

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Polyomavirus nephropathy (PVN) is primarily caused by a productive intra-renal BK virus infection. It is often an iatrogenic complication due to long term over immunosuppression and frequently leads to chronic kidney dysfunction and failure. Post renal transplantation, PVN has emerged as a major problem affecting up to 10% of all kidney grafts, most commonly within the first 6-18 months post surgery. Recent advances in our understanding of the development and progression of PVN have resulted in the definition of clinically significant disease stages: A (early), B (florid) and C (sclerosing). This chapter provides a detailed description of polyomavirus induced acute and chronic renal injury. Morphologic changes are correlated with the clinical presentation and graft outcome. Key biologic aspects that are pertinent to patient management are highlighted. New ‘morphology based’ strategies, i.e., urine electron microscopy and improved decoy cell analyses, to reliably diagnose PVN non-invasively are discussed and incorporated into an updated diagnostic algorithm for patient screening and monitoring.
Introduction
Polyomaviruses are of no clinical significance in the immune competent individual. Under immunocompromised conditions, however, the BK- and JC-polyomavirus strains can enter into “organ-destructive” replicative cycles and cause disease. Clinically significant infections/ disease caused by BK-viruses are primarily seen in the urinary tract, i.e., the bladder and kidneys, whereas JC-viruses mainly cause neurologic disorders, i.e., progressive multifocal leukoencephalopathy. Early reports that had linked productive infections with BK viruses to the development of ureteral necrosis and stenosis1 could not be confirmed in a recent series which demonstrated BK viruses in only 8% (2/25) of histologically analyzed necrotic ureters.2
Polyomavirus allograft nephropathy (PVN) post kidney transplantation was first described nearly 3 decades ago as a single case report by the pathologist Mackenzie.3 It was, however, not until the introduction of potent third generation immunosuppressive drugs, mainly high dose tacrolimus and mycophenolate mofetil, into clinical management that transplant centers world-wide experienced “an outbreak” of PVN as a mostly iatrogenic complication.4-10 Currently, PVN has a prevalence of 1% to 10% and is associated with a graft failure rate of 50% in some centers. It exceeds productive CMV graft infections by approximately 100 times.9,11-15 Likely, the incidence of PVN will decrease in the future under altered and optimized immunosuppression. A recent report from the Mayo Clinic found a highly significant reduction of the incidence of PVN from 10.5% to 2.5% following the clinical introduction of “low dose” maintenance tacrolimus immunosuppression.9 At present, however, PVN still constitutes a major clinical challenge. Specific and potent antiviral drugs to treat productive polyomavirus infections are not available. Consequently, much emphasis is placed on patient screening and a diagnosis of PVN in an early disease stage that often responds favorably to our limited therapeutic options mainly consisting of a reduction of the maintenance immunosuppression.
Most of our current disease knowledge has been gained by studying patients; thus far, reliable animal models mirroring PVN in humans have not been developed.16 Many aspects of PVN and infections with polyomaviruses including risk factors and treatment options have been discussed elsewhere in this book (see chapter by Ahsan). In the following paragraphs, we will primarily focus on morphologic changes and biologic concepts that help to understand and manage PVN. We will place special emphasis on recently developed ‘morphologic’ strategies to accurately diagnose PVN non-invasively that will be critically compared to conventional PCR techniques. In the future, chronic graft failure due to PVN should be the exception rather than the rule.
Background
Infections with polyomaviruses and PVN are characterized by several key features (see Table 1):
Table 1
Polyomavirus infections (modified from ref. 13).
- PVN is caused by the re-activation of latent intra-renal/ intra-graft, i.e., donor derived, polyomaviruses under long-lasting and intense immunosuppression, such as high dose tacrolimus or mycophenolate-mofetil. High latent polyomavirus loads are detected in approximately 5% of all normal/donor kidneys. It is presumably this sub-group of organs that is at increased risk for the development of PVN post transplantation. Latent infections with polyomaviruses do not cause any functional or morphologic changes; they can only be detected by molecular techniques. Of note: circulating antibodies against BK virus, which are found at different titer levels in 66%-90% of healthy adults,17-19 do not accurately reflect latent intra-renal BK virus loads. In a pilot analysis of 30 deceased nontransplant patients without BKN we compared plasma anti-BK virus antibody titers (by the hemaglutinine-inhibition assay, HAI) to the corresponding latent intra-renal BK virus load levels. Sero-negativity (HAI titers below 1:128) was found in 1/30 patients (3%); this patient showed a corresponding high, latent intra-renal BK virus load. Normal antibody titer ranges (HAI: 1:128-1:1024) were observed in 26/30 patients (87%), of which 16 were without latent intra-renal BK-virus infections. The remaining 3/30 patients (10%) demonstrated high plasma anti-BK virus antibody titers (HAI ≥1:2048); in one patient without a latent intra-renal BK virus infection (personal observation, 20). Thus, it seems that dormant intra-renal BK-virus load levels cannot be extrapolated from plasma antibody titers. Consequently, kidneys from sero-negative donors may contain latent BK-viruses that can predispose the recipient for the development of PVN post grafting (also see Table 1 in the chapter by Ahsan).
- Slight changes in the immune status can lead to transient, mostly asymptomatic and self-limiting activation of latent polyomaviruses, 21 especially in the urothelium, which harbors latent BK virus infections in 43% of individuals (personal observation). Such activation is characterized by the detection of free viral particles in the urine (by electron microscopy or PCR techniques) and viral inclusion bearing cells, so-called “decoy cells” in urine cytology specimens (see below). Signs of transient, asymptomatic viral activation can occasionally also be detected in serum samples by real time PCR. Such polyomavirus (re)activation is commonly not accompanied by PVN.4,22,23 PVN, on the other hand, is always associated and typically preceded by the activation of polyomaviruses.6,19,22,24-26
- PVN is typically diagnosed 10-12 months post transplantation with only anecdotal cases reported as early as 6 days and as late as 6 years post grafting.14,26 Depending on the extent of virally induced tubular injury (Table 2), patients clinically present with varying degrees of allograft dysfunction. Serum creatinine levels vary from normal (PVN stage A) to markedly increased (PVN stages B and C).26-30 Not surprisingly, PVN has been diagnosed in surveillance protocol biopsies,11,28,30 and thus, clinical evidence of graft dysfunction is only ill suited to optimally time a diagnostic graft biopsy in order to establish an early diagnosis.
- PVN is best diagnosed histologically in a graft biopsy, which additionally provides prognostically relevant information on the disease stage, concomitant rejection and potential other abnormalities, such as calcineurin inhibitor induced toxicity. Since PVN affects the kidney in a focal fashion, adequate samples are needed to guarantee an optimal diagnostic yield, i.e., two biopsy cores obtained with a 15 or 16 gauge needle. The diagnosis may be missed in 25%-37% of cases if only one small core of renal cortex is sampled.13,31 A new strategy for accurately diagnosing PVN non-invasively is urine electron microscopy and the detection of three-dimensional, cast-like polyomavirus clusters32 (see below).
