was present (this was less prominent in dying animals), albeit they lacked pulmonary necrosis and caseation, which is the hallmark of tuberculosis in the guinea pig.31 Significantly, DNA vaccination of guinea pigs with Ag85A did not result in positive tuberculin skin responses,31 suggesting that this vaccination strategy could be implemented alongside tuberculin-based surveillance mechanisms. Cattle vaccinated with this plasmid did not produce any demonstrable immune responses after vaccination and therefore the vaccine's ability to protect cattle against bovine tuberculosis has not been tested.32 It should be noted that members of the Ag85 complex also impart protection against mycobacterial diseases other than tuberculosis (see below).
Phosphate Transport Receptors (PstS-1, PstS-2, PstS-3)
Analysis of the M. tuberculosis genome identified three putative phosphate binding protein homologues to the periplasmic ATP-cassette (ABC) phosphate-binding receptor PstS of Escherichia coli. These M. tuberculosis proteins were called PstS-1 (identical to the well-characterized 38 kDa antigen), PstS-2, and PstS-3.33 The 38 kDa antigen PstS-1 was, alongside HSP65 and Ag85A, amongst the first antigens tested as DNA vaccine to protect mice against tuberculosis.20 Results by Zhu and coworkers demonstrated strong cellular immune responses characterized by IFN-γ production and CD8-mediated cytotoxicity. Strikingly, the specificity of both CD4 and CD8 T cells was found to be different between M. tuberculosis-infected and DNA-vaccinated mice with respect to several epitopes. Vaccinated mice were significantly protected against M. tuberculosis challenge up to 12 weeks postinfection.20 In contrast, although Tanghe et al, demonstrated high levels of Th1-type IL-2 and IFN-γ responses in mice vaccinated with DNA expressing either PstS-1, PstS-2 or PstS-3, no protection was observed in the PstS-1-vaccinated mice.30 At present it is not possible to reconcile these conflicting results with PstS-1 as different mouse strains, different challenge routes, and different resting periods between vaccination and infection (2 vs. 10 weeks) were used. Vaccination with PstS-2 resulted in modest reduction in bacterial numbers in spleen whereas the PstS-3 vaccination caused significant and sustained protection both in lungs and spleens over a period of at least 12 weeks.30 These results make PstS-3 a particular interesting vaccine antigen and it is hoped that it will be tested soon in other models.
ESAT-6
ESAT-6 is a low-molecular protein found in short-term culture filtrate of organisms of the M. tuberculosis complex (M. tuberculosis, M. bovis, M. africanum) and only a few other mycobacterial species (i.e., M. kansasii or M. marinum).34,35 It is one of the most prominent targets of the cellular immune system in all hosts tested so far (mice, guinea pigs, humans, cattle: e.g., refs. 36–39). Apart from its immunogenicity and relative specificity for tubercle bacilli, its main attraction is the presence of its gene in the RD1 region of the genome that is deleted in all strains of BCG. These attributes have made it an ideal candidate as a specific diagnostic reagent to distinguish between infected and BCG vaccinated individuals (e.g., see ref. 38). Nevertheless, due to its pronounced immunogenicity it has also been evaluated as a vaccine candidate. Besides being tested in classic protein-based vaccine approaches it has also been tested frequently as a DNA vaccine in mice. These diverse studies have resulted in widely variable efficacies. Whilst some studies demonstrated no, or only marginal protection, others demonstrated highly significant and strong protection.22,23 Interestingly, the best protective effects were observed with DNA vaccine constructs where a tPA secretion signal peptide was included.22–24,40 The ability of this antigen to protect other host species, like the guinea pig, against tuberculosis, is unknown at the time of the preparation of this manuscript.
The long list of antigens conferring some degree of protection against tuberculosis shows that many diverse proteins can protect against tuberculous infections. However, a number of antigens have also been described that did not confer any significant degree of protection (e.g., the 19 kDa protein Rv3763,41,42 16 kDa alpha-crystallin-like chaperonin protein Rv2031c,22 Ag85C Rv0129c,29 PstS-2 Rv0932c,30 Rv1796, Rv2428 or Rv2945c42–44).
DNA Vaccines as Post-Exposure Vaccines or Immunotherapeutic Agents
As we have pointed out earlier, there is also a need to develop novel tuberculosis vaccines that are effective in latently infected individuals (post-exposure vaccine), or are effective as immunotherapeutic reagents to assist conventional chemotherapy. Effective treatment with chemotherapy requires large doses of costly antibacterial drugs that have to be taken for at least 6 months, which is difficult to achieve in many developing countries. In addition, immunotherapy could improve the treatment of multidrug-resistant tuberculosis. Addressing these objectives, Lowrie and colleagues demonstrated that DNA vaccines that were effective as prophylactic vaccines against tuberculosis also had therapeutic effects against an established, chronic M. tuberculosis infection by reducing the bacterial burdens in both the lungs and spleens of treated mice.45 They tested DNA vaccines expressing M. leprae HSP65, M. tuberculosis HSP70, ESAT-6, and another secreted antigen, MPT70 (see Table 1 for details of antigens) and could show strong therapeutic activity, mainly with HSP65 and MPT70. Interestingly, injection of DNA encoding IL-12 alone was as beneficial as injection of HSP65 or MPT70 although coinjection of IL-12 and HSP65 DNA resulted in reduced therapeutic efficacy.45 The administration of BCG had no effect in these experiments. The authors' experiments suggested that the herapeutic effect of HSP65 was due to a reprogramming of the immune responses characterized by a switch from IL-4 responses towards predominant IFN-γ responses.45
A frequent result of incomplete drug treatment of tuberculosis is the regrowth of bacteria that have persisted in a nonreplicating and physiologically drug-resistant form. Lowrie and colleagues used a mouse relapse model to show that HSP65 DNA vaccination given at the end of incomplete chemotherapy could prevent the corticosteroid-induced regrowth of M. tuberculosis in the lungs of 8/8 mice and in the spleens of 6/8 mice.45 Significantly, BCG was ineffective in this experimental model of relapsing tuberculosis. These results raised the possibility that therapeutic DNA vaccination could be used as a postexposure vaccine in man to eliminate latent or persistent infection prior to disease.
Turner et al46 conducted similar immunotherapeutic vaccination experiments in mice with an Ag85A DNA vaccine. They were unable to demonstrate any effect of vaccination on the course of infection in the lung of aerosol-infected mice but could demonstrate a reduction in bacterial loads in their spleens.
DNA Vaccines Protecting Against Mycobacterial Diseases Other Than Tuberculosis
Mycobacteria are the causative agents of several other human diseases apart from tuberculosis. For example, M. leprae is the causative agent of leprosy in humans, M. avium is a common opportunistic pathogen of immuno-compromised individuals like AIDS patients, and buruli ulcer, an emerging health problem of developing countries is caused by M. ulcerans. DNA vaccines are being developed to protect against all three of these diseases. This is being facilitated by the availability of suitable murine models to test vaccine efficacy. For example, DNA vaccination with M. tuberculosis Ag85B has been shown to impart protection against M. leprae growth on challenge of mouse footpads,47 as did a DNA vaccine expressing the M. leprae 35 kDa antigen (ORF designation ML084147,48). The degree of M. leprae growth reduction was similar to that achieved with BCG, which appears to confer protection against leprosy in man. DNA vaccination with the M. avium homologues of either the M. leprae 35 kDa antigen,47,49 Ag85A,50 or of HSP6550 protected mice against M. avium infection to a degree similar to, but not exceeding, BCG vaccination. In addition, DNA vaccination with M. tuberculosis Ag85A protected mouse footpads against M. ulcerans infection almost as well as BCG vaccination.51 Interestingly, “vaccination” of mice, either before or after M. avium infection using a construct expressing IL-18 reduced the bacterial loads and increased the survival period of treated mice.52
Strategies to Improve the Efficacy of Tuberculosis DNA Vaccines
As shown in Table 1 and emphasized throughout this chapter, protection levels with DNA vaccination against challenge with tuberculosis has been generally less effective than BCG vaccination alone. Our own DNA vaccination studies in cattle, which, unlike mice and guinea pigs, are natural hosts for M. bovis, have confirmed this observation. We have tested 5 different DNA vaccine constructs (M. leprae HSP65, M. tuberculosis HSP70, Apa, MPB83, MPB70), either individually or as cocktails, and, to date, we have not been able to protect cattle against M. bovis infection (Buddle, Vordermeier, Hewinson, unpublished data) despite the proven effectiveness of these vaccines in rodents.32,52–54 Therefore, it is important to improve the efficacy of DNA vaccines against tuberculosis and several strategies have been applied to address this objective.
Use of Vaccine Cocktails
Several groups have applied DNA vaccines in pools of up to 4 different vaccines (e.g., ESAT-6, MPT43, Ag85B, or ESAT-6, MPT63, MPT64, KatG,22,55). Some combinations gave improvements in protection compared to the individual vaccines. However, BCG was still the most protective vaccine in these studies (Table 2A). Interestingly, formulating Ag85B and MPT64 together with the cytokine GM-CSF did not further improve protection, despite increases in cellular immunity.56
Table 2
Strategies to increase efficacies of tuberculosis DNA vaccines.
Protein Modifications
DNA vaccines have been constructed to give either enhanced antigen secretion from mammalian cells (by the addition of the tPA secretion signal peptide23,28), or to give enhanced proteosome-dependent degradation of the endogenously produced proteins (by conjugation to ubiquitin57). Both strategies have resulted in improved protective efficacies (Table 2B), although the modified vaccines were still inferior to BCG.
Heterologous Prime-Boost Strategies
Heterologous prime-boost strategies have been adopted in the quest for improved TB vaccines and sometimes resulted in protection superior to BCG vaccination. These protocols were based on the use of DNA vaccination to prime immune responses, followed by recombinant proteins, recombinant viruses, or BCG itself to boost the immune response. Examples of such experiments are summarized in Table 2C.
Boosting Ag85A DNA vaccinated mice with recombinant Ag85A resulted in improved protection compared to DNA or protein alone.58 In these studies, two different adjuvants were used to deliver the protein boost: MPL and SBAAS2A. Boosting with the latter adjuvant resulted in the same level of protection as conferred by BCG vaccination, whilst the protection achieved by boosting with protein combined with MPL, although better than DNA alone, was not as good as BCG (Table 2C). This improved protective efficacy was characterized by increased IFN-γ and IL-2 responses.58 Similarly, when we vaccinated cattle with HSP65 DNA and then boosted the responses with recombinant protein in incomplete Freund's adjuvant (IFA) there were increased and more homogenous IFN-γ and T cell proliferative responses and a more balanced IgG1/IgG2 ratio (Vordermeier and Hewinson, unpublished results). Challenge infections to assess the protective efficacy of this vaccine protocol in cattle are now underway (Table 2C).
McShane and co-workers59 have used recombinant viruses to boost DNA primed responses. Mice that had been primed by 3 vaccinations with DNA expressing a fusion protein of ESAT-6 and another M. tuberculosis antigen, MPT63, were boosted with the same fusion protein expressed by recombinant modified vaccinia virus Ankara (MVA). This resulted in significantly increased frequencies of CD4 IFN-γ secreting cells and protection equivalent to BCG vaccination (Table 2C).
