

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

The coronin gene family comprises seven vertebrate paralogs and at least five unclassified subfamilies in nonvertebrate metazoa, fungi and protozoa, but no representatives in plants or distant protists. All known members exhibit elevated structural conservation in two unique domains of unknown function (DUF1899 and DUF1900) interspaced by three canonical WD40 domains (plus additional pseudo domains) that form part of a 7-bladed β-propeller scaffold, plus a C-terminal variable “coiled coil domain” responsible for oligomerization. Phylogenetic analysis of the N-terminal conserved region in known members (i.e. 420 aa in 250 taxa) established the origin of the founding monomeric unit and a dimeric paralog in unicellular eukaryotes. The monomeric ancestor duplicated to two distinct lineages in basal metazoa and later propagated during the whole genome duplications in primitive chordates 450-550 million years ago to form six vertebrate-specific genes. The delineation of 12 subfamily clades in distinct phyla provided a rational basis for proposing a simplified, universal nomenclature for the coronin family in accordance with evolutionary history, structural relationships and functional divergence.
Comparative genomic analysis of coronin subfamily locus maps and gene organization provided corroboratory evidence for their chromosomal dispersal and structural relatedness. Statistical analysis of evolutionary sequence conservation by profile hidden Markov models (pHMM) and the prediction of Specificity Determining Positions (SDPpred) helped to characterize coronin domains by highlighting structurally conserved sites relevant to coronin function and subfamily divergence. The incorporation of such evolutionary information into 3D models facilitated the distinction between candidate sites with a structural role versus those implicated in dynamic, actin-related cytoskeletal interactions. A highly conserved “KGD” motif identified in the coronin DUF1900 domain has been observed in other actin-binding proteins such as annexins and is a potential ligand for integrins and C2 domains known to be associated with structural and signalling roles in the membrane cytoskeleton. Molecular evolution studies provide a comprehensive overview of the structural history of the coronin gene family and a systematic methodology to gain deeper insight into the function(s) of individual members.
Introduction
Biological and molecular sequence data describing the coronin gene family provide intriguing but limited information about their species distribution, expression profiles and structural features relevant to function.1-3 A comprehensive phylogenetic analysis can be instructive to document gene family history, rationalize its nomenclature and fully appreciate the diversity and relatedness of individual members. Phylogenetic tree reconstruction is most reliable when supported by a broad representation of authenticated homologs from completed genomes to ensure the identification of all paralogous genes and their cognate orthologs in different species. Computational rigor can be achieved by sequence analysis using various models of molecular evolution that generate maximum likelihood or bootstrap confidence values for the branching topology in which branch order and lengths are consistent with current knowledge about species evolution.
The focus on gene family evolution is of central importance to understanding gene and protein function because it assembles much relevant information from comparative genomics and proteomics. It yields statistically verifiable results for associating patterns and processes of structural change with functional determination and adaptation in the natural selection of species. In particular, the ability to define structure-function relationships among orthologous groups at the level of individual gene subfamilies is vital for comprehending the functional diversity and specifcity of biological interactions for all members within a gene family. A multitude of algorithms in computational biology afford comprehensive, detailed views of genomic and proteomic data whilst others extend to comparative and investigative analyses. These can provide original insight into the structural features important for gene function because the targets of functional constraint can be deciphered from the statistics of evolutionary conservation and divergence. The elaboration of profile hidden Markov models (pHMM) and 3D structures that incorporate this information can be of predictive value and help to conceptualize and validate structure-function hypotheses worthy of empirical testing.
The vast quantity of molecular data being made available by ongoing genome sequencing projects provides a valuable resource that can be transformed, through computational biology techniques, into a knowledgebase relating evolutionary information to structure and function. The primary aims of the present study are to reconstruct the evolutionary relationships among all coronin subfamilies and use this as a framework to define their structural profiles with models that highlight functional relevance. Similar approaches can be contemplated for future studies to examine the molecular basis of coronin gene regulation or to evaluate coronin molecular interactions and conformational changes using in silico docking models.