- PVN is nearly always caused by a productive infection with the BK-virus strain. Only a minority of cases (approximately one third) show activation of polyoma-BK- and JC-viruses simultaneously with, as yet, undetermined biological significance. 33,34 Polyomavirus nephropathies that are only induced by a productive JC- or SV-40 virus infection are exceptionally rare.35,36 PVN is hardly ever seen in association with a concurrent second viral infection.15,37 Morphologic changes induced by productive BK-, JC-, or SV-40 polyomavirus infections are identical; ancillary techniques such as immunohistochemistry, in-situ hybridization, or PCR are required for the exact identification of viral strains.
- PVN is typically limited to the transplant and “the worst case scenario” is graft failure. Systemic signs of an infection including involvement of the native kidneys38 are generally not seen (with only exceptional case reports of systemic viral disease, a potential case of BK virus associated hemophagocytic syndrome and PVN associated hemorrhagic cystitis39-43). Thus, patients are usually not at risk for generalized disease or lethal viral spread.
Table 2
PVN: Mode of kidney injury and dysfunction.
PVN: Morphologic Findings
PVN is defined as an intraparenchymal productive polyomavirus infection (BK-virus >> JC-virus) of the kidney with light microscopic (i.e., intra-nuclear viral inclusion bodies), or immunohistochemical or in-situ hybridization evidence of viral replication accompanied by varying degrees of parenchymal damage ranging from minimal to marked (Tables 1-3).
Table 3
Polyomavirus allograft nephropathy: Disease stages (modified from refs. 13,47).
Gross Pathology
Transplant nephrectomy specimens from patients with PVN show uncharacteristic changes closely resembling fibrotic lesions seen in other chronic diseases leading to graft failure. In PVN the removed allografts are generally slightly decreased in size, firm, with an ill-defined corticomedullary junction and thinned, sclerosed cortex. The renal surface is either smooth or granular. Infarction and large scar formation are not features of PVN.
Light Microscopy
Histologic signs caused by a productive polyomavirus infection of the kidney are characteristically found in epithelial cells lining collecting ducts, tubules, Bowman's capsule (parietal epithelial cells) and the renal pelvis (transitional cells). Viral replication and the assembly of daughter virions result in the formation of intra-nuclear inclusion bodies, cell injury and cell lysis.5-7,24,26 Most important for the clinical presentation and course of PVN are the tubular changes as severe, virally induced tubular injury and the denudation of tubular basement membranes can ultimately lead to atrophy and fibrosis, i.e., PVN disease stage C (Tables 2 and 3). Despite marked epithelial damage, however, the tubular basement membranes usually remain intact. During early stages of disease (i.e., PVN stages A and B1) they can serve as the structural skeleton for regeneration and restitution once the viral replication ceases. Parietal epithelial cells lining Bowman's capsule can also show signs of viral replication and on occasion form “pseudo-crescents”.24,44,45 Viral inclusion bodies are also found in the transitional cell layer lining the renal pelvis, the (graft) ureter and potentially even the recipient's urinary bladder.24,41 Polyomavirus replication in the urothelium, however, is not a defining histologic hallmark characterizing PVN since it can also be seen in patients without viral nephropathy as a transient and asymptomatic sign of viral activation46 -or- in the setting of hemorrhagic cystitis (without PVN) following bone marrow transplantation (Table 1). Glomerular capillary tufts, blood vessels, inflammatory and mesenchymal cells are generally nonpermissive for the replication of BK-/polyomaviruses.
Staging
Based on the degree of tubulo-interstial damage, PVN is histologically classified into three disease stages: early “A”, florid “B” and late sclerosing “C” (Table 3).13,24,29,31,47-50 Pertinent to staging are the severity of virally induced epithelial cell injury, interstitial fibrosis and tubular atrophy rather than the extent of interstitial inflammation. Progression from disease stage A to B or C can occur within few months.12,49
PVN stage A represents the earliest disease phase with only very focal ‘nonlytic' viral replication and inconspicuous denudation of tubular basement membranes; it is often limited to the renal medulla and likely represents re-activated foci of latent BK virus infections (Fig. 1). The interstitium is normal or only shows minimal inflammation and fibrosis. Intra-nuclear viral inclusion bodies are generally found (albeit sometimes few in number). In rare, presumably very early phases of a productive polyomavirus infection, characteristic intra-nuclear viral inclusion bodies may be absent; in these cases, the diagnosis of PVN is solely based on immunohistochemical or in-situ hybridization staining signals. The detection of one intra-nuclear viral inclusion body or alternatively a crisp staining signal in one nucleus (with or without an inclusion body) suffices for proving ‘active’ viral replication and rendering the diagnosis of PVN stage A.

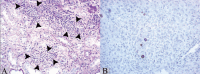
Florid PVN (stage B) is characterized by marked virally induced tubular injury and epithelial cell lysis, conspicuous denudation of tubular basement membranes and interstitial edema with a mixed, mild to marked inflammatory cell infiltrate (B- and T-lymphocytes, plasma cells and histiocytes) (Fig. 2). Polymorphonuclear leukocytes can be prominent adjacent to severely injured tubules, presumably due to urinary back-leak into the interstial compartment. On occasion, tubulo-centric granulomas rich in histiocytes form in and around virally injured tubules (Fig. 3).
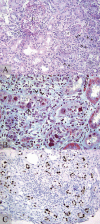
PVN stage C, the sclerosing phase, shows marked interstitial fibrosis and tubular atrophy involving, per definition more than 50% of the tissue sample. Stage C is associated with varying degrees of inflammation and viral replication (Fig. 4).

The burnt-out stage of PVN following the resolution of viral replication demonstrates nonspecific “chronic” changes: varying degrees of interstitial fibrosis and tubular atrophy (ranging from minimal to marked) and occasionally mild lymphocytic inflammation in areas of parenchymal scarring. Any signs of polyomavirus replication by light microscopy and immunohistochemistry/in-situ hybridization are, per definition, no longer detectable. The PVN burnt-out stage closely mimicks so-called “chronic allograft nephropathy”; it can only be suspected based on the patient's history.
The staging of PVN is robust and it carries clinical significance (see below). Additional attempts may be made (and recorded for each disease stage) to semi-quantitatively assess the extent of viral replication/ infection in the tubular compartment from cy0 (no replication), cy1 (<10% infected tubules with viral replication in the biopsy sample), cy2 (10%-≤25% infected tubules), cy3 (26%-≤50% infected tubules) and cy4 (>50% of tubules with viral replication;48). Most commonly, PVN stage A presents with cy1, PVN stage B with cy 2, 3, or 4 and PVN stage C with cy 1, 2, or 3. Of note: the ‘cy’ scoring does not determine the PVN disease stage. Definitive rules to adequately assess and compare “cy” scores have so far not been set, i.e., light microscopy (detection of intra-nuclear inclusion bodies) versus immunohistochemistry (detection of the large T-antigen associated with viral replication) or in-situ hybridization (detection of amplified viral DNA/mature daughter virions). Thus, depending on the approach taken, “cy” scores may vary. In our experience, the presence and extent of a “productive polyomavirus infection/PVN” are best documented by the immunohistochemical detection of the “large T-antigen” expression (see below).