Sequential immunization with an Ag85B expressing DNA vaccine followed by BCG boosting has been found to be more effective than BCG vaccination alone.60 The protective effect of this heterologous prime-boost protocol was superior to the effect seen in mice that were vaccinated twice with BCG (“homologous boost”). The improved protection was at least partially mediated by CD8 T cells (Table 1C). We have recently performed an experiment in which cattle were primed twice with a mixture of DNA plasmids expressing HSP65, HSP70 and Apa) before BCG vaccination and found that the degree of pathology in these animals after M. bovis challenge was reduced compared to BCG vaccination alone. The DNA vaccination alone did not result in significant protection (Table 2C, Vordermeier, Hewinson and Buddle, unpublished results).
It is likely that advances will be made with additional strategies, such as optimizing CpG motifs,61 identification of new antigens by sequence comparisons with mycobacterial genome sequences,62,63 analysis of in vivo antigen expression and using epitope prediction algorithms.64 Codon-optimized epitopes of novel antigens could be readily incorporated into existing DNA vaccines.
Characterization of Immune Responses after DNA Vaccination
It is not known with certainty what immune responses equate with protection in tuberculosis. As highlighted in Table 1, DNA vaccination generally induces strong antigen-specific T cell proliferative and type 1 cytokine responses and IFN-γ and class I-restricted CD8 CTL responses tend to be prominent. The level of IFN-γ produced early after infection is certainly one parameter observed in protective immunity resulting from DNA vaccination of mice.65,66 The breadth of the profile of epitopes recognized by both CD4 and CD8 T cells may be another. For example, CD4 T cells from PstS-1 DNA vaccinated BALB/c mice recognized epitopes on 7 different peptides compared to 5 recognized following M. tuberculosis infection;20,67 CD4 T cells from M. tuberculosis-infected mice recognized a single Ag85A-derived epitope whereas 8 were recognized in DNA vaccinated mice.68 Similarly, the CD8 T cell epitope repertoire of DNA vaccinated mice was also strikingly different and broader than that of M. tuberculosis-infected mice (Ag85A: 3 vs. 0 epitopes, PstS-1: 3 vs. 1 epitope, in DNA vaccinated and M. tuberculosis-infected mice, respectively20,67,68). The induction, through DNA vaccination, of T cells that are specific for epitopes that are subdominant or cryptic during natural infection, can have a significant impact on protective immunity. For example, it has been recently shown that effective protection could be obtained after vaccination with a peptide comprising a subdominant epitope of ESAT-6 that gave only negligible responses during M. tuberculosis infection, whereas vaccination with a peptide that was a dominant epitope of ESAT-6 during infection was not protective.69
Several studies have attempted to define the T cell subsets responsible for protecting mice after DNA vaccination. D'Souza and co-workers70 vaccinated a range of CD4, β2-microglobulin and IFN-γ gene-disrupted mouse strains with an Ag85A DNA vaccine. They demonstrated that mice lacking CD4 T cells or unable to produce IFN-γ could not be protected against tuberculosis, whereas β2-microglobulin knock-out mice effectively controlled the disease when vaccinated with the DNA vaccine. These experiments therefore confirmed the importance of IFN-γ produced by CD4 T cells in providing protective immunity against tuberculous infection in mice. The authors concluded that such cells mediate the protective effect of this vaccine independently of CD8 T cells. However, their experiments did not rule out a role for CD8 T cells in Ag85A-mediated protection. Mice with an MHC H-2b were used and no Ag85A-specific CD8 T cell responses were demonstrable, even in wild-type mice.70 This experiment should therefore be repeated in mice of H-2d background where strong CD8 T cell responses were reported after Ag85A vaccination.
The importance of CD8 T cells that both secrete IFN-γ and lyse M. tuberculosis-infected cells in vitro, has been demonstrated in a series of experiments analyzing T cell clones prepared from M. leprae HSP65 DNA or J774-HSP65 vaccinated mice.71–73 These CD8CD44hi activated/memory T cells were able to adoptively transfer protection into naïve recipients. IFN-γ producing, cytolytic CD4CD44hi clones were also able to transfer protection, although they were less potent than the CD8CD44hi cells. In contrast, CD8CD44hi T cells that produced IFN-γ but were not cytolytic were unable to transfer protection. Furthermore, adoptive transfer of protection correlated with the ability of the clones to use the cytotoxic granule-mediated pathway to lyse infected targets; in other systems, T cells that lyse targets by apoptosis may also contribute to mycobacterial killing.74
A key question to be addressed is how long protective immunity will persist after DNA vaccination. The rest periods between vaccination and mycobacterial infection, in most cases, do not exceed 10 weeks, and are more frequently around 4–6 weeks. However, a study by Silva and colleagues indicated that mice were still protected after long periods post-DNA vaccination prior to challenge.73 They vaccinated mice with a DNA vaccine expressing mycobacterial HSP65 and challenged them after rest periods of 1, 4, 8, and 15 months. Encouragingly, they demonstrated that mice were significantly protected even if they were challenged after a 15 months rest period. The same was observed for BCG vaccinated mice but the degree of protection after either DNA or BCG vaccination was lower than expected at all intervals. However, CD44hi T cells that produced high levels of IFN-γ and were cytolytic were found in these mice throughout the 15 months rest period in the DNA vaccinated mice. It is encouraging to demonstrate such long-lived memory in rodent models, but it will obviously be important to determine how DNA-vaccine induced memory responses can be sustained in target species. We have addressed this recently in cattle by studying the kinetics of the immune response following immunization with DNA encoding the M. leprae HSP65. In these studies we determined T cell proliferation and IFN-γ production after vaccination and demonstrated potent proliferative T cell and IFN-γ responses in vitro 2 weeks after the final DNA vaccination (Fig. 2). However, after a 6 weeks rest period, we detected only low frequencies of IFN-γ producing cells and low levels of in vitro produced IFN-γ, whereas we were still able to demonstrate strong HSP65-specific T cell proliferation. This seemingly paradoxical finding is completely in line with current concepts of memory T cell development in mice and humans (e.g., see refs. 75–77): The frequency of IFN-γ producing effector cells declines with the decrease in antigen stimulus over time, to be replaced with low frequencies of effector memory cells producing cytokines like IFN-γ, and central memory cells producing mainly IL-2. It is likely that the latter subset of T cells were those that proliferated but did not produce IFN-γ after the prolonged rest periods. Consistent with this hypothesis, IFN-γ producing effector cells were induced within 3 days of the subcutaneous injection of HSP65 protein (as judged by the amount of and frequency of IFN-γ cells found in the draining lymph nodes of these cows, Fig. 2). Therefore DNA vaccination against tuberculosis in rodent models as well as in a target species is able to induce long-lasting memory responses of the expected type. In conclusion, DNA vaccination against mycobacterial infections has been a potent tool to define protective antigens and to study aspects of protective immunity. It has now reached a stage of development where studies in target species are opportune. In particular we look forward to the exploitation in target host species of strategies like heterologous prime-boost protocols that have improved vaccine efficacies compared to BCG and to the immunotherapeutic applications of DNA vaccines in which role BCG vaccination is not effective.

DNA Vaccines against Bacterial Diseases Other Than Those Caused by Mycobacterial Species
Bacillus anthracis
Bacillus anthracisis the causative agent of anthrax, a potentially lethal gastrointestinal and respiratory disease of domesticated and wild animals, as well as humans. Virulent strains of B. anthracis are characterized by their expression of a polyglutamic acid capsule and the production of a protein toxin. The anthrax toxin has three components (protective antigen, lethal factor and edema factor), although toxicity can be mediated experimentally using a combination of protective antigen (PA) and lethal factor (LF) alone. The PA of anthrax toxin binds to the target cell permitting access of the lethal and edema factors to the cytosol. The current licensed vaccine against anthrax contains PA adsorbed onto aluminum hydroxide. Antibodies to PA prevent binding to the target cell and confer protection from anthrax.
To our knowledge there has only been one report of a DNA immunization approach to anthrax vaccination.78 Mice immunized with a plasmid encoding the immunogenic portion of PA-generated splenocytes that secreted IFN-γ and IL-4, consistent with the generation of a mixed Th1/Th2-like response. Immunized mice produced a serum IgG response to PA able to neutralize the biological activity of the protein in vitro and were protected from lethal challenge with a combination of PA and LF. The level of protection varied from 75–100%, depending on the dose of lethal factor administered. Complete protection to five lethal doses of toxin was obtained. It is hoped that a DNA vaccine against anthrax may be safer and more efficacious than the current licensed vaccine.
Borrelia burgdorferi
Borrelia burgdorferi is one of the members of the Borrelia genus of spirochetes responsible for Lyme disease, a chronic disease with arthritic and neurological sequelae if not diagnosed and treated adequately early in infection. The Outer Surface Protein A (OspA) of B. burgdorferi is the most frequent antigen component of vaccines under development for Lyme disease. Mice immunized with DNA expressing OspA generated a serum immunoglobulin response79,80 that was dependent on both the dose of DNA administered and the promoter used to drive expression of OspA.80 Interestingly, antibodies to OspA were generated following immunization with plasmid containing OspA in the reverse orientation,80 suggesting that the upstream control region of OspA was active in eukaryotic cells, a fact confirmed in vitro.80 All isotypes of murine IgG were generated to OspA DNA immunization, consistent with the proliferative response of spleen and lymph node T-cells to OspA and the production of IFN-γ.80 Vaccinated mice were protected from spirochete challenge.79,80 Immune serum was demonstrated to inhibit growth of B. burgdorferi in culture,79 as well as to confer protection to SCID mice against the onset of clinical arthritis following spirochete challenge.80 Compared with OspA, DNA vaccination with OspB stimulated a lower titer of serum immunoglobulin and failed to protect mice from challenge, correlating with the inability of the immune serum to inhibit spirochete growth in culture.79
Brucella abortus
Brucella sp. are Gram-negative facultative intracellular coccobacilli that infect both animals and humans. Brucella may enter the body via the skin, respiratory tract, or digestive tract. Once there, they can enter the blood and the lymphatics where they multiply inside phagocytes and eventually cause bacteraemia. Symptoms of Brucellosis vary from patient to patient but can include high fever, chills, and sweating. The four species of this genus that can infect humans are principally named after the animal in which they are most commonly found: B. abortus (cattle), B. suis (swine), B. melitensis (goats), B. canis (dogs). B. abortus causes spontaneous abortion and infertility in cattle and is the most common species associated with Brucellosis.
There is currently only one report of a DNA vaccination approach to Brucellosis.81 In that study, mice were vaccinated once with DNA expressing the ribosomal protein L7/L12, the immunodominant protein of B. abortus identified in primed cattle.82 Mice responded to vaccination by splenocyte proliferation and a serum IgG response specific for L7/L12. When challenged 28 days later, vaccinated mice had 0.47 to 1.26 log less bacteria in their spleens compared with controls, depending on the time postinfection. Thirty days postinfection, the protection induced by DNA vaccination was equivalent to that achieved with the live Brucella vaccine-B. abortus strain 19.