Homolog Search and Assembly
Annotated protein databases such as UniProt (http://www.ebi.ac.uk/uniprot/) and PFAM (http://www.sanger.ac.uk/Sofware/Pfam/) and major sequence repositories such as National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/), Ensembl (http://www.ensembl.org) and KEGG (http://www.genome.ad.jp/kegg/) provide direct access to many known, full-length coronin sequences. Additional sequences were retrieved using bioinformatic search tools such as BLAST, specialized databases such as Homologene (http://www.ncbi.nlm.nih.gov/sites/entrez?db=homologene) and other resources to search, assemble and cross-compare novel sequences from expressed transcripts (dbEST) and whole genome shotgun (WGS) traces. Organism specialized sequence databases from the Joint Genome Institute (http://www.jgi.doe.gov/), the Broad Institute (http://www.broad.mit.edu) and Washington University (http://genome.wustl. edu/) were accessed to reconstruct coronins of special interest for evolutionary placement. These included preliminary sequence assemblies of Trichoplax adherens (placozoa), Monosiga brevicollis (choanoflagellate), Nematostella vectensis (sea anemone), Strongylocentrotus purpuratus (purple urchin), Branchiostoma floridae (amphioxus), Petromyzon marinus (sea lamprey) and Callorhincus milii (elephant shark). Completed genome assemblies of key model vertebrates such as Danio rerio (zebrafish), Xenopus tropicalis (western clawed frog), Gallus gallus (chicken) and mammals such as platypus, shrew, bat, mouse, rat, cow, dog and various primates were also searched for annotated or predicted homologs. PSI-BLAST and HMMER search tools were used to confirm the absence of coronin domains in Giardia intestinalis and the plant kingdom.4
Approximately 250 full-length protein sequences spanning all major phyla were assembled into a multiple sequence alignment using the ClustalW utility in Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0.5 Phylogenetic tree construction was initially performed by MEGA4 neighbor-joining analysis of maximum likelihood distances for 5000 bootstrap alignments of 400 conserved sites (i.e., excluding the coiled coil region) in approximately 250 coronin proteins from a broad range of species. These results were validated by Bayesian analysis using Mr Bayes6 and finally by maximum likelihood (ML) analysis using IQPNNI7 v3.2 to yield the robust family trees for animal coronins (Fig. 1A) and those in unicellular eukaryotes (Fig. 1B).
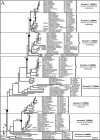
Phylogenetic Analysis
The most recent diverging clades of monomeric coronins comprise six “vertebrate” subfamilies with species orthologs ranging from primitive fishes such as jawless Agnatha (e.g., sea lamprey), cartilaginous Chondrichthyes (e.g., elephant shark) and teleosts (e.g., zebrafish) to mammals, including human (Fig. 1A). They are designated here in reverse phylogenetic order as new coronins 1 through 6 (see nomenclature proposal section below) accompanied by their currently official nomenclature symbols CORO1B, 1C, 6, 1A, 2A and 2B. The common ancestry and species distribution of these subfamilies point to their probable divergence during the two postulated whole genome duplications (WGD) in primitive chordate ancestors. Duplicate coronin genes frequently observed in teleosts are a consequence of the extra tetraploidization event early in this lineage approximately 300-350 million years ago (Mya); examples shown include Tetraodon nigroviridis (green spotted pufferfish) coronin 2, Gasterosteus aculeatus (stickleback) and Oryzias latipes (Japanese medaka fish) coronin 3 and Danio rerio (zebrafish) coronin 6. The recently discovered coronin 3 (CORO6) gene is phylogenetically close to coronin 4 (CORO1A) but also shares some aspects of gene organization with the coronin 5 (CORO2A) ancestor (see later). It is pertinent to note that subfamily orthologs branch in the expected speciation order, except for occasional displacements due to the inclusion of some partial sequences. The branch lengths from bifurcation nodes together with the observed species distribution for each subfamily permit an estimation of the age and timing of these gene duplication and speciation events based on the horizontal time scale, which is quasi-linear due to rate variations in individual gene evolution. The formation of these expanded clades occurred in rapid succession around the time of emergence of the first vertebrate about 550 Mya, represented by the sea lamprey Petromyzon marinus. The earliest metazoan coronin monomers originated in two distinct clades of newly assigned invertebrate subfamilies 8 and 9 further to the left on the horizontal scale (Fig. 1A). These are represented at their basal position by the only two coronins detected in the 50 Mb genome assembly of Trichoplax adherens, a placozoan considered to represent the most primitive animal cell. Other invertebrates such as the Cnidarian Nematostella vectensis, the echinoderm Strongylocentrotus purpuratus, the cephalochordate Branchiostoma floridae and various insects all similarly possess two monomeric coronins within these two founding clades. The branching order shown in Figure 1A clarifies that invertebrate coronin 9 probably shared its most recent common ancestor with vertebrate coronins 5 and 6 (CORO2A and 2B), whilst a distinct common ancestor of invertebrate coronin 8 probably gave rise to vertebrate coronins 1-4.