Inflammation
PVN stages A-C can be associated with varying degrees of interstitial inflammation (Figs. 1-4) that is only poorly understood:6,12,51
Scenario #1: The inflammatory response may potentially be beneficial and indicate cellular containment of viral replication by BK virus specific cytotoxic T-lymphocytes52-54 and the release of gamma interferon.55 Immunologic reconstitution under decreased immunosuppression during attempts to treat PVN may possibly provide the right window of opportunity for such a cellular anti-viral immune response. However, data to support this concept are currently scant.56 In one small series the stimulation of BK virus specific CD8+ T-cells was not beneficial but rather detrimental and associated with graft failure.54
Scenario #2: In cases of severe, virally induced acute tubular injury, edema and inflammation including the attraction of polymorphonuclear leukocytes may represent a “reactive” phenomenon that is caused by urine back-leak through denuded basement membranes into the interstitial compartment. Similar changes can be seen in native kidneys with severe nephrotoxic forms of acute tubular injury (ATN). Such a “reactive” inflammatory response driven by tubular damage is seen in disease stages B or C.
Scenario #3: Viral replication in markedly damaged tubules can induce the formation of expansile, intra- and peritubular, nonnecrotizing histiocytic granulomas (Fig. 3). More commonly, however, it triggers a diffuse mononuclear (lymphocytic/histiocytic) and plasma cell rich interstitial inflammatory cell infiltrate, edema and tubulitis (Fig. 2). Such inflammatory responses including granuloma formation might be classified as a “type IV hypersensitivity reaction”; they can be seen in PVN disease stages B and C.
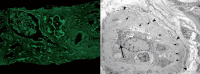
Scenario #4: Inflammation rich in lymphocytes can occasionally reflect a cellular (and antibody) mediated immune reaction driven by the release of virions and characterized by the formation of immune complex type deposits in tubular basement membranes (Fig. 5). In such cases, immunofluorescence microscopy and electron microscopy typically show granular, immunoglobulin deposits in tubular basement membranes reminiscent of ‘anti-TBM disease/primary tubulointerstitial nephritis with immune complexes’ seen in native kidneys.45,57 This immune mediated inflammation might be classified as a “type III hypersensitivity reaction”; it is reported in PVN disease stages B or C.
Scenario #5: CD8+ lymphocytic allo-responses and acute rejection episodes with tubulitis, transplant endarteritis, or glomerulitis, occasionally C4d positive, have been reported in cases of PVN.6,26,58-60 It seems that acute allograft rejection in the setting of PVN is an independent, second disease process that is not primarily triggered by viral replication (see chapter by Ahsan).6,19,26,60 Acute rejection can be seen in PVN stages A-C. It is most easily diagnosed in stage A that should, per definition, lack a significant inflammatory cell infiltrate.13,47,60
Except for the potentially transient “immune reconstitution associated” inflammatory reaction (hypothetical scenario #1), all other persistent inflammatory responses are detrimental since they can result in progressive fibrosis and chronic graft failure.6,24,26,51
The interpretation of “inflammation” in PVN is challenging, in particular, the diagnosis of acute allograft rejection. Careful histologic analysis and the detection of scattered polymorphonuclear leukocytes adjacent to severely injured and denuded tubules (scenario#2), granulomas (scenario# 3), granular immune deposits along tubular basement membranes (scenario#4), or transplant endarteritis, glomerulitis, tubular MHC-class II expression or capillary C4d deposits (scenario#5) may help to guide the diagnostic decision making process. Clinically and prognostically most important is the diagnosis of concurrent allograft rejection, since it should alter the therapeutic strategies.26,60 Since cases of PVN with marked tubulitis generally fare poorly,31 unrecognized tubulo-interstitial cellular rejection (Banff type I) may potentially contribute to PVN induced graft demise more frequently than commonly suspected.60,61 The immunohistochemical phenotyping of the inflammatory cells in PVN has shown plasma cell (CD138) as well as B (CD20) or T-cell (CD3) dominant infiltrates with currently undetermined pathophysiological significance. It is not diagnostically helpful for distinguishing different forms of “viral nephritis” or to diagnose concurrent acute cellular allograft rejection.51,62,63
Ancillary techniques
In tissue specimens, polyomavirus replication is readily detected with commercially available antibodies directed against the SV-40 T-antigen (large T-antigen) that cross-react with all polyomaviruses pathogenic in humans (i.e., BK-, JC- and SV-40). They reliably work in frozen and paraffin sections and give a crisp, purely intra-nuclear staining signal (Figs. 1B, 2C, 3B, 4B). The expression of the T-antigen marks the initial phase of polyomavirus replication. It can precede the formation of intra-nuclear viral inclusion bodies and crisp staining signals may be seen in normal nuclei/tissue6,24,26,29 as the earliest morphologic sign of viral replication and disease (Tables 1 and 3). Polyomaviruses can also be identified by in-situ hybridization or with antibodies directed against viral capsid proteins. These latter techniques mostly detect “mature” virions and, therefore, not only mark intra-nuclear but additionally also dense intratubular “cast-like” viral aggregates (Fig. 6). Strain specific antibodies directed against BK-, JC-, or SV-40 viruses are also available, although most of them are primarily used by the research community; their use for diagnostic clinical purposes does not carry any advantage over antibodies to SV-40 T-antigen since PVN is nearly always driven by the replication of BK-viruses. Of note: depending on the technique used, staining results may vary greatly; suboptimal staining protocols can result in an artificially low number of “positive signals” rather than a marked decrease of the staining intensity in individual nuclei. Antibodies directed against the BK-virus T-antigen (clone BK.T-1) cross react with unrelated nuclear proteins (Ku86) and result in nonspecific staining signals.64 Epithelial cells with signs of polyomavirus replication express p53 tumor suppressor proteins43 and the proliferation marker KI-67 as signs of altered (viral) DNA assembly.

PCR techniques may also be utilized to demonstrate viral DNA or RNA in tissue samples and to confirm the diagnosis of PVN.65-67 However, PCR results must be interpreted with great caution. Only very strong amplification signals of viral DNA (greater than 10 viral copies per cell equivalent), in the setting of histologically or immunohistochemically demonstrable virally induced cytopathic changes, can be used to confirm the diagnosis of PVN, to identify the viral strain and to distinguish clinically significant productive from clinically insignificant latent polyomavirus infections (see above).65,66,68-72 The detection of viral RNA in renal biopsy cores by PCR clearly indicates viral replication/PVN. RNA extraction and amplification methods, however, are challenging techniques, susceptible to error and do not provide information which exceeds the results obtained with standard immunohistochemistry (such as the detection of the SV-40 T-antigen).66,67
In PVN, standard immunofluorescence microscopy with a common panel of antibodies directed against IgG, IgA, IgM, kappa and lambda light chains, fibrinogen and complement factor C3 generally does not show any diagnostic staining pattern in tubules, glomeruli, or blood vessels. In some patients, PVN is associated with a type III hypersensitivity reaction, resembling “anti-TBM disease” in native kidneys. These latter cases are characterized by granular, immune deposits along thickened tubular basement membranes that are easily discernible by immunofluorescence microscopy (using various antibodies directed against immunoglobulins and complement factors) or by electron microscopy (Fig. 5). Tubular immune deposits do not seem to contain viral capsid proteins.57
PVN and virally induced tubular injury are not associated with marked and diffuse upregulation of MHC-class II (i.e., HLA-DR) in tubular epithelial cells or with the deposition of the complement degradation product C4d along peritubular capillaries.6,12,73 Both, the detection of C4d and tubular HLA-DR expression can help to establish a diagnosis of (concurrent) allograft rejection.60
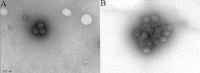
By electron microscopy, polyomaviruses present as viral particles of 30 to 50 nm in diameter that occasionally form crystalloid aggregates (Fig. 6B).22,24 Polyomaviruses are ultrastructurally identified by size and their icosahedral capsid structure (Fig. 8); polyomavirus strains cannot be distinguished. Virions are primarily found in the nucleus and rarely in the cytoplasm. Following viral replication, intra-nuclear viral assembly and the subsequent lysis of the host cell, crystalloid arrays of mature daughter virions are released into tubular lumens rich in Tamm-Horsfall protein (Fig. 6). Tamm-Horsfall protein likely serves as an “adhesive” that facilitates the formation of densely packed, cast-like viral clusters (Fig. 8). These viral clusters are flushed out of injured tubules and excreted with the urine. They can be detected in voided urine samples as pathognomonic markers for PVN (see below).