Chlamydia spp
There are four species of Chlamydia recognized to date: Chlamydia pecorum, Chlamydia pneumoniae, Chlamydia psittaci and Chlamydia trachomatis. All are obligate intracellular pathogens, causing disease primarily through induction of the host immune response. This feature has hampered efforts to develop a protective vaccine against chlamydial infection. Humans are the predominant host for C. pneumoniae and C. trachomatis, whereas C. pecorum and C. psittaci can infect a variety of hosts, including humans. At least C. trachomatis and C. psittaci can be subdivided into biovariants, depending on both the disease they cause and the host they infect. The four biovariants of C. trachomatis cause trachoma or sexually transmitted infections in humans, mouse pneumonitis, or infections of swine, respectively. Similarly, four biovariants of C. psittaci have been defined, responsible for infections in guinea pigs, cats, ruminants, or birds, respectively. Most chlamydial species can infect the respiratory tract, although C. pneumoniae is the most common agent of chlamydial pneumonia, a potentially life-threatening condition with systemic sequelae. Infection of birds and farm mammals with Chlamydia can result in significant economic losses, as well as presenting an occupational zoonotic hazard. The prevalence of Chlamydia, together with the emergence of antibiotic-resistant isolates, provides the impetus to develop vaccines against chlamydial disease. Candidate antigens for incorporation into a vaccine, include the Major Outer Membrane Protein (MOMP), a cysteine rich outer membrane protein (Omp2), Heat Shock Protein 60 (HSP60), and ADP/ATP translocase (Npt1Cp). Determination of the genome sequences of C. trachomatis and C. pneumoniae 55,83 has resulted in opportunities for the empirical testing of open reading frames (ORFs) in the context of DNA immunization.84
C. pneumoniae
Intranasal vaccination of C57Bl/6 mice with a DNA plasmid encoding the HSP60 of C. pneumoniae did not generate an anti-HSP60 serum IgG response. However, seven days after intranasal infection with C. pneumoniae, the vaccinated mice had 5–20-fold less bacteria in their lungs as well as milder pneumonia, compared with controls.85 In contrast, intradermal immunization with the same plasmid elicited a serum IgG response to HSP60 but failed to protect mice from intranasal challenge.85 Mice immunized with the HSP60 plasmid and subsequently challenged showed enhanced expression of IFN-γ in both the lung and from cultured splenocytes, compared with non-immunized challenged controls. Protection was further enhanced by the co-administration of plasmid expressing IFN-γ or IL-12, but not GM-CSF, at the time of vaccination. A protective role for IFN-γ in this model was further demonstrated using IFN-γ receptor-deficient mice. Protection was also shown to be dependent on CD4 and CD8 positive T-cells. Induction of CD4 cells by DNA immunization in the absence of CD8 cells led to a worsening of the outcome following challenge.
A similar intranasal challenge model against the BALB/c background was used to screen eight ORFs of C. pneumoniae expressed individually as DNA vaccines.84 Two constructs expressing the ORFs encoding MOMP and Npt1Cp gave protection, whereas those expressing ytfF, GltL, pml116, DnaK, ndk, and dagA did not. The utility of this approach was revealed in the confirmation of MOMP as a protective antigen in chlamydial infections,86–91 as well as the identification of a novel target antigen, Npt1Cp.
A recent study of intramuscular DNA immunization with plasmids encoding HSP60, MOMP, or Omp2 confirmed the lack of correlation between a serum immunoglobulin response and protection against intranasal challenge with C. pneumoniae.92 This study is noteworthy in that it looked at the effect of immunization with a cocktail of all three plasmids. Although a proliferative response was seen to each individual antigen, no significant protection was obtained with the cocktail. In fact, significant protection was demonstrated only following immunization with DNA encoding MOMP and HSP60, and then in only one experiment.
C. psittaci
Two studies describe the efficacy of DNA immunization for C. psittaciinfection of turkeys.86,87 In both, animals were immunized with DNA encoding the MOMP of an avian C. psittaci strain (Serovar A). Two inoculation strategies were evaluated: combined parenteral and mucosal delivery, and gene-gun based epidermal delivery. Both modes of delivery elicited T and B cell responses, as evidenced by the production of anti-MOMP serum immunoglobulin and enhanced proliferation of peripheral blood lymphocytes. All animals that received the DNA vaccine were protected against a high-dose aerosol challenge with C. psittaci. Protection was expressed in terms of a reduction or absence of signs of clinical disease, and of nasal shedding and tissue replication of organisms. No difference was observed in the relative protection afforded by the different routes of immunization. Antibody responses in vaccinated animals were weak and variable. Protection occurred in all vaccinated turkeys that had a serological response before challenge. The best protection was seen in turkeys that did not mount a secondary antibody response to challenge. Both modes of immunization generated antibodies of IgM, IgG and IgA isotype. Although IgG was the predominant isotype, levels of anti-MOMP IgG did not correlate with the level of protection, supporting observations made with C. pneumoniae in mice.85,92 The immunogenicity and efficacy of intraepidermal vaccination was dependent on the size of the gold beads used for gene-gun delivery and the quantity of plasmid DNA delivered.
C. trachomatis
The majority of studies of DNA vaccination for C. trachomatis have used the MOMP gene product as the vaccine antigen and the biovariant of C. trachomatis that causes mouse pneumonitis (MoPn) as the challenge organism.88–91 These studies reported variable levels of induction of an anti-MOMP serum immunoglobulin response. Where serum IgG responses were detected there was a bias to induction of the IgG2a isotype suggesting induction of a more Th1-like response. No serum IgA response was detected in the two studies where it was assayed.88,90 The induction of cellular immune responses was similarly variable, although studies reported the induction of splenocyte proliferation,90,91 IFN-γ production,90 and cutaneous DTH reactions following injection of C. trachomatis elementary bodies, the infectious extracellular forms of the organism.89,90 Induction of DTH was considered important since it has been reported to correlate with protection in the MoPn model.93 Measurement of the relative levels of expression of IL-10 and IFN-γ further supported the notion that DNA immunization with MOMP favored a Th1-type response.90 Following MOMP DNA vaccination, mice were most frequently challenged with MoPn intranasally. Generally, the levels of protection induced by MOMP DNA vaccination were encouraging, reaching as high as a 4 log reduction in lung bacterial burden88 and significant protection from weight loss.89,90 The level of protection was shown to depend on the route of immunization and the dose of DNA used.88,90 Interestingly, protection in the lung was also obtained when an auxotroph of Salmonella was used to deliver the MOMP DNA plasmid orally.88
In one study, three different strains of vaccinated mice were challenged via the physiologically relevant genital route.91 In this case DNA immunization induced weak cellular and humoral responses. No anti-MOMP vaginal antibodies were detected and vaccination had no effect on vaginal shedding of MoPn.91 The failure of DNA vaccination in this case could be attributable to the route of challenge, dose of DNA, or even the expression plasmid used. Two other C. trachomatis antigens have been evaluated in the MoPn model: cytosine triphosphate (CTP) and serine threonine kinase (STK). Protection was reported with the use of STK94 but not CTP.89
Only one study has been published using a human strain of C. trachomatis: serovar L2, responsible for the invasive sexually transmitted disease, lymphogranuloma venereum. The MOMP antigen was used again but humoral responses were weak to absent, even when the DNA plasmid had been engineered for increased expression of MOMP protein in vitro. Mice vaccinated by either the intramuscular, intradermal or intranasal route were reported to resist intranasal challenge with C. trachomatis longer than controls, although the data were not analyzed statistically and all vaccinated mice eventually succumbed to infection.95
Clostridium tetani
Two studies report the evaluation of DNA immunization for protection against tetanus toxin, the potent neurotoxin synthesized by the anaerobic bacterium Clostridium tetani.96,97 In both studies, BALB/c mice were immunized intramuscularly with plasmids encoding the non-toxic C-terminal domain of tetanus toxin (fragment C). Anti-fragment C serum immunoglobulin responses were obtained, as well as proliferative responses by cultured splenocytes. Evaluation of the IgG subclasses and cytokines produced by the splenocyte cultures suggested that DNA immunization induced a Th1-like response. In contrast, immunization with tetanus toxoid or a polypeptide of fragment C induced a Th2-like response. In most cases, the serum immunoglobulin response following DNA immunization was sufficient to protect 70–100% of mice from lethal challenge with tetanus toxin. The level of protection was dependent on the amount of DNA used for immunization and the challenge dose. Whilst encouraging, the level of protection conferred by DNA immunization was inferior to that achieved with conventional toxoid or a polypeptide of fragment C.
Corynebacterium pseudotuberculosis
Corynebacterium pseudotuberculosis is the causative agent of caseous lymphadenitis (CLA); a chronic infectious disease of sheep and goats that results in abscesses of the lymph nodes, and occasionally, the internal organs. C. pseudotuberculosis may also cause ulcerative lymphangitis, a mildly contagious disease of horses characterized by inflammation of the lymphatic vessels of the lower limbs. CLA is highly contagious and causes significant economic loss to affected premises. Infection of humans with C. pseudotuberculosis is rare and is most often associated with skinning infected animals or the consumption of unpasteurized milk from such animals.
A DNA vaccination trial against CLA was recently conducted in sheep98 and used the novel approach of directing the antigen to sites of immune induction by expressing it as a fusion protein with bovine CTLA-4. This approach had previously been shown to enhance both the speed and magnitude of the immune response to human immunoglobulin.99 The phospholipase D gene of C. pseudotuberculosis was genetically detoxified (PLD) and cloned into two DNA plasmids, designed (and confirmed in vitro) to secrete PLD with or without targeting to CD80/86-positive, antigen-presenting cells. A significant antibody response was generated only in those sheep vaccinated with DNA expressing the targeted CTLA-4-PLD fusion. Animals were challenged above the coronet with 106 C. pseudotuberculosis six weeks after the primary immunization and killed six weeks later. The lymph nodes draining the site of challenge were inspected post mortem for the presence of abscesses. A proportion of animals that received either PLD construct as DNA vaccine were completely protected from abscess formation: 45% in the case of the non-targeted vaccine; 70% with the targeted vaccine. This compared favorably to the level of protection (90%) obtained with a commercial vaccine comprised of formalinin-activated toxins and PLD.
Enterotoxigenic Escherichia coli
Enterotoxigenic Escherichia coli (ETEC) is one of the most common causes of acute diarrhea in children in developing countries and in travelers who visit those areas. ETEC is also one of the most important pathogens of pigs, causing diarrhea, edema disease or colisepticaemia. Essential virulence factors for the pathogenicity of ETEC include the enterotoxins and the Colonisation Factor Antigens (CFAs), which comprise fimbrial and nonfimbrial adhesins responsible for the adhesion of ETEC to enterocyte receptors. Studies of DNA vaccination for ETEC are restricted to the CFA/I fimbriae of human ETEC serogroups (encoded by cfaB) and FaeG, the major protein subunit of K88 fimbriae expressed by a subset of pig ETEC serogroups. Mice given a single inoculation of DNA expressing the CFA/I fimbrial adhesin mounted a serum IgG response, although the antibodies were unable to agglutinate bacterial cells or inhibit the haemagglutination (HA) promoted by CFA/I-bearing ETEC.100 The serological response to CFA/I DNA vaccination was subsequently shown to be predominantly of the IgG2a isotype, compared with the predominantly IgG1 response induced by immunization with purified CFA/I protein subunits.101 This group went on to show that the magnitude, isotype, and specificity of the serological response to CFA/I was dependent on the number of DNA inocula administered102 as well as the plasmid used. Fusion of the cfaB gene to the tissue plasminogen activator protein (tPA) signal sequence resulted in the secretion of CFA/I protein from transfected cells103 and vaccination with this construct induced serum IgG capable of inhibiting the HA of CFA/Ipositive bacteria.103,104 Cellular responses to CFA/I protein were demonstrable with all plasmids, but not IgA in gut mucosa or feces.104 In order to generate mucosal IgA mice were primed with CFA/I plasmid DNA, followed by intragastric inoculation with recombinant Salmonella expressing CFA/I protein. The combined regime of DNA prime-Salmonella boost, generated serum IgG and fecal IgA responses greater than achieved with either DNA or recombinant Salmonella alone.105
In the pig ETEC study,106 mice immunized with DNA encoding the faeG gene of ETEC K88ab produced a serum IgG2a response. Three pregnant sows were subsequently immunized with 200μg, 400μg or 1128μg total DNA. Neither of the sows receiving the lower doses of DNA seroconverted, although an antibody response was reported in the colostrum of the first sow. A serum IgG response was detected in the sow that received the highest dose of DNA, although it aborted 48 hours after the second DNA inoculation. Antibody titers in the surviving offspring were reportedly higher than those of their respective mothers, although these data were not presented in the paper.
Francisella tularensis
Francisella tularensis is the causative agent of tularaemia. There are five forms of clinical tularaemia. The most common (80% of cases) is ulceroglandular tularaemia with symptoms of fever, headache, lymphadenopathy and characteristic ulceration of the site of inoculation. Tularaemia carries a mortality rate of 5–15%, even higher with the typhoidal form, but antibiotics lower this rate to about 1 %. Many wild animals and some domestic animals can harbor F. tularensis, although the rabbit is the species most often involved in transmission. Infection can be acquired through numerous routes. The most common routes include: inoculation of the skin or mucous membranes with blood or tissue while handling, dressing or skinning infected animals, contact with fluids from infected flies or ticks, the bite of infected ticks or handling or eating insufficiently cooked contaminated meat.
An attenuated F. tularensis live vaccine strain (LVS) is available for those most at risk of infection, so there has been little effort to develop a DNA vaccine for tularaemia. However, lethal infection of mice with F. tularensis LVS has been used as a model with which to study the protective host response to noneukaryotic DNA.107 BALB/c mice were injected with 0.01–20 μg chromosomal DNA isolated from F. tularensis and then challenged with different doses of F. tularensis at various time points after immunization. Protection was dependent on the dose of DNA administered, the challenge dose and time-to-challenge, but not the route of immunization. Protection was shown to be dependent on the unmethylated state of the bacterial DNA. Synthetic unmethylated CpG-rich oligonucleotides (CpG.ODN) are common in prokaryotic genomes but rare in eukaryotes. Their ability to induce the innate immune response is well documented, and as such, CpG.ODN have attracted much interest as mediators of vaccine enhancement (reviewed in 108; see section on Listeria monocytogenes). The importance of unmethylated CpG in F. tularensis DNA for the induction of short-term protection was demonstrated using CpG-containing ODN. Immunization with unmethylated CpG-ODN, but not methylated, conferred 100% protection against lethal challenge with either F. tularensis or L. monocytogenes. Using IFN-γ gene-knockout (GKO) mice, the protection obtained with either unmethylated F. tularensis DNA or CpG-ODN was shown to be dependent on IFN-γ, T-cells and critically, B-cells. Importantly, long-term pathogen-specific immunity was demonstrated in mice that had survived a previous lethal challenge through immunization with bacterial DNA.
Helicobacter pylori
Helicobacter pylori colonizes the gastric mucosa of more than half the world's population, causing asymptomatic gastritis in the majority. However, up to 10% of infected individuals develop clinical disease, ranging from gastric and duodenal ulcers to adenocarcinoma of the stomach and lymphoma of mucosaassociated lymphoid tissue. The current standard treatment for H. pylori infection consists of antibiotics administered with an inhibitor of gastric acid secretion. The increasing resistance of H. pylori to antibiotics makes vaccination an attractive alternative, although complete elimination of the organism may be undesirable given the possible association of H. pylori colonization with reduced incidence of esophageal diseases. The latter observation has likely tempered enthusiasm for H. pylori vaccination and, in fact, only one study has been published on a DNA approach to vaccination.109 Immunization of mice with plasmid encoding either the Heat Shock Protein (Hsp) A or B of H. pylori stimulated an antigen-specific serum IgG and IgA response. Differences in the relative levels of IgG to IgA and of IgG2a to IgG1 isotypes were observed between the response to Hsp A and Hsp B. DNA vaccination conferred protection against H. pylori replication in the stomachs of mice infected three months after vaccination and examined six months after challenge. Vaccinated mice also had less antral gastritis, as determined histologically. The protection afforded by Hsp B was greater than with Hsp A by both criteria.
Leptospira interrogans
Leptospirosis is the most widespread bacterial infection in the world, affecting both humans and animals. Zoonotic infection of humans with Leptospira interrogans responds well to treatment, but rarely infection may cause the acute life-threatening (Weil's) disease, resulting in jaundice and severe damage to the body organs. The protection conferred to guinea pigs by DNA vaccination with the seroreactive P68 antigen110 is, to our knowledge, the only published report of a DNA vaccine approach to leptospirosis. In that study, 77% of vaccinated guinea pigs were protected from death and 85% from pulmonary diffuse hemorrhaging (PDH), following challenge with L. interrogans. In contrast, 75% of guinea pigs vaccinated with vector plasmid alone died and nine out of ten had PDH post mortem.
Listeria monocytogenes
Listeria monocytogenes is the only one of seven species of Listeria that is pathogenic to humans. Diarrhea can follow ingestion of organisms via contaminated food, although infection is rare in normal populations. However, in individuals made susceptible by age, pregnancy, or immunosuppression, infection can lead to encephalitis, meningitis, stillbirth, and high mortality.
Infection of mice with L. monocytogenes is an established model for studying host antibacterial defense mechanisms (reviewed in ref. 111). The most extensively studied protective antigen in murine listeriosis is Listeriolysin O (LLO). Protection of BALB/c mice against challenge with L. monocytogenes can be mediated by CD8 T cytotoxic lymphocytes that recognize an H-2Kd restricted epitope from amino acids 91 – 99 of LLO.112 It is not surprising, therefore, that the majority of studies on DNA vaccination against L. monocytogenes have included this antigen. However, with the possible exception of Mycobacteria, the strategies adopted for DNA vaccination against L. monocytogenes are amongst the most diverse and novel.
The first publication of a DNA vaccination approach to L. monocytogenes described the novel use of an attenuated strain of Salmonella to deliver DNA vaccine plasmids via the oral route.113 In this way, plasmids encoding truncated L. monocytogenes genes actA and hly (encoding LLO) were most likely transferred from the Salmonella organisms to host antigen-presenting cells, resulting in CD8CTL activity, CD4 T-cell proliferation and the release of IFN-γ, as well as serum IgG and IgA responses.113 Mice vaccinated with recombinant Salmonella containing the hly plasmid, but not the actA plasmid, were protected from a 10 × LD50 challenge with L. monocytogenes.
Despite the protective nature of the 91–99 epitope of LLO, vaccination of BALB/c mice with DNA encoding the epitope failed to induce an immune response or confer protection against challenge.114 However, when the plasmid was reconstructed so that the codon usage of the epitope was optimized for expression in mice, a CTL response was induced, mediated by IFN-γ-secreting CD8 T lymphocytes.114 Furthermore, immunization with the optimized 91–99 epitope plasmid gave partial protection against sublethal challenge with L. monocytogenes. This study emphasized that the optimization of codon usage to improve the efficiency of antigen gene translation in the target species is an important consideration in constructing DNA vaccines against nonviral pathogens. Using DNA constructs containing other epitopes of LLO, these workers demonstrated further that the magnitude of protection was related to the level of CTL induction.115
In contrast to the protective efficacy observed in the studies using Salmonella as a surrogate vector for DNA vaccines described above, a study using the whole hly gene for intramuscular DNA vaccination found the immune response to be poor and unable to confer significant protection against challenge.116 Since LLO encoded by hly is a cytotoxin, these workers reasoned that expression of whole LLO by antigen-presenting cells might result in autotoxicity and failure to induce protective immunity. A variety of plasmids were therefore constructed, expressing either mutant versions of LLO with reduced toxicity, or engineered to secrete the wild-type or mutant LLO (by virtue of fusion to the murine tPA signal sequence). In adoptive transfer protection experiments, the mutant form of LLO gave a ˜0.6 log improvement in protection, the secreted wild-type form a ˜0.3 log improvement, and the secreted mutant form a ˜1.9 log improvement compared with the nonsecreted wild-type form of LLO.116 However, in the vaccinated animal itself, only the secreted mutant form of LLO conferred protection to challenge.
Gene-gun immunization using LLO and p60 of L. monocytogenes has been used to examine the ability of CpG-ODN to act as an adjunct to DNA immunization, in addition to boosting or co-administration of plasmid encoding GM-CSF.117 An optimal vaccination schedule utilized LLO with or without p60 plus co-injection of CpG-ODN. With this schedule over 3 log protection was obtained, representing sterile elimination of L. monocytogenes from the spleen of challenged mice117 and demonstrating that CpG-ODN can improve suboptimal vaccination protocols. In another study, administration of CpG-ODN alone to mice was sufficient to confer 100 % protection against lethal challenge with L. monocytogenes three days later.107
Mycoplasma pulmonis
Mycoplasma are minute pleomorphic Gram-negative microorganisms without cell walls that are intermediate in some respects between viruses and bacteria.Mycoplasma pneumoniae is an important human pathogen and mycoplasmoses among farm animals are responsible for considerable economic losses. Murine respiratory mycoplasmosis caused by M. pulmonis is one of the most important pathologies for laboratory rodents and is comparable to the pathological manifestations of M. pneumoniae in humans and of other animal respiratory mycoplasmoses. The experimental infection of mice with M. pulmonis has been used as a model for two novel approaches to DNA vaccination.118,119 The first was termed Expression Library Immunization (ELI), in which an expression library of M. pulmonis DNA was used to vaccinate mice in order to identify protective antigens in an empirical fashion. The small genome size of M. pulmonis was ideal to prove the principle behind this approach. The second, and only other report of DNA vaccination against Mycoplasma, was to our knowledge the first to demonstrate the therapeutic potential of DNA immunization.
Using the ELI approach, the entire M. pulmonis genome was represented in each reading frame by nine libraries of about 3000 members. Injection of two of the nine libraries intradermally into groups of mice resulted in serum antibody and DTH responses to M. pulmonis antigens. Complete protection to M. pulmonis challenge was obtained with either library.118 A Listeria monocytogenes ELI library was also constructed and used a negative control. Protection against L. monocytogenes challenge was not examined. The advantage of the ELI approach is that a DNA vaccine can be constructed without any prior knowledge of the pathogen's biology or of the suitable target genes to use for immunization. However, the approach may only be suitable for pathogens with genomes of manageable size and ultimately the genes responsible for protection must be dissected from the library and tested individually.
The therapeutic potential of DNA immunization in bacterial infections was reported twice, first for M. pulmonis119 and subsequently, for Mycobacterium tuberculosis.45,46 The latter papers are reviewed earlier in this chapter. In the M. pulmonis study, cellular and humoral immune responses were generated in mice immunized with DNA encoding M. pulmonis antigens A7-1 or A8-1, administered either singly or in combination. For the protection study, mice received a combination of A7-1 and A8-1 either one week prior to M. pulmonis challenge (vaccination) or one week after (therapy). Further immunizations were administered two more times at two-week intervals after the first immunization. Mice were killed monthly after infection and the extent of disease determined histopathologically and by enumerating bacteria in tracheo-lung lavage. Following either vaccination regime, the number of M. pulmonis recovered by lavage decreased monthly until no bacteria were cultured at four months. The continued reduction in CFU was accompanied by reduction in the severity of lung inflammation.