Course of PVN
PVN contributes directly to chronic graft failure and loss.6,74 Unfortunately, treatment options are currently limited since highly specific anti-polyomavirus drugs are not available75 (also see chapter by Ahsan). Outcome of PVN is largely determined by three factors: (i) the disease stage at time of the initial diagnosis, (ii) the duration of PVN with persistent, virally driven, pro-fibrotic tubulo-interstitial injury (Table 2), and (iii) the extent of pre-existing graft damage, e.g., hypertension induced arterionephrosclerosis, structural calcineurin-inhibitor toxicity etc. Allograft rejection can contribute to graft demise.6,12,26 The ultimate therapeutic goal is to diagnose PVN early (stage A) and to rapidly stop viral replication, thereby limiting tubular injury and preventing disease progression to irreversible scarring.11,28,30 Once PVN has resolved and the infection has resumed a latent state, signs of viral activation in the tissue, plasma and urine cease.6,25,31,59 The sclerosing PVN stage C with greater than 50% interstitial scarring is typically associated with severe allograft dysfunction and rapid loss.12,24,27,29,31,49
The early PVN disease stages, namely A and B1, can best respond to therapeutic intervention with favorable long-term outcome. 6,11,19,26,28-31,59,76 Drachenberg and colleagues diagnosed PVN stage A on average 8.7 months and stages B and C 15.9 months post transplantation.76 Under optimal conditions, viral clearance can be achieved within 3 weeks (personal observation in a patient presenting with PVN stage B1) and can result in complete functional restitution. 59 Usually, however, viral resolution that has been observed in up to 53%-78% of patients12,30,76 is a rather slow process taking many months. Donor kidneys of cadaveric origin, high viral loads at time of diagnosis, i.e., plasma BK levels of >1.5×105 copies/ml, and >5% of tubules with signs of polyomavirus infection have been identified as potential conditions protracting viral clearance.30 In diagnostic repeat biopsies, disease progression was observed in 37%-75% of patients initially presenting with PVN stage A and in 13%-60% with stage B.12,49 In some cases, PVN can progress rapidly. One of our patients advanced from disease stage A to C in three months (Figs. 1 and 4); a similar case was reported by Gaber and colleagues.12 If, ultimately, PVN resolves (after disease progression), kidney biopsies show increased interstitial fibrosis and tubular atrophy, i.e., the PVN-burnt out stage (Table 3).6,12 Depending on the extent of irreversible chronic injury, renal allograft function at time of viral resolution can vary from “back to baseline” (best case scenario in the setting of no or mild fibrosis), to chronic renal dysfunction (moderate fibrosis), or graft failure (worst case scenario in cases with marked fibrosis and atrophy).6,12,30,76
Although, in recent years, treatment options for PVN have remained limited, disease outcome has nevertheless improved. This is largely due to a high level of clinical suspicion and the implementation of patient screening procedures facilitating an early diagnosis.11,19,25,26,48,76,77
Diagnostic strategies: patient screening and monitoring
The clinical risk assessment for PVN is based on the observation that all patients with disease show signs of polyomavirus activation in the urine and plasma that often precede viral nephropathy by weeks to months. Over the last years, the systematic introduction of patient screening and monitoring protocols into clinical practice has resulted in significant improvements in the detection and outcome of PVN; the basic principles of these clinical surveillance protocols have been extensively described and reviewed elsewhere (see chapter by Ahsan).6,13,19,22,25-27,48,78-81 All currently employed screening strategies, i.e., decoy cell quantification by urine cytology (Fig. 7), PCR assays to detect viral DNA or RNA in the urine and plasma, or urine electron microscopy to quantify the shedding of free viral particles (Fig. 8A), have an excellent negative predictive value of 95%-100% to exclude PVN, i.e., no signs of viral activation = no PVN. Far less affirmative, however, are positive screening results ranging in their predictive values from 27% (quantification of decoy cells6) to approximately 80% (quantitative plasma PCR assays to detect BK virus DNA with a defined threshold level of 1×104 copies/ml19). ‘False’ positive screening tests for PVN are mainly due to polyomavirus activation in extra renal sites of viral latency, in particular in the urothelium, and can include high levels of BK viremia and/or viruria.13,46,79,82-86 Significant BK viremia in the absence of PVN is seen in patients with polyomavirus associated inflammation of the urothelium following bone marrow transplantation83 and occasionally also in nonkidney solid organ transplant recipients.87 In order to better identify patients at high risk for disease, BK virus cut-off load levels for viruria (PCR readings >1×107 BK copies/ml urine) and viremia (PCR readings >1×104 BK copies/ml plasma) have been proposed.48,78 These “threshold levels”, however, have not been confirmed in large multi-center trials, vary from laboratory to laboratory due to nonstandardized PCR assays, are only significant at time of the initial diagnosis and not during persistent PVN to detect viral clearance, and do not reliably distinguish between nondiseased viral activators and patients with PVN.13,30,79,83-85 Thus, the predictive diagnostic power of PCR readings in the plasma and urine is often weaker than expected. Of note: a multicenter comparative trial to quantify BK virus load levels by PCR in defined test sample sets demonstrated inter-laboratory differences of the BK DNA load readings of up to 2 logs.88 At present, clinical strategies for risk assessment, diagnosis and monitoring of PVN can be regarded as helpful, but not ideal.

In the following paragraphs we will briefly discuss two new methods to identify patients with PVN more accurately (Figs. 7-9): a) urine cytology and the quantification of different decoy cell phenotypes and b) urine electron microscopy and the detection of cast-like polyomavirus aggregates. Both studies can conveniently be performed non- invasively in a voided urine sample and can render a definitive diagnosis of PVN based on a single collection. Pending further validation, these methods hold great promise to provide powerful diagnostic information.
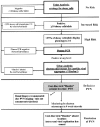
- Urine cytology (Fig. 7): identification and quantification of different decoy cell phenotypes.So-called “decoy cells” are intra-nuclear, polyomavirus inclusion bearing epithelial cells found in voided urine specimens. Decoy cells are a morphologic sign of polyomavirus activation. They are commonly thought to be primarily of transitional/urothelial origin and can easily be detected and counted in standard Papanicolaou stained ThinPrep® cytology preparations.22,82 It was already noted during the early days of the PVN epidemic that the shedding of large numbers of decoy cells was associated with an increased risk for the development of PVN post renal transplantation.4-6 The quantitative assessment of decoy cells is fast, easy, economical and readily available in every major transplant center affiliated with a pathology division. It is commonly used as the first screening method to identify patients at (increased) risk for PVN with an overall positive predictive value of 25%-30% (using a cut-off level of ≥10 decoy cells per ThinPrep® slide to call a sample “decoy positive”).6,13,19,22,25,26,48,79 The predictive yield can—in our experience—be easily increased by assessing not only the total number of decoy cells in a preparation but, additionally, also the different decoy cell variants (Fig. 7). If the common ground-glass decoy cell variant (phenotype 1, Fig. 7A) accounts for <75% of the total decoy cell count in a ThinPrep® specimen and the uncommon clumped variants (phenotypes 2-4, Fig. 7B-D) total >25%, then a patient is at high risk for PVN (positive predictive value >75%,89 Fig. 9). The increased shedding of clumped decoy cell variants is seen in all PVN disease stages (A-C) and persists during ongoing viral nephropathy.89 In our experience, the uncommon, clumped decoy cell variants are not typically seen in the urine of patients with polyomavirus associated lower urinary tract infections following bone marrow transplantation, i.e., hemorrhagic cystitis. It, therefore, is appealing to assume that decoy cell phenotypes 2-4 primarily originate from intra-renal/intra-tubular sites where polyomavirus replication often leads to the formation of different intra-nuclear inclusion bodies in tubular cells (see refs. 6, 26, 47). Thus, the easy and rapid identification and quantification of different decoy cell phenotypes may help to mark PVN more accurately, thereby rendering urine cytology a highly attractive and powerful screening tool. Future prospective studies are required to further validate the clinical significance of “decoy cell phenotyping”; until then, plasma PCR tests should additionally be used to adequately assess a patient's risk for PVN (Fig. 9).
- Urine electron microscopy (Fig. 8): identification of cast-like polyomavirus aggregates, so-called “Haufen”.In renal allograft recipients with PVN, urine is drained through infected nephrons with virally induced tubular injury. As illustrated above (Fig. 6) post replication daughter virions are released, often in the form of crystalloid viral arrays, from necrotic epithelial cells into tubular lumens. The specific intra tubular microenvironment allows for the formation of three-dimensional, cast-like polyomavirus aggregates that are flushed out of the infected kidneys. Consequently, viral clusters, named “Haufen” (German for cluster or stack), can be detected in voided urine samples from patients with PVN by conventional electron microscopy employing negative staining techniques (Fig. 8B). In our experience, “Haufen” are highly sensitive and specific for kidney disease. Their appearance in and disappearance from the urine closely follows the histologic course of PVN in all disease stages (positive and negative predictive values: >95%32). A protracted prodromal phase with shedding of “Haufen” prior to the development of PVN does not occur. We could never find “Haufen” in any patient with asymptomatic polyomavirus activation including high levels of viruria (and viremia) or in patients suffering from lower urinary tract polyomavirus infections post bone marrow transplantation (n = 50 control patients). Thus, the qualitative assessment of cast-like polyomavirus clusters in the urine by electron microscopy can be regarded as the first non-invasive test to accurately diagnose PVN (Fig. 9).
Negative staining electron microscopy as a diagnostic tool to facilitate patient management requires “thinking outside the box” for most transplant physicians. Until now, only few investigators have used this technique on urine samples in the setting of polyomavirus infections, 8,79,90,91 and the assessment of “Haufen” to diagnose PVN is a very recent development not yet in common practice. However, negative staining electron microscopy is a very well established and commonly used technique to search for viral pathogens in body fluids. It has a rapid turn around time of approximately 3 hours post sample collection and can be performed in any pathology division with a diagnostic electron microscopy unit at low costs.79 Negative staining electron microscopy simply has to be added to the list of required tests. We advocate its use and the specific search for cast-like polyomavirus aggregates, “Haufen”, in voided urine samples in all patients at high risk in order to rule PVN in or out (Fig. 9).
Conclusion
Polyomavirus nephropathy (PVN) is a challenging and serious complication mainly affecting renal allografts. Recent advances in risk assessment, the development of novel non-invasive diagnostic techniques, and disease staging will result in improvements of patient management and graft survival. Future research efforts should be directed towards the development of potent and specific anti-polyomavirus drugs and the better definition of risk factors promoting PVN. In addition, improved multi-center cooperations are required in order to validate and standardize clinical tests more efficiently, such as quantitative PCR assays, decoy cell analyses including cell phenotyping, and urine electron microscopy as a non-invasive method to diagnose PVN. We also have to gain better insight into the pathobiological significance of T-cell rich inflammatory cell infiltrates in florid cases of PVN as well as the potential role of JC-virus co-activation. How often does allograft rejection in PVN remain unrecognized, how aggressive should it be treated, and does JC-virus coreplication alter the presentation and course of PVN?
References
- 1.
- Coleman DV, Mackenzie EF, Gardner SD. et al. Human polyomavirus (BK) infection and ureteric stenosis in renal allograft recipients. J Clin Pathol. 1978;31:338–347. [PMC free article: PMC1145271] [PubMed: 205555]
- 2.
- Karam G, Maillet F, Parant S. et al. Ureteral necrosis after kidney transplantation: risk factors and impact on graft and patient survival. Transplantation. 2004;78:725–729. [PubMed: 15371676]
- 3.
- Mackenzie EF, Poulding JM, Harrison PR. et al. Human polyoma virus (HPV)—a significant pathogen in renal transplantation. Proc Eur Dial Transplant Assoc. 1978;15:352–360. [PubMed: 216990]
- 4.
- Binet I, Nickeleit V, Hirsch HH. et al. Polyomavirus disease under new immunosuppressive drugs: a cause of renal graft dysfunction and graft loss. Transplantation. 1999;67:918–922. [PubMed: 10199744]
- 5.
- Drachenberg CB, Beskow CO, Cangro CB. et al. Human polyoma virus in renal allograft biopsies: morphological findings and correlation with urine cytology. Hum Pathol. 1999;30:970–977. [PubMed: 10452511]
- 6.
- Nickeleit V, Hirsch HH, Zeiler M. et al. BK-virus nephropathy in renal transplants-tubular necrosis, MHC-class II expression and rejection in a puzzling game. Nephrol Dial Transplant. 2000;15:324–332. [PubMed: 10692517]
- 7.
- Randhawa PS, Finkelstein S, Scantlebury V. et al. Human polyoma virus-associated interstitial nephritis in the allograft kidney. Transplantation. 1999;67:103–109. [PubMed: 9921805]
- 8.
- Howell DN, Smith SR, Butterly DW. et al. Diagnosis and management of BK polyomavirus interstitial nephritis in renal transplant recipients. Transplantation. 1999;68:1279–1288. [PubMed: 10573064]
- 9.