Pseudomonas aeruginosa
Pseudomonas aeruginosa is an opportunistic pathogen, commonly associated with cystic fibrosis patients, immunocompromised individuals, including AIDS patients, and humans and animals that have a breach in their skin barrier caused by burn or wounding. Infection with P. aeruginosa is a significant cause of death in domestic animals. A number of virulence factors are secreted by Pseudomonas. The most toxic protein produced by Pseudomonas is Exotoxin A, which shares its mode of action (ADP-ribosylation) with diphtheria toxin. Neutralization of Exotoxin A toxicity by DNA immunization was the aim of two recent studies in which the coding sequence for non-toxic mutant forms of P. aeruginosa Exotoxin A were cloned into DNA expression vectors.120,121 Both studies reported a serum immunoglobulin response to Exotoxin A following DNA immunization. Analysis of the IgG isotype and cytokine secretion by splenocytes suggested that vaccination favored a Th1-type response.121 DNA vaccinated mice in both studies were completely protected from the lethal effect of intraperitoneal injection with wild-type Exotoxin A, consistent with the demonstration of Exotoxin A neutralization by immune sera in vitro.121
While neutralization of Exotoxin A activity following DNA immunization would not be expected to prevent infection with P. aeruginosa, it might prevent certain pathology associated with infection. The coding sequence for a non-toxic mutant form of Exotoxin A could, therefore, be an important constituent of a multicomponent P. aeruginosa DNA vaccine. An additional candidate for such a vaccine cocktail could be the oprF gene of P. aeruginosa, encoding Outer Membrane Protein F. Immunization with this protein has been shown to confer some protection in a variety of rodent models of P. aeruginosa infection.122–124 Mice immunized with a DNA plasmid encoding oprF generated an IgG1 serum response to both OprF and OprH of the homologous strain of P. aeruginosa, and to heterologous immunotypes.125 The immune serum also mediated opsonophagocytic uptake of P. aeruginosa by peripheral blood mononuclear cells in culture. In a chronic pulmonary infection model, DNA vaccinated mice showed a significant reduction in the number of severe lung lesions and the number of mice with lungs yielding >5000 CFU of P. aeruginosa compared with controls.125
Salmonella typhi
Salmonella typhi causes typhoid fever, a severe systemic disease characterized by fever and abdominal symptoms, including hemorrhage, which is the principle cause of death in such infections. If untreated, typhoid fever has a 10–20% mortality rate. S. typhi infects only humans and chimpanzees, making study of the infection difficult. However, under certain experimental conditions it is possible to infect animals such as mice and mimic some aspects of the natural infection. The murine model has been used to evaluate a DNA immunization approach to prevent S. typhi infection.126 In these studies, BALB/c mice injected with DNA expressing the Outer Membrane Protein C (OmpC) Porin of S. typhi produced a serum IgG response specific to the protein. Responses were greater to OmpC expressed with its leader sequence than without. Since vaccination of mice with recombinant OmpC induces protection from S. typhi in mice.127 The preliminary DNA immunization results were taken as encouraging, although to our knowledge, no further studies have been reported.
Staphylococcus aureus
Contamination of food by toxins produced by Staphylococcus aureus is the major cause of food-borne intoxication. Symptoms are associated with acute gastroenteritis and mortality is less than 1%. Since S. aureus is part of the normal respiratory and skin flora of approximately 30–40% of people collectively, contamination of food with S. aureus most commonly originates from the foodhandler. More significantly, such transmission may occur nosocomially from hospital personnel to susceptible patients by direct contact. This is of particular concern with regard to the transmission of methicillin-resistant S. aureus (MRSA) to immunocompromised individuals. More than 90% of MRSA isolates produce the penicillin-binding protein PBP2', encoded by the mecA gene and carried on a foreign region of DNA integrated into the chromosome of MRSA. The source of mecA is not known but it may originate from other genera of bacteria or from within the Staphylococci.128 Possession of mecA and expression of PBP2' distinguishes MRSA from methicillin-susceptible isolates.
A DNA vaccination approach to prevent MRSA infection was reported recently.129 In this study, DNA encoding the mecA gene was used to immunize BALB/c mice three times at two-week intervals. Immunization resulted in a serum immunoglobulin response to PBP2', demonstrated by immunoblotting and ELISA. The immune serum enhanced the phagocytosis of PBP2'-positive MRSA by peritoneal macrophages. Fourteen days after the final immunization, the mice were challenged intravenously with 108 CFU MRSA 1191. Eight days after challenge, the kidneys were removed and the bacterial load determined. Vaccination with mecA plasmid gave a 0.4 log reduction in kidney CFU compared with controls, although this difference was significant statistically. This preliminary study suggests that PBP2' might be a target molecule for vaccination against MRSA.
Streptococcus pneumoniae
Despite the introduction of antibiotics, infection with Streptococcus pneumoniae still causes significant human mortality and morbidity through the induction of pneumococcal pneumonia, septicaemia, meningitis and otitis media. Most clinical isolates of S. pneumoniae express capsular polysaccharide that protects the organism from phagocytosis and represents the major virulence determinant of the organism. Ninety chemically and antigenically distinct capsular polysaccharides have been identified, forming the basis of the serotypic classification of S. pneumoniae. Recent efforts at vaccination have focused predominantly on the production of conjugate vaccines in which polysaccharide antigens are linked to a protein carrier in order to generate protective antibodies in recipients. DNA immunization against S. pneumoniae has not been rigorously pursued, although one study has reported the protection of mice from lethal pneumococcal septicaemia following immunization with a plasmid expressing pneumococcal surface protein A (PspA).130 PspA is expressed on the surface of all serotypes of S. pneumoniae and is required for full virulence. Only 30% of BALB/c mice immunized with the plasmid expressing PspA produced a serum immunoglobulin response to PspA protein, although the isotype of immunoglobulin was not reported. Interestingly, the generation of this response did not correlate with protection from challenge with 20 lethal doses of virulent S. pneumoniae. None of the mice generated a mucosal response to PspA, as determined serologically using saliva and feces. Immunization with the plasmid protected 40% of mice from death for up to 150 hours after challenge. All of the control mice were dead before 100 hours following challenge. Protection was also expressed in a significant reduction in the number of S. pneumoniae cultured from the blood. The efficacy of DNA immunization was less than that achieved with PspA protein, which had been shown to protect >90% of mice from challenge with more than one serotype of S. pneumoniae.131,132
Yersinia spp
The genus Yersinia contains three pathogenic species: Yersinia enterocolitica, Yersinia pseudotuberculosis and Yersinia pestis. The first two species cause food-borne illness. Y. enterocolitica causes gastroenteritis characterized by fever and abdominal pain often accompanied by diarrhea and/or fever. Y. pseudotuberculosis typically causes an acute mesenteric lymphadenitis. Y. pestis is the causative agent of ‘the plague’, one of the most devastating infectious diseases known to man. Even though the three species use different routes to infect their host and cause diseases of different severity, they share a common tropism for the lymphoid tissue and are able to resist the primary immune defense mechanisms of the host. Only one immunogenicity study has been reported using DNA encoding the V antigen of Y. pestis.133 Two challenge studies have been reported for Y. enterocolitica,134,135 the latter study also investigated the outcome of challenge with Y. pseudotuberculosis.135
Y. enterocolitica and Y. pseudotuberculosis
The Heat-shock Protein 60 (HSP60) of Y. enterocolitica is a protective antigen in mouse models of yersiniosis and invokes both the humoral and cellular arms of the immune response.136 Vaccination of mice with a DNA plasmid encoding HSP60 induced a Th1-type response characterized by an IgG2a serum antibody response, antigen-specific splenocyte proliferation and the liberation of IFN-γ, but not IL-4 or IL-10.134,135 Mice immunized with the HSP60 DNA vaccine were protected from challenge with a lethal dose of Y. enterocolitica administered either intravenously134,135 or orally135 For those mice infected orally, protection was expressed at the level of a reduction in bacterial burden in the spleen but not locally at the site of mucosal entry. By using knockout mice, protection was found to be dependent on both CD4 and CD8 T-cells.135
Protection of mice against yersiniosis requires a Th1-type response, characterized by the interplay of T-cells and macrophages, and the production of IFN-γ. Therefore, in an effort to augment the response of mice to DNA vaccination, the HSP60 DNA plasmid was engineered to coexpress the gene for IL-2, IL-4 or IFN-γ.134 In each case, the cytokine gene was separated from hsp60 by an internal ribosomal entry sequences (IRES) to facilitate equimolar expression of both the cytokine and HSP60. Coexpression of IFN-γ with HSP60 enhanced both splenocyte proliferation and the production of IgG. Coexpression of IFN-γ, but not IL-2 or IL-4, also enhanced protection against challenge by a further 1.58 log, compared with the HSP60 plasmid alone.
Since the HSP60 is highly conserved across bacterial genera, mice vaccinated with the HSP60 of Y. enterocolitica were also challenged with a variety of different bacteria, including Y. pseudotuberculosis, to determine whether any cross-protection had been generated following vaccination. No cross-protection was observed, even to Y. pseudotuberculosis.135
Y. pestis
The protective V antigen of Y. pestis137 was used recently in a DNA immunogenicity study,133 primarily as a model to examine the relative merits of gene-gun immunization over conventional needle inoculation for a DNA vaccine encoding a bacterial subunit protein. As such, immunization was not followed by challenge with Y. pestis. Immunization of mice by either method generated a serological response to the V antigen, although genegun mediated delivery was found to be more efficient.
General Conclusions
It may be considered remarkable that in almost every reported instance where DNA vaccination has been tested for protection against bacterial disease it has indeed given protection. That this is so, irrespective of the nature of the bacterium, its lifestyle and its mode of causing disease testifies to the power and potential of this approach. Obviously there may be bacterial targets against which the approach has been tested and so far failed. Negative results tend not to get published. Yet it would clearly be wrong to suppose that the technique will only be useful against intracellular pathogens because of an inherent bias towards cell-mediated immunity. Antibody responses to DNA vaccination have given good protection by opsonizing against S. aureus and by neutralizing anthrax, tetanus and pseudomonas toxins in mouse models. If and when the approach does fail one may anticipate that this will be due either to a lack of suitable antigens or to their inaccessibility in the target rather than to an inability of DNA vaccination to generate an appropriate response. In any case, techniques to selectively enhance responses to DNA vaccination in different directions are being developed, playing on the greatest strength of DNA vaccination, its versatility and the ease with which the DNA and the consequent responses can be modified.
As yet few DNA vaccines against bacteria have been evaluated in the final target host species, but encouraging results have been observed against psittacosis in turkeys and against tuberculosis in cattle. The big question, of course, is whether DNA vaccination will give responses that are strong enough or long-lasting enough to be practical in the real target species. Whether the prime-boost strategy will remain the best approach for strong, long-lasting responses, or if newer more sophisticated forms of DNA vaccination alone will eventually suffice remains to be seen.
One final note is due on therapeutic vaccination. Vaccines are not usually therapeutic and the implications of the observations that DNA vaccines could cure mycoplasma and tuberculosis infections in mice have yet to be fully explored. We do not fully understand how DNA vaccines do this, but at the very least they serve as proof of principle; even the established inadequate or detrimental immune responses in heavily infected animals can be turned around. Thus there is some hope that in future immune therapy against bacterial disease, whether based on DNA vaccination or not, will complement and reduce the use of antibacterial drugs. The multidrug-resistant bacteria threat will always be with us and DNA immune therapy deserves further investigation.
References
- 1.
- WHO Tuberculosis Fact Sheet No 104(http://www.who.org.)2000.
- 2.
- Maher D, Raviglione MC. The global epidemic of tuberculosis: A World Health Organization perspective In: D.Schlossberg, ed. Tuberculosis and nontuberculous mycobacterial infections Philadelphia, London, Toronto, Montreal, Sydney, Tokyo: W.B. Saunders Company,1999104–113.