- Cosio FG, Amer H, Grande JP. et al. Comparison of low versus high tacrolimus levels in kidney transplantation: assessment of efficacy by protocol biopsies. Transplantation. 2007;83:411–416. [PubMed: 17318073]
- 10.
- Mengel M, Marwedel M, Radermacher J. et al. Incidence of polyomavirus-nephropathy in renal allografts: influence of modern immunosuppressive drugs. Nephrol Dial Transplant. 2003;18:1190–1196. [PubMed: 12748354]
- 11.
- Ramos E, Drachenberg C, Hirsch HH. et al. BK polyomavirus allograft nephropathy (BKPVN): eight-fold decrease in graft loss with prospective screening and protocol biopsy. Am J Transplant (supplement) WTC 2006 congress abstracts. 2006:121.
- 12.
- Gaber LW, Egidi MF, Stratta RJ. et al. Clinical utility of histological features of polyomavirus allograft nephropathy. Transplantation. 2006;82:196–204. [PubMed: 16858282]
- 13.
- Nickeleit V, Mihatsch MJ. Polyomavirus nephropathy in native kidneys and renal allografts: an update on an escalating threat. Transpl Int. 2006;19:960–973. [PubMed: 17081225]
- 14.
- Sachdeva MS, Nada R, Jha V. et al. The high incidence of BK polyoma virus infection among renal transplant recipients in India. Transplantation. 2004;77:429–431. [PubMed: 14966420]
- 15.
- Nada R, Sachdeva MU, Sud K. et al. Co-infection by cytomegalovirus and BK polyoma virus in renal allograft, mimicking acute rejection. Nephrol Dial Transplant. 2005;20:994–996. [PubMed: 15741209]
- 16.
- Nickeleit V. Animal models of polyomavirus nephropathy: hope and reality. Am J Transplant. 2006;6:1507–1509. [PubMed: 16827848]
- 17.
- Knowles WA, Pipkin P, andrews N. et al. Population-based study of antibody to the human polyomaviruses BKV and JCV and the simian polyomavirus SV40. J Med Virol. 2003;71:115–123. [PubMed: 12858417]
- 18.
- Bohl DL, Storch GA, Ryschkewitsch C. et al. Donor Origin of BK Virus in Renal Transplantation and Role of HLA C7 in Susceptibility to Sustained BK Viremia. Am J Transplant. 2005;5:2213–2221. [PubMed: 16095500]
- 19.
- Hirsch HH, Knowles W, Dickenmann M. et al. Prospective study of polyomavirus type BK replication and nephropathy in renal-transplant recipients. N Engl J Med. 2002;347:488–496. [PubMed: 12181403]
- 20.
- Nickeleit V, Gordon J, Thompson D. et al. Antibody titers and latent polyoma-BK-virus (BKV) loads in the general population: potential donor risk assessment for the development of BK-virus nephropathy (BKN) post transplantation. J Am Soc Nephrol (abstracts issue). 2004;15:524A.
- 21.
- Polo C, Perez JL, Mielnichuck A. et al. Prevalence and patterns of polyomavirus urinary excretion in immunocompetent adults and children. Clin Microbiol Infect. 2004;10:640–644. [PubMed: 15214877]
- 22.
- Singh HK, Bubendorf L, Mihatsch MJ. et al. Urine cytology findings of polyomavirus infections, in Polyomaviruses and Human Diseases, edited by Ahsan N, 1st ed, New York, NY. Georgetown, TX: Springer Science+Business Media, Landes Bioscience/Eurekah.com. 2006:201–212. [PubMed: 16626038]
- 23.
- Tani EM, Montenegro MR, Viana de Camargo JL. Urinary bladder washings in autopsies. A cytopathologic study of 63 cases. Acta Cytol. 1983;27:128–132. [PubMed: 6301184]
- 24.
- Nickeleit V, Hirsch HH, Binet IF. et al. Polyomavirus infection of renal allograft recipients: from latent infection to manifest disease. J Am Soc Nephrol. 1999;10:1080–1089. [PubMed: 10232695]
- 25.
- Nickeleit V, Klimkait T, Binet IF. et al. Testing for polyomavirus type BK DNA in plasma to identify renal allograft recipients with viral nephropathy. N Engl J Med. 2000;342:1309–1315. [PubMed: 10793163]
- 26.
- Nickeleit V, Steiger J, Mihatsch MJ. BK Virus Infection after Kidney Transplantation. Graft. 2002;5(suppl):S46–S57.
- 27.
- Nickeleit V, Singh HK, Mihatsch MJ. Polyomavirus nephropathy: morphology, pathophysiology and clinical management. Curr Opin Nephrol Hypertens. 2003;12:599–605. [PubMed: 14564196]
- 28.
- Buehrig CK, Lager DJ, Stegall MD. et al. Influence of surveillance renal allograft biopsy on diagnosis and prognosis of polyomavirus-associated nephropathy. Kidney Int. 2003;64:665–673. [PubMed: 12846764]
- 29.
- Nickeleit V, Mihatsch MJ. Polyomavirus nephropathy: pathogenesis, morphological and clinical aspects, in Verh Dtsch Ges Pathol, 88. Tagung, edited by Kreipe HH: Muenchen, Jena, Urban & Fischer. 2004:69–84. [PubMed: 16892536]
- 30.
- Wadei HM, Rule AD, Lewin M. et al. Kidney transplant function and histological clearance of virus following diagnosis of polyomavirus-associated nephropathy (PVAN). Am J Transplant. 2006;6:1025–1032. [PubMed: 16611340]
- 31.
- Drachenberg CB, Papadimitriou JC, Hirsch HH. et al. Histological patterns of polyomavirus nephropathy: correlation with graft outcome and viral load. Am J Transplant. 2004;4:2082–2092. [PubMed: 15575913]
- 32.
- Singh HK, Madden V, Detwiler R. et al. Detection of three dimensional polyomavirus clusters (“Haufen”) by negative staining urine electron microscopy: a sensitive and specific marker of polyomavirus-BK nephropathy. J Am Soc Nephrol (abstracts issue). 2006;17:760A–761A.
- 33.
- Baksh FK, Finkelstein SD, Swalsky PA. et al. Molecular genotyping of BK and JC viruses in human polyomavirus-associated interstitial nephritis after renal transplantation. Am J Kidney Dis. 2001;38:354–365. [PubMed: 11479162]
- 34.
- Trofe J, Cavallo T, First MR. et al. Polyomavirus in kidney and kidneypancreas transplantation: a defined protocol for immunosuppression reduction and histologic monitoring. Transplant Proc. 2002;34:1788–1789. [PubMed: 12176577]
- 35.
- Kazory A, Ducloux D, Chalopin JM. et al. The first case of JC virus allograft nephropathy. Transplantation. 2003;76:1653–1655. [PubMed: 14702550]
- 36.
- Milstone A, Vilchez RA, Geiger X. et al. Polyomavirus simian virus 40 infection associated with nephropathy in a lung-transplant recipient. Transplantation. 2004;77:1019–1024. [PubMed: 15087764]
- 37.
- Bruno B, Zager RA, Boeckh MJ. et al. Adenovirus nephritis in hematopoietic stem-cell transplantation. Transplantation. 2004;77:1049–1057. [PubMed: 15087771]
- 38.