- 3.
- Colditz GA. et al. Efficacy of BCG vaccine in the prevention of tuberculosis. Meta analysis of the published literature. JAMA. 1994;271(9):698–702. [PubMed: 8309034]
- 4.
- Brewer TF. Preventing tuberculosis with bacillus Calmette-Guerin vaccine: a meta analysis of the literature. Clin Infect Dis. 2000;31(Suppl 3):S64–7. [PubMed: 11010824]
- 5.
- Fine PE. The BCG story: lessons from the past and implications for the future. Rev Infect Dis. 1989;11(Suppl 2):S35–39. [PubMed: 2652252]
- 6.
- Rodrigues LC, Diwan VK, Wheeler JG. Protective effect of BCG against tuberculous meningitis and miliary tuberculosis: a meta-analysis. Int J Epidemiol. 1993;22(6):1154–1158. [PubMed: 8144299]
- 7.
- Cosivi O. et al. Epidemiology of Mycobacterium bovis infection in animals and humans, with particular reference to Africa. Rev Sci Tech. 1995;14(3):733–746. [PubMed: 8593405]
- 8.
- Cosivi O. et al. Zoonotic tuberculosis due to Mycobacterium bovis in developing countries. Emerg Infect Dis. 1998;4(1):59–70. [PMC free article: PMC2627667] [PubMed: 9452399]
- 9.
- Krebs JR. Bovine Tuberculosis in cattle and badgers London: Ministry of Agriculture, Fisheries and Food Publications,.1997. [PMC free article: PMC178798]
- 10.
- Andersen P. TB vaccines: progress and problems. Trends Immunol. 2001;22(3):160–168. [PubMed: 11286732]
- 11.
- Flynn JL, Ernst JD. Immune responses in tuberculosis. Curr Opin Immunol. 2000;12(4):432–436. [PubMed: 10899019]
- 12.
- Chan J, Kaufmann S H E. Immune mechanisms of protection In: B.R. Bloom, ed.Tuberculosis: Pathogenesis, protection, disease. Washington, DC: ASM Press,1994389–415. [PMC free article: PMC178708]
- 13.
- Orme IM, Cooper AM. Cytokine/chemokine cascades in immunity to tuberculosis. Immunol Today. 1999;20(7):307–312. [PubMed: 10379048]
- 14.
- Dannenberg A M Jr. Pathogenesis of pulmonary Mycobacterium bovis infection: basic principles established by the rabbit model. Tuberculosis. 2001;81(12):87–96. [PubMed: 11463228]
- 15.
- Vordermeier HM. et al. Increase of tuberculous infection in the organs of B cell deficient mice. Clin Exp Immunol. 1996;106(2):312–316. [PMC free article: PMC2200584] [PubMed: 8918578]
- 16.
- Silva CL, Lowrie DB. A single mycobacterial protein (hsp 65) expressed by a transgenic antigenpresenting cell vaccinates mice against tuberculosis. Immunology. 1994;82(2):244–248. [PMC free article: PMC1414830] [PubMed: 7927495]
- 17.
- Lowrie DB. et al. Towards a DNA vaccine against tuberculosis. Vaccine. 1994;12(16):1537–1540. [PubMed: 7879421]
- 18.
- Huygen K. et al. Immunogenicity and protective efficacy of a tuberculosis DNA vaccine. Nat Med. 1996;2(8):893–898. [PubMed: 8705859]
- 19.
- Tascon RE. et al. Vaccination against tuberculosis by DNA injection. Nat Med. 1996;2(8):888–92. [PubMed: 8705858]
- 20.
- Zhu X. et al. Functions and specificity of T cells following nucleic acid vaccination of mice against Mycobacterium tuberculosis infection. J Immunol. 1997;158(12):5921–5926. [PubMed: 9190945]
- 21.
- Orme IM, Andersen P, Boom WH. T cell response to Mycobacterium tuberculosis. J Infect Dis. 1993;167(6):1481–1497. [PubMed: 8501346]
- 22.
- Morris S. et al. The immunogenicity of single and combination DNA vaccines against tuberculosis. Vaccine. 2000;18(20):2155–2163. [PubMed: 10715531]
- 23.
- Li Z. et al. Immunogenicity of DNA vaccines expressing tuberculosis proteins fused to tissue plasminogen activator signal sequences. Infect Immun. 1999;67(9):4780–4786. [PMC free article: PMC96809] [PubMed: 10456931]
- 24.
- Lowrie DB. et al. Protection against tuberculosis by a plasmid DNA vaccine. Vaccine. 1997;15(8):834–838. [PubMed: 9234527]
- 25.
- Turner OC. et al. Lack of protection in mice and necrotizing bronchointerstitial pneumonia with bronchiolitis in guinea pigs immunized with vaccines directed against the hsp60 molecule of Mycobacterium tuberculosis. Infect Immun. 2000;68(6):3674–3679. [PMC free article: PMC97658] [PubMed: 10816527]
- 26.
- Svanholm C. et al. Enhancement of antibody responses by DNA immunization using expression vectors mediating efficient antigen secretion. J Immunol Methods. 1999;228(12):121–130. [PubMed: 10556549]
- 27.
- Ragno S. et al. Protection of rats from adjuvant arthritis by immunization with naked DNA encoding for mycobacterial heat shock protein 65. Arthritis Rheum. 1997;40(2):277–283. [PubMed: 9041939]
- 28.
- Baldwin SL. et al. Immunogenicity and protective efficacy of DNA vaccines encoding secreted and nonsecreted forms of Mycobacterium tuberculosis Ag85A. Tuber Lung Dis. 1999;79(4):251–259. [PubMed: 10692994]
- 29.
- Lozes E. et al. Immunogenicity and efficacy of a tuberculosis DNA vaccine encoding the components of the secreted antigen 85 complex. Vaccine. 1997;15(8):830–833. [PubMed: 9234526]
- 30.
- Tanghe A. et al. Immunogenicity and protective efficacy of tuberculosis DNA vaccines encoding putative phosphate transport receptors. J Immunol. 1999;162(2):1113–1119. [PubMed: 9916741]
- 31.
- Baldwin SL. et al. Evaluation of new vaccines in the mouse and guinea pig model of tuberculosis. Infect Immun. 1998;66(6):2951–2959. [PMC free article: PMC108294] [PubMed: 9596772]
- 32.
- Vordermeier HM. et al. Effective DNA vaccination of cattle with the mycobacterial antigens MPB83 and MPB70 does not compromise the specificity of the comparative intradermal tuberculin skin test. Vaccine. 2000;19(910):1246–1255. [PubMed: 11137264]
- 33.
- Lefevre P. et al. Three different putative phosphate transport receptors are encoded by the Mycobacterium tuberculosis genome and are present at the surface of Mycobacterium bovis BCG. J Bacteriol. 1997;179(9):2900–2906. [PMC free article: PMC179052] [PubMed: 9139906]
- 34.
- Sorensen AL. et al. Purification and characterization of a low-molecular-mass T-cell antigen secreted by Mycobacterium tuberculosis. Infect Immun. 1995;63(5):1710–1717. [PMC free article: PMC173214] [PubMed: 7729876]
- 35.
- Harboe M. et al. Evidence for occurrence of the ESAT-6 protein in Mycobacterium tuberculosis and virulent Mycobacterium bovis and for its absence in Mycobacterium bovis BCG. Infect Immun. 1996;64(1):16–22. [PMC free article: PMC173721] [PubMed: 8557334]
- 36.
- Andersen P. et al. Recall of longlived immunity to Mycobacterium tuberculosis infection in mice. J Immunol. 1995;154(7):3359–3372. [PubMed: 7897219]
- 37.
- Pollock JM, Andersen P. The potential of the ESAT-6 antigen secreted by virulent mycobacteria for specific diagnosis of tuberculosis. J Infect Dis. 1997;175(5):1251–1254. [PubMed: 9129098]
- 38.
- Vordermeier HM. et al. Development of diagnostic reagents to differentiate between Mycobacterium bovis BCG vaccination and M. bovis infection in cattle. Clin Diagn Lab Immunol. 1999;6(5):675–682. [PMC free article: PMC95753] [PubMed: 10473516]
- 39.
- Lalvani A. et al. Enumeration of T cells specific for RD1encoded antigens suggests a high prevalence of latent Mycobacterium tuberculosis infection in healthy urban Indians. J Infect Dis. 2001;183(3):469–477. [PubMed: 11133379]
- 40.
- Kamath AT. et al. Differential protective efficacy of DNA vaccines expressing secreted proteins of Mycobacterium tuberculosis. Infect Immun. 1999;67(4):1702–1707. [PMC free article: PMC96517] [PubMed: 10085007]
- 41.
- Yeremeev VV. et al. The 19kD antigen and protective immunity in a murine model of tuberculosis. Clin Exp Immunol. 2000;120(2):274–279. [PMC free article: PMC1905638] [PubMed: 10792376]
- 42.
- Erb KJ. et al. Identification of potential CD8 T-cell epitopes of the 19 kDa and AhpC proteins from Mycobacterium tuberculosis. No evidence for CD8 T-cell priming against the identified peptides after DNA vaccination of mice. Vaccine. 1998;16(7):692–697. [PubMed: 9562688]
- 43.
- Garapin A. et al. Mixed immune response induced in rodents by two naked DNA genes coding for mycobacterial glycosylated proteins. Vaccine. 2001;19(2022):2830–2841. [PubMed: 11282194]
- 44.
- Lefevre P. et al. Cloning of the gene encoding a 22-kilodalton cell surface antigen of Mycobacterium bovis BCG and analysis of its potential for DNA vaccination against tuberculosis. Infect Immun. 2000;68(3):1040–7. [PMC free article: PMC97246] [PubMed: 10678905]
- 45.
- Lowrie DB. et al. Therapy of tuberculosis in mice by DNA vaccination. Nature. 1999;400(6741):269–271. [PubMed: 10421369]
- 46.
- Turner J. et al. Effective preexposure tuberculosis vaccines fail to protect when they are given in an immunotherapeutic mode. Infect Immun. 2000;68(3):1706–1709. [PMC free article: PMC97334] [PubMed: 10678993]
- 47.
- Britton WJ. et al. Immunoprophylaxis against Mycobacterium leprae infection with subunit vaccines. Lepr Rev. 2000;71(Suppl:):S176–81. [PubMed: 11201878]
- 48.
- Martin E. et al. DNA encoding a single mycobacterial antigen protects against leprosy infection. Vaccine. 2001;19(1112):1391–1396. [PubMed: 11163661]
- 49.
- Martin E. et al. Protection against virulent Mycobacterium avium infection following DNA vaccination with the 35-kilodalton antigen is accompanied by induction of gamma interferon-secreting CD4(+) T cells. Infect Immun. 2000;68(6):3090–3096. [PMC free article: PMC97536] [PubMed: 10816448]
- 50.
- VelazFaircloth M. et al. Protection against Mycobacterium avium by DNA vaccines expressing mycobacterial antigens as fusion proteins with green fluorescent protein. Infect Immun. 1999;67(8):4243–4250. [PMC free article: PMC96731] [PubMed: 10417198]
- 51.
- Tanghe A. et al. Protective efficacy of a DNA vaccine encoding antigen 85A from Mycobacterium bovis BCG against Buruli ulcer. Infect Immun. 2001;69(9):5403–5411. [PMC free article: PMC98650] [PubMed: 11500410]
- 52.
- Kim SH, Cho D, Kim TS. Induction of in vivo resistance to Mycobacterium avium infection by intramuscular injection with DNA encoding interleukin18. Immunology. 2001;102(2):234–241. [PMC free article: PMC1783174] [PubMed: 11260329]
- 53.