- Funk GA, Steiger J, Hirsch HH. Rapid dynamics of polyomavirus type BK in renal transplant recipients. J Infect Dis. 2006;193:80–87. [PubMed: 16323135]
- 39.
- Petrogiannis-Haliotis T, Sakoulas G, Kirby J. et al. BK-related polyomavirus vasculopathy in a renal-transplant recipient. N Engl J Med. 2001;345:1250–1255. [PubMed: 11680445]
- 40.
- Sandler ES, Aquino VM, Goss-Shohet E. et al. BK papova virus pneumonia following hematopoietic stem cell transplantation. Bone Marrow Transplant. 1997;20:163–165. [PMC free article: PMC7101754] [PubMed: 9244421]
- 41.
- Singh D, Kiberd B, Gupta R. et al. Polyoma virus-induced hemorrhagic cystitis in renal transplantation patient with polyoma virus nephropathy. Urology. 2006;67:423 e411–423. [PubMed: 16461105]
- 42.
- Esposito L, Hirsch H, Basse G. et al. BK virus-related hemophagocytic syndrome in a renal transplant patient. Transplantation. 2007;83:365. [PubMed: 17297418]
- 43.
- Weinreb DB, Desman GT, Burstein DE. et al. Expression of p53 in virally infected tubular cells in renal transplant patients with polyomavirus nephropathy. Hum Pathol. 2006;37:684–688. [PubMed: 16733208]
- 44.
- Celik B, Randhawa PS. Glomerular changes in BK virus nephropathy. Hum Pathol. 2004;35:367–370. [PubMed: 15017594]
- 45.
- Nair R, Katz DA, Thomas CP. Diffuse glomerular crescents and peritubular immune deposits in a transplant kidney. Am J Kidney Dis. 2006;48:174–178. [PubMed: 16797403]
- 46.
- Herawi M, Parwani AV, Chan T. et al. Polyoma virus-associated cellular changes in the urine and bladder biopsy samples: a cytohistologic correlation. Am J Surg Pathol. 2006;30:345–350. [PubMed: 16538054]
- 47.
- Colvin RB, Nickeleit V. Renal transplant pathology, in Pathology of the Kidney (vol 2), edited by Jennette JC, Olson JL, Schwartz MM, Silva FG, 6 ed, Philadelphia, Baltimore, New York, London: Lippincott Williams & Wilkins. 2007. pp. 1347–1490.
- 48.
- Hirsch HH, Brennan DC, Drachenberg CB. et al. Polyomavirus-associated nephropathy in renal transplantation: interdisciplinary analyses and recommendations. Transplantation. 2005;79:1277–1286. [PubMed: 15912088]
- 49.
- Drachenberg RC, Drachenberg CB, Papadimitriou JC. et al. Morphological spectrum of polyoma virus disease in renal allografts: diagnostic accuracy of urine cytology. Am J Transplant. 2001;1:373–381. [PubMed: 12099383]
- 50.
- van Gorder MA, Della Pelle P, Henson JW. et al. Cynomolgus polyoma virus infection: a new member of the polyoma virus family causes interstitial nephritis, ureteritis and enteritis in immunosuppressed cynomolgus monkeys. Am J Pathol. 1999;154:1273–1284. [PMC free article: PMC1866551] [PubMed: 10233865]
- 51.
- Mannon RB, Hoffmann SC, Kampen RL. et al. Molecular evaluation of BK polyomavirus nephropathy. Am J Transplant. 2005;5:2883–2893. [PubMed: 16303001]
- 52.
- Chen Y, Trofe J, Gordon J. et al. Interplay of cellular and humoral immune responses against BK virus in kidney transplant recipients with polyomavirus nephropathy. J Virol. 2006;80:3495–3505. [PMC free article: PMC1440396] [PubMed: 16537617]
- 53.
- Krymskaya L, Sharma MC, Martinez J. et al. Cross-reactivity of T-lymphocytes recognizing a human cytotoxic T-lymphocyte epitope within BK and JC virus VP1 polypeptides. J Virol. 2005;79:11170–11178. [PMC free article: PMC1193623] [PubMed: 16103168]
- 54.
- Hammer MH, Brestrich G, andree H. et al. HLA type-independent method to monitor polyoma BK virus-specific CD4 and CD8 T-cell immunity. Am J Transplant. 2006;6:625–631. [PubMed: 16468975]
- 55.
- Abend JR, Low JA, Imperiale MJ. Inhibitory effect of gamma interferon on BK virus gene expression and replication. J Virol. 2007;81:272–279. [PMC free article: PMC1797268] [PubMed: 17035315]
- 56.
- Comoli P, Azzi A, Maccario R. et al. Polyomavirus BK-specific immunity after kidney transplantation. Transplantation. 2004;78:1229–1232. [PubMed: 15502726]
- 57.
- Bracamonte E, Leca N, Smith KD. et al. Tubular basement membrane immune deposits in association with BK polyomavirus nephropathy Am J Transplant 2007 . (in press) [PubMed: 17425622]
- 58.
- Han Lee ED, Kemball CC, Wang J. et al. A mouse model for polyomavirus-associated nephropathy of kidney transplants. Am J Transplant. 2006;6:913–922. [PubMed: 16611327]
- 59.
- Mayr M, Nickeleit V, Hirsch HH. et al. Polyomavirus BK nephropathy in a kidney transplant recipient: critical issues of diagnosis and management. Am J Kidney Dis. 2001;38:E13. [PubMed: 11532715]
- 60.
- Nickeleit V, Mihatsch MJ. Polyomavirus allograft nephropathy and concurrent acute rejection: a diagnostic and therapeutic challenge. Am J Transplant. 2004;4:838–839. [PubMed: 15084184]
- 61.
- Celik B, Shapiro R, Vats A. et al. Polyomavirus allograft nephropathy: sequential assessment of histologic viral load, tubulitis and graft function following changes in immunosuppression. Am J Transplant. 2003;3:1378–1382. [PubMed: 14525598]
- 62.
- Jeong HJ, Hong SW, Sung SH. et al. Polyomavirus nephropathy in renal transplantation: a clinicopathological study. Transpl Int. 2003;16:671–675. [PubMed: 12768232]
- 63.
- Ahuja M, Cohen EP, Dayer AM. et al. Polyoma virus infection after renal transplantation. Use of immunostaining as a guide to diagnosis. Transplantation. 2001;71:896–899. [PubMed: 11349723]
- 64.
- Zambrano A, Villarreal LP. A monoclonal antibody specific for BK virus large T-antigen (clone BK.T-1) also binds the human Ku autoantigen. Oncogene. 2002;21:5725–5732. [PubMed: 12173042]
- 65.
- Randhawa PS, Vats A, Zygmunt D. et al. Quantitation of viral DNA in renal allograft tissue from patients with BK virus nephropathy. Transplantation. 2002;74:485–488. [PubMed: 12352906]
- 66.
- Schmid H, Burg M, Kretzler M. et al. BK virus associated nephropathy in native kidneys of a heart allograft recipient. Am J Transplant. 2005;5:1562–1568. [PubMed: 15888070]
- 67.
- Schmid H, Nitschko H, Gerth J. et al. Polyomavirus DNA and RNA detection in renal allograft biopsies: results from a European multicenter study. Transplantation. 2005;80:600–604. [PubMed: 16177632]
- 68.
- Nickeleit V, Singh HK, Gilliland MGF. et al. Latent Polyomavirus Type BK Loads in Native Kidneys Analyzed by TaqMan PCR: What Can Be Learned To Better Understand BK Virus Nephropathy? J Am Soc Nephrol (abstracts issue). 2003;14:424A.
- 69.
- Boldorini R, Veggiani C, Barco D. et al. Kidney and urinary tract polyomavirus infection and distribution: molecular biology investigation of 10 consecutive autopsies. Arch Pathol Lab Med. 2005;129:69–73. [PubMed: 15628910]
- 70.
- Randhawa P, Shapiro R, Vats A. Quantitation of DNA of polyomaviruses BK and JC in human kidneys. J Infect Dis. 2005;192:504–509. [PubMed: 15995966]
- 71.
- Chesters PM, Heritage J, McCance DJ. Persistence of DNA sequences of BK virus and JC virus in normal human tissues and in diseased tissues. J Infect Dis. 1983;147:676–684. [PubMed: 6302172]
- 72.
- Limaye AP, Smith KD, Cook L. et al. Polyomavirus nephropathy in native kidneys of nonrenal transplant recipients. Am J Transplant. 2005;5:614–620. [PubMed: 15707418]
- 73.
- Nickeleit V, Zeiler M, Gudat F. et al. Detection of the complement degradation product C4d in renal allografts: diagnostic and therapeutic implications. J Am Soc Nephrol. 2002;13:242–251. [PubMed: 11752044]
- 74.
- Ramos E, Drachenberg CB, Papadimitriou JC. et al. Clinical course of polyoma virus nephropathy in 67 renal transplant patients. J Am Soc Nephrol. 2002;13:2145–2151. [PubMed: 12138148]
- 75.
- Rinaldo CH, Hirsch HH. Antivirals for the treatment of polyomavirus BK replication. Expert Rev Anti Infect Ther. 2007;5:105–115. [PubMed: 17266458]
- 76.
- Drachenberg CB, Papadimitriou JC, Wali R. et al. Improved outcome of polyoma virus allograft nephropathy with early biopsy. Transplant Proc. 2004;36:758–759. [PubMed: 15110653]
- 77.
- Brennan DC, Agha I, Bohl DL. et al. Incidence of BK with tacrolimus versus cyclosporine and impact of preemptive immunosuppression reduction. Am J Transplant. 2005;5:582–594. [PubMed: 15707414]
- 78.
- Hirsch HH, Drachenberg CB, Steiger J. et al. Polyomavirus associated nephropathy in renal transplantation: critical issues of screening and management, in Polyomaviruses and Human Diseases, edited by Ahsan N, 1st ed, New York, NY. Georgetown, TX. Springer Science+Business Media, Landes Bioscience/Eurekah.com. 2006:160–173. [PubMed: 16626034]
- 79.
- Singh HK, Madden V, Shen YJ. et al. Negative-staining electron microscopy of the urine for the detection of polyomavirus infections. Ultrastruct Pathol. 2006;30:329–338. [PubMed: 17090512]
- 80.
- Ding R, Medeiros M, Dadhania D. et al. Noninvasive diagnosis of BK virus nephritis by measurement of messenger RNA for BK virus VP1 in urine. Transplantation. 2002;74:987–994. [PubMed: 12394843]
- 81.
- Nickeleit V, Steiger J, Mihatsch MJ. Re: Noninvasive diagnosis of BK virus nephritis by measurement of messenger RNA for BK virus VP1 Transplantation 2003752160–2161. (letter) [PubMed: 12829935]
- 82.
- Koss LG. Chapter 22: the lower urinary tract in the absence of cancer, in Koss' diagnostic cytology and its histopathologic basis (vol 1), edited by Koss LG, Melamed MR, 5 ed, Philadelphia, Baltimore, New York: Lippincott Wiliams and Wilkins. 2006. pp. 738–776.
- 83.
- Erard V, Kim HW, Corey L. et al. BK DNA viral load in plasma: evidence for an association with hemorrhagic cystitis in allogeneic hematopoietic cell transplant recipients. Blood. 2005;106:1130–1132. [PMC free article: PMC1895165] [PubMed: 15845896]
- 84.
- Erard V, Storer B, Corey L. et al. BK virus infection in hematopoietic stem cell transplant recipients: frequency, risk factors and association with postengraftment hemorrhagic cystitis. Clin Infect Dis. 2004;39:1861–1865. [PubMed: 15578413]
- 85.
- Leung AY, Suen CK, Lie AK. et al. Quantification of polyoma BK viruria in hemorrhagic cystitis complicating bone marrow transplantation. Blood. 2001;98:1971–1978. [PubMed: 11535537]
- 86.
- Bressollette-Bodin C, Coste-Burel M, Hourmant M. et al. A prospective longitudinal study of BK virus infection in 104 renal transplant recipients. Am J Transplant. 2005;5:1926–1933. [PubMed: 15996241]
- 87.
- Razonable RR, Brown RA, Humar A. et al. A longitudinal molecular surveillance study of human polyomavirus viremia in heart, kidney, liver and pancreas transplant patients. J Infect Dis. 2005;192:1349–1354. [PubMed: 16170751]
- 88.
- Gordon J, Brennan D, Limaye AP. et al. Multicenter validation of polyomavirus BK quantification for screening and monitoring of renal transplant recipients. Am J Transplant. 2005;(suppl 11 5):381A–382A.
- 89.
- Singh HK, Shen YJ, Detwiler R. et al. Polyomavirus inclusion bearing decoy cell phenotypes: role in the management of patients with polyomavirus-BK nephropathy (BKN). J Am Soc Nephrol (abstracts issue). 2006;17:761A.
- 90.
- Tong CY, Hilton R, MacMahon EM. et al. Monitoring the progress of BK virus associated nephropathy in renal transplant recipients. Nephrol Dial Transplant. 2004;19:2598–2605. [PubMed: 15292464]
- 91.
- Alexander RT, Langlois V, Tellier R. et al. The prevalence of BK viremia and urinary viral shedding in a pediatric renal transplant population: a single-center retrospective analysis. Pediatr Transplant. 2006;10:586–592. [PubMed: 16856995]
- Polyomavirus Allograft Nephropathy: Clinico-Pathological Correlations - Madame C...Polyomavirus Allograft Nephropathy: Clinico-Pathological Correlations - Madame Curie Bioscience Database
- STATs and Infection - Madame Curie Bioscience DatabaseSTATs and Infection - Madame Curie Bioscience Database
- Regulation of Medical Devices - Madame Curie Bioscience DatabaseRegulation of Medical Devices - Madame Curie Bioscience Database
- Calreticulin in Cytotoxic Lymphocyte-Mediated Cytotoxicity - Madame Curie Biosci...Calreticulin in Cytotoxic Lymphocyte-Mediated Cytotoxicity - Madame Curie Bioscience Database
- Predicting the Spread of a Transgene in African Malaria Vector Populations: Curr...Predicting the Spread of a Transgene in African Malaria Vector Populations: Current Knowledge and Limitations - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...