- Chambers MA. et al. Vaccination of mice and cattle with plasmid DNA encoding the Mycobacterium bovis antigen MPB83. Clin Infect Dis. 2000;30(Suppl 3):S283–287. [PubMed: 10875801]
- 54.
- Chambers MA. et al. Vaccination of guinea pigs with DNA encoding the mycobacterial antigen MPB83 influences pulmonary pathology but not hematogenous spread following aerogenic infection with Mycobacterium bovis. Infect Immun. 2002;70:2159–65. [PMC free article: PMC127856] [PubMed: 11895982]
- 55.
- Kalman S. et al. Comparative genomes of Chlamydia pneumoniae and C.trachomatis. Nat Genet. 1999;21(4):385–389. [PubMed: 10192388]
- 56.
- Kamath AT. et al. Coimmunization with DNA vaccines expressing granulocyte-macrophage colony-stimulating factor and mycobacterial secreted proteins enhances Tcell immunity, but not protective efficacy against Mycobacterium tuberculosis. Immunology. 1999;96(4):511–516. [PMC free article: PMC2326784] [PubMed: 10233735]
- 57.
- Delogu G. et al. DNA vaccination against tuberculosis: expression of a ubiquitin-conjugated tuberculosis protein enhances anti-mycobacterial immunity. Infect Immun. 2000;68(6):3097–3102. [PMC free article: PMC97537] [PubMed: 10816449]
- 58.
- Tanghe A. et al. Improved immunogenicity and protective efficacy of a tuberculosis DNA vaccine encoding Ag85 by protein boosting. Infect Immun. 2001;69(5):3041–3047. [PMC free article: PMC98258] [PubMed: 11292722]
- 59.
- McShane H. et al. Enhanced immunogenicity of CD4(+) tcell responses and protective efficacy of a DNA-modified vaccinia virus Ankara primeboost vaccination regimen for murine tuberculosis. Infect Immun. 2001;69(2):681–686. [PMC free article: PMC97939] [PubMed: 11159955]
- 60.
- Feng CG. et al. Priming by DNA immunization augments protective efficacy of Mycobacterium bovis Bacille Calmette-Guerin against tuberculosis. Infect Immun. 2001;69(6):4174–4176. [PMC free article: PMC98488] [PubMed: 11349095]
- 61.
- Verthelyi D. et al. Human peripheral blood cells differentially recognize and respond to two distinct CPG motifs. J Immunol. 2001;166(4):2372–2377. [PubMed: 11160295]
- 62.
- Cole ST. et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393(6685):537–544. [PubMed: 9634230]
- 63.
- Cole ST. et al. Massive gene decay in the leprosy bacillus. Nature. 2001;409(6823):1007–1011. [PubMed: 11234002]
- 64.
- Meister GE. et al. Two novel T cell epitope prediction algorithms based on MHCbinding motifs; comparison of predicted and published epitopes from Mycobacterium tuberculosis and HIV protein sequences. Vaccine. 1995;13(6):581–591. [PubMed: 7483779]
- 65.
- Lyadova IV. et al. Intranasal BCG vaccination protects BALB/c mice against virulent Mycobacterium bovis and accelerates production of IFN-γ in their lungs. Clin Exp Immunol. 2001;125:274–279. [PMC free article: PMC1906185] [PubMed: 11703371]
- 66.
- Kamath AT. et al. Protective effect of DNA immunization against mycobacterial infection is associated with the early emergence of interferon-γ (IFN-γ) secreting lymphocytes. Clin Exp Immunol. 2000;120(3):476–482. [PMC free article: PMC1905572] [PubMed: 10844526]
- 67.
- Zhu X. et al. Specificity of CD8 T cells from subunit-vaccinated and infected H2b mice recognizing the 38 kDa antigen of Mycobacterium tuberculosis. Int Immunol. 1997;9(11):1669–1676. [PubMed: 9418128]
- 68.
- Denis O. et al. Vaccination with plasmid DNA encoding mycobacterial antigen 85A stimulates a CD4 and CD8 T-cell epitopic repertoire broader than that stimulated by Mycobacterium tuberculosis H37Rv infection. Infect Immun. 1998;66(4):1527–1533. [PMC free article: PMC108084] [PubMed: 9529077]
- 69.
- Olsen AW. et al. Efficient protection against Mycobacterium tuberculosis by vaccination with a single subdominant epitope from the ESAT-6 antigen. Eur J Immunol. 2000;30(6):1724–1732. [PubMed: 10898510]
- 70.
- D'Souza S. et al. CD4T cells contain Mycobacterium tuberculosis infection in the absence of CD8 T cells in mice vaccinated with DNA encoding Ag85A. Eur J Immunol. 2000;30(9):2455–2459. [PubMed: 11009076]
- 71.
- Silva CL. et al. Protection against tuberculosis by passive transfer with Tcell clones recognizing mycobacterial heat-shock protein 65. Immunology. 1994;83(3):341–346. [PMC free article: PMC1415049] [PubMed: 7835957]
- 72.
- Silva CL. et al. Characterization of T cells that confer a high degree of protective immunity against tuberculosis in mice after vaccination with tumor cells expressing mycobacterial hsp65. Infect Immun. 1996;64(7):2400–2407. [PMC free article: PMC174089] [PubMed: 8698458]
- 73.
- Silva CL. et al. Characterization of the memory/activated T cells that mediate the long lived host response against tuberculosis after bacillus Calmette-Guerin or DNA vaccination. Immunology. 1999;97(4):573–581. [PMC free article: PMC2326889] [PubMed: 10457209]
- 74.
- Silva CL, Lowrie DB. Identification and characterization of murine cytotoxic T cells that kill Mycobacterium tuberculosis. Infect Immun. 2000;68(6):3269– 3274. [PMC free article: PMC97577] [PubMed: 10816472]
- 75.
- Kim CH, Campbell Dj, Butcher EC. Nonpolarized memory T cells. Trends Immunol. 2001;22(10):527–530. [PubMed: 11574259]
- 76.
- Sallusto F. et al. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401(6754):708–712. [PubMed: 10537110]
- 77.
- Wang X, Mosmann T. In vivo priming of CD4 T cells that produce interleukin (IL)2 but not IL-4 or interferon (IFN)gamma, and can subsequently differentiate into IL-4 or IFN-γ-secreting cells. J Exp Med. 2001;194(8):1069–1080. [PMC free article: PMC2193514] [PubMed: 11602637]
- 78.
- Gu ML, Leppla SH, Klinman DM. Protection against anthrax toxin by vaccination with a DNA plasmid encoding anthrax protective antigen. Vaccine. 1999;17(4):340–344. [PubMed: 9987172]
- 79.
- Luke CJ. et al. An OspA-based DNA vaccine protects mice against infection with Borrelia burgdorferi. J Infect Dis. 1997;175(1):91–97. [PubMed: 8985201]
- 80.
- Simon MM. et al. Protective immunization with plasmid DNA containing the outer surface lipoprotein A gene of Borrelia burgdorferi is independent of an eukaryotic promoter. Eur J Immunol. 1996;26(12):2831–2840. [PubMed: 8977275]
- 81.
- Kurar E, Splitter GA. Nucleic acid vaccination of Brucella abortus ribosomal L7/L12 gene elicits immune response. Vaccine. 1997;15(1718):1851–1857. [PubMed: 9413093]
- 82.
- Brooks-Worrell BM, Splitter GA. Antigens of Brucella abortus S19 immunodominant for bovine lymphocytes as identified by one and twodimensional cellular immunoblotting. Infect Immun. 1992;60(6):2459–2464. [PMC free article: PMC257181] [PubMed: 1587614]
- 83.
- Stephens RS. et al. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science. 1998;282(5389):754–759. [PubMed: 9784136]
- 84.
- Murdin AD. et al. Use of a mouse lung challenge model to identify antigens protective against Chlamydia pneumoniae lung infection. J Infect Dis. 2000;181(Suppl 3):S544–S551. [PubMed: 10839756]
- 85.
- Svanholm C. et al. Protective DNA immunization against Chlamydia pneumoniae. Scand J Immunol. 2000;51(4):345–353. [PubMed: 10736106]
- 86.
- Vanrompay D. et al. Turkeys are protected from infection with Chlamydia psittaci by plasmid DNA vaccination against the major outer membrane protein. Clin Exp Immunol. 1999;118(1):49–55. [PMC free article: PMC1905387] [PubMed: 10540159]
- 87.
- Vanrompay D. et al. Protection of turkeys against Chlamydia psittaci challenge by gene gun-based DNA immunizations. Vaccine. 1999;17(2021):2628–2635. [PubMed: 10418912]
- 88.
- Brunham RC, Zhang D. Transgene as vaccine for chlamydia. Am Heart J. 1999;138(5 Pt 2):S519–522. [PubMed: 10539864]
- 89.
- Zhang D. et al. DNA vaccination with the major outermembrane protein gene induces acquired immunity to Chlamydia trachomatis (mouse pneumonitis) infection. J Infect Dis. 1997;176(4):1035–1040. [PubMed: 9333163]
- 90.
- Zhang DJ. et al. Characterization of immune responses following intramuscular DNA immunization with the MOMP gene of Chlamydia trachomatis mouse pneumonitis strain. Immunology. 1999;96(2):314–321. [PMC free article: PMC2326737] [PubMed: 10233711]
- 91.
- Pal S. et al. Vaccination of mice with DNA plasmids coding for the Chlamydia trachomatis major outer membrane protein elicits an immune response but fails to protect against a genital challenge. Vaccine. 1999;17(5):459–465. [PubMed: 10073724]
- 92.
- Penttila T. et al. Immunity to Chlamydia pneumoniae induced by vaccination with DNA vectors expressing a cytoplasmic protein (Hsp60) or outer membrane proteins (MOMP and Omp2). Vaccine. 2000;19(910):1256–1265. [PubMed: 11137265]
- 93.
- Yang X, HayGlass KT, Brunham RC. Genetically determined differences in IL-10 and IFN-γ responses correlate with clearance of Chlamydia trachomatis mouse pneumonitis infection. J Immunol. 1996;156(11):4338–4344. [PubMed: 8666805]
- 94.
- Brunham RC. et al. The potential for vaccine development against chlamydial infection and disease. J Infect Dis. 2000;181(Suppl 3):S538–43. [PubMed: 10839755]
- 95.
- Strugnell RA. et al. DNA vaccines for bacterial infections. Immunol Cell Biol. 1997;75(4):364–369. [PubMed: 9315479]
- 96.
- Anderson R. et al. Immune response in mice following immunization with DNA encoding fragment C of tetanus toxin. Infect Immun. 1996;64(8):3168–3173. [PMC free article: PMC174203] [PubMed: 8757849]
- 97.
- Saikh KU. et al. Are DNAbased vaccines useful for protection against secreted bacterial toxins? Tetanus toxin test case. Vaccine. 1998;16(910):1029–1038. [PubMed: 9682355]
- 98.
- Chaplin PJ. et al. Targeting improves the efficacy of a DNA vaccine against Corynebacterium pseudotuberculosis in sheep. Infect Immun. 1999;67(12):6434–6438. [PMC free article: PMC97052] [PubMed: 10569760]
- 99.
- Boyle JS, Brady JL, Lew AM. Enhanced responses to a DNA vaccine encoding a fusion antigen that is directed to sites of immune induction. Nature. 1998;392(6674):408–411. [PubMed: 9537327]
- 100.
- Alves AM. et al. Epitope specificity of antibodies raised against enterotoxigenic Escherichia coli CFA/I fimbriae in mice immunized with naked DNA. Vaccine. 1998;16(1):9–15. [PubMed: 9607003]
- 101.
- Alves AM. et al. Immunoglobulin G subclass responses in mice immunized with plasmid DNA encoding the CFA/I fimbria of enterotoxigenic Escherichia coli. Immunol Lett. 1998;62(3):145–149. [PubMed: 9698112]
- 102.
- Alves AM. et al. Antibody response in mice immunized with a plasmid DNA encoding the colonization factor antigen I of enterotoxigenic Escherichia coli. FEMS Immunol Med Microbiol. 1999;23(4):321–330. [PubMed: 10225292]
- 103.
- Alves AM. et al. DNA immunisation against the CFA/I fimbriae of enterotoxigenic Escherichia coli (ETEC). Vaccine. 2000;19(78):788–795. [PubMed: 11115700]
- 104.
- Alves AM. et al. New vaccine strategies against enterotoxigenic Escherichia coli. I: DNA vaccines against the CFA/I fimbrial adhesin. Braz J Med Biol Res. 1999;32(2):223–229. [PubMed: 10347758]
- 105.
- Lasaro MO. et al. New vaccine strategies against enterotoxigenic Escherichia coli. II: Enhanced systemic and secreted antibody responses against the CFA/I fimbriae by priming with DNA and boosting with a live recombinant Salmonella vaccine. Braz J Med Biol Res. 1999;32(2):241–246. [PubMed: 10347761]
- 106.
- Turnes CG. et al. DNA inoculation with a plasmid vector carrying the faeG adhesin gene of Escherichia coli K88ab induced immune responses in mice and pigs. Vaccine. 1999;17(1516):2089–95. [PubMed: 10217611]
- 107.
- Elkins KL. et al. Bacterial DNA containing CpG motifs stimulates lymphocyte dependent protection of mice against lethal infection with intracellular bacteria. J Immunol. 1999;162(4):2291–2298. [PubMed: 9973506]
- 108.
- Krieg AM, Davis HL. Enhancing vaccines with immune stimulatory CpG DNA. Curr Opin Mol Ther. 2001;3(1):15–24. [PubMed: 11249727]
- 109.
- Todoroki I. et al. Suppressive effects of DNA vaccines encoding heat shock protein on Helicobacter pylori-induced gastritis in mice. Biochem Biophys Res Commun. 2000;277(1):159–163. [PubMed: 11027657]
- 110.
- Dai B. et al. [Immunoprotection in guinea pigs using DNA recombinant plasmid rpDJt and expressed protein P68 in L. interrogans serovar lai] Hua Xi Yi Ke Da Xue Xue Bao. 1998;29(3):248–251. [PubMed: 10684084]
- 111.
- North RJ, Dunn PL, Conlan JW. Murine listeriosis as a model of antimicrobial defense. Immunol Rev. 1997;158:27–36. [PubMed: 9314071]
- 112.
- Harty JT, Bevan MJ. CD8 T cells specific for a single nonamer epitope of Listeria monocytogenes are protective in vivo. J Exp Med. 1992;175(6):1531–1538. [PMC free article: PMC2119257] [PubMed: 1375265]
- 113.
- Darji A. et al. Oral somatic transgene vaccination using attenuated S. typhimurium. Cell. 1997;91(6):765–775. [PubMed: 9413986]
- 114.
- Uchijima M. et al. Optimization of codon usage of plasmid DNA vaccine is required for the effective MHC class Irestricted T cell responses against an intracellular bacterium. J Immunol. 1998;161(10):5594–5599. [PubMed: 9820537]
- 115.
- Koide Y. et al. [DNA vaccines for infections with intracellular bacteria] Nippon Saikingaku Zasshi. 1999;54(4):773–793. [PubMed: 10643297]
- 116.
- Cornell KA. et al. Genetic immunization of mice against Listeria monocytogenes using plasmid DNA encoding listeriolysin O. J Immunol. 1999;163(1):322–329. [PubMed: 10384131]
- 117.
- Fensterle J. et al. Effective DNA vaccination against listeriosis by prime/boost inoculation with the gene gun. J Immunol. 1999;163(8):4510–4518. [PubMed: 10510394]
- 118.
- Barry MA, Lai WC, Johnston SA. Protection against mycoplasma infection using expressionlibrary immunization. Nature. 1995;377(6550):632–635. [PubMed: 7566175]
- 119.
- Lai WC. et al. Therapeutic effect of DNA immunization of genetically susceptible mice infected with virulent Mycoplasma pulmonis. J Immunol. 1997;158(6):2513–2516. [PubMed: 9058780]
- 120.
- DenisMize KS. et al. Analysis of immunization with DNA encoding Pseudomonas aeruginosa exotoxin A. FEMS Immunol Med Microbiol. 2000;27(2):147–154. [PubMed: 10640610]
- 121.
- Shiau JW. et al. Mice immunized with DNA encoding a modified Pseudomonas aeruginosa exotoxin A develop protective immunity against exotoxin intoxication. Vaccine. 2000;19(910):1106–1112. [PubMed: 11137245]
- 122.
- MatthewsGreer JM, Gilleland H E Jr. Outer membrane protein F (porin) preparation of Pseudomonas aeruginosa as a protective vaccine against heterologous immunotype strains in a burned mouse model. J Infect Dis. 1987;155(6):1282–1291. [PubMed: 3033095]
- 123.
- Gilleland H E Jr. et al. Use of a purified outer membrane protein F (porin) preparation of Pseudomonas aeruginosa as a protective vaccine in mice. Infect Immun. 1984;44(1):49–54. [PMC free article: PMC263467] [PubMed: 6323316]
- 124.
- Gilleland H E Jr, Gilleland LB, Matthews-Greer JM. Outer membrane protein F preparation of Pseudomonas aeruginosa as a vaccine against chronic pulmonary infection with heterologous immunotype strains in a rat model. Infect Immun. 1988;56(5):1017–1022. [PMC free article: PMC259755] [PubMed: 2833440]
- 125.
- Price BM. et al. Protection against Pseudomonas aeruginosa chronic lung infection in mice by genetic immunization against outer membrane protein F (OprF) of P. aeruginosa. Infect Immun. 2001;69(5):3510–3515. [PMC free article: PMC98322] [PubMed: 11292786]
- 126.
- LopezMacias C. et al. Induction of antibodies against Salmonella typhi OmpC porin by naked DNA immunization. Ann N Y Acad Sci. 1995;772:285–288. [PubMed: 8546410]
- 127.
- Isibasi A. et al. Role of porins from Salmonella typhi in the induction of protective immunity. Ann N Y Acad Sci. 1994;730:350–352. [PubMed: 7521604]
- 128.
- Archer GL, Niemeyer DM. Origin and evolution of DNA associated with resistance to methicillin in staphylococci. Trends Microbiol. 1994;2(10):343–347. [PubMed: 7850198]
- 129.
- Ohwada A. et al. DNA vaccination by mecA sequence evokes an antibacterial immune response against methicillin-resistant Staphylococcus aureus. J Antimicrob Chemother. 1999;44(6):767–774. [PubMed: 10590277]
- 130.
- McDaniel LS. et al. Immunization with a plasmid expressing pneumococcal surface protein A (PspA) can elicit protection against fatal infection with Streptococcus pneumoniae. Gene Ther. 1997;4(4):375–377. [PubMed: 9176525]
- 131.
- McDaniel LS. et al. PspA, a surface protein of Streptococcus pneumoniae, is capable of eliciting protection against pneumococci of more than one capsular type. Infect Immun. 1991;59(1):222–228. [PMC free article: PMC257730] [PubMed: 1987036]
- 132.
- Tart RC. et al. Truncated Streptococcus pneumoniae PspA molecules elicit cross-protective immunity against pneumococcal challenge in mice. J Infect Dis. 1996;173(2):380–386. [PubMed: 8568299]
- 133.
- Bennett AM. et al. Gene gun mediated vaccination is superior to manual delivery for immunisation with DNA vaccines expressing protective antigens from Yersinia pestis or Venezuelan Equine Encephalitis virus. Vaccine. 1999;18(78):588–596. [PubMed: 10547416]
- 134.
- Hornef MW. et al. DNA vaccination using coexpression of cytokine genes with bacterial gene encoding a 60-kDa heat shock protein. Med Microbiol Immunol (Berl). 2000;189(2):97–104. [PubMed: 11138643]
- 135.
- Noll A. et al. DNA immunization confers systemic, but not mucosal, protection against enteroinvasive bacteria. Eur J Immunol. 1999;29(3):986–996. [PubMed: 10092103]
- 136.
- Noll A. et al. Protective role for heat shock protein-reactive alpha beta T cells in murine yersiniosis. Infect Immun. 1994;62(7):2784–2791. [PMC free article: PMC302882] [PubMed: 7911784]
- 137.
- Williamson ED. et al. A new improved subunit vaccine for plague: the basis of protection. FEMS Immunol Med Microbiol. 1995;12(34):223–230. [PubMed: 8745007]
- 138.
- Flynn JL, Chan J. Immunology of tuberculosis. Annu Rev Immunol. 2001;19:93–129. [PubMed: 11244032]
- 139.
- Tanghe A. et al. Tuberculosis DNA vaccine encoding Ag85A is immunogenic and protective when administered by intramuscular needle injection but not by epidermal gene gun bombardment. Infect Immun. 2000;68(7):3854–3860. [PMC free article: PMC101658] [PubMed: 10858194]
- 140.
- Mollenkopf HJ. et al. Protective efficacy against tuberculosis of ESAT-6 secreted by a live Salmonella typhimurium vaccine carrier strain and expressed by naked DNA. Vaccine. 2001;19(2829):4028–4035. [PubMed: 11427279]
- 141.
- Coler RN. et al. Vaccination with the T cell antigen Mtb 8.4 protects against challenge with Mycobacterium tuberculosis. J Immunol. 2001;166(10):6227–6235. [PubMed: 11342645]
- 142.
- Dillon DC. et al. Molecular characterization and human T cell responses to a member of a novel Mycobacterium tuberculosis mtb39 gene family. Infect Immun. 1999;67(6):2941–2950. [PMC free article: PMC96604] [PubMed: 10338503]
- 143.
- Skeiky YA. et al. T cell expression cloning of a Mycobacterium tuberculosis gene encoding a protective antigen associated with the early control of infection. J Immunol. 2000;165(12):7140–7149. [PubMed: 11120845]
- 144.
- Delogu G, Brennan MJ. Comparative immune response to PE and PE_PGRS antigens of Mycobacterium tuberculosis. Infect Immun. 2001;69(9):5606–5611. [PMC free article: PMC98675] [PubMed: 11500435]
- DNA Vaccines against Bacterial Pathogens - Madame Curie Bioscience DatabaseDNA Vaccines against Bacterial Pathogens - Madame Curie Bioscience Database
- Genetic Risk Factors for Disseminated Intravascular Coagulation - Madame Curie B...Genetic Risk Factors for Disseminated Intravascular Coagulation - Madame Curie Bioscience Database
- Regulation of Cardiomyocyte Proliferation and Apoptosis - Madame Curie Bioscienc...Regulation of Cardiomyocyte Proliferation and Apoptosis - Madame Curie Bioscience Database
- S-Modulin - Madame Curie Bioscience DatabaseS-Modulin - Madame Curie Bioscience Database
- Development of Biological Potential - Madame Curie Bioscience DatabaseDevelopment of Biological Potential - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...