The N- and C-terminal halves of all coronin 7 dimer representatives were aligned as separate taxa, beginning with their respective DUF1899 “domains of unknown function”. They grouped into two adjacent clades rooted in the amoebae and fungal monomeric coronins (Fig. 1B), confirming that they originated from tandem duplication and fusion in a distant common ancestor, as opposed to the heterologous fusion of distinct monomers. The broad species distribution among protists, fungi and metazoa confirmed that this ubiquitous dimeric subfamily originated very early in coronin phylogeny and presumably conserves some basic cellular role. The relatively long branch lengths of some coronin 7 representatives reflect more extensive adaptive evolution (on horizontal scale) since the divergence from monomeric coronins. The basal branches in the coronin tree comprised monomeric coronins from fungi and diverse protozoa, the common ancestor of which formed coronin 7 via tandem duplication. Both maximum likelihood and independent bootstrap values at the bifurcation nodes of duplication or speciation gave generally strong support for the branching order. Minor variations in branch swapping were observed among basal taxa with low NJ-bootstrap support, such as the Dictyostelium representatives, but three well-defined clades distinguished the coronins from Alveolata, Fungi and Euglenozoa, which were assigned to new subfamilies 10, 11 and 12, respectively. The tree was rooted in the single coronin detected in Naegleria gruberi, a basal lineage of amoebae that transforms through a unique three-stage life cycle as amoeba, flagellate (with microtubule cytoskeleton) and cyst. Isolated cases of duplicate monomeric coronins in Entamoeba, Cryptosporidium and Trichomonas all appear to be relatively recent lineage-specific duplications. A unique duplicative fusion event in a common ancestor of Dictyostelium and Entamoeba gave rise to a novel gene fusion product of coronin with villin to form villidin (VilA, e.g., GenBank accession XP_636652) which is phylogenetically close to the coronin 7 C-terminal and coronin 10 Alveolata clades.
Nomenclature Proposal for the Coronin Gene Family
A successful nomenclature scheme for gene families is a priority objective of fundamental importance for the annotation and information exchange of emerging genomes. Ideally, it should be based on a comprehensive overview of each family and incorporate ample information about gene relationships in a clear, intuitive manner (Table 1). Original nomenclature compilations unified diverse terminologies derived from independent discoveries under the common name of coronins 1 through 5 assigned by a simple numbering scheme.3 The currently official system from the Human Gene Nomenclature Committee (HGNC, http://www.genenames.org/) established the important precedent of an abbreviated gene symbol (CORO) as the primary unique identifier for information management and retrieval by electronic databases and attempted to define known gene relationships with enumeration by number-letter combinations (i.e., CORO1A, 1B, 1C, 2A, 2B, 6, 7). The apparent failure of the latter to be generally adopted stems from its intention of imparting more information by invoking awkward designations and by doing so prematurely such that later discovered coronins 6 and 7 appear dissociated and mislabeled. The lack of a broader overview of the family also precluded both systems from formally classifying many known invertebrate and protozoan coronins into a universal, systematic gene family nomenclature able to convey context, order and diversity.
Table 1
Nomenclature proposal for the coronin gene family, based on phylogenetic relationshipsa.
The reconstruction of an ostensibly complete and properly rooted phylogenetic tree provides all the necessary information to devise a rational and universal nomenclature scheme that respects the principles and standards of systematic nomenclature and satisfies user needs for a simple, intuitive and informative system (Table 1). The delineation of major paralogous gene duplications and orthologous speciation events now makes it practicable to incorporate key evolutionary relationships as the scientific basis for associating structure with function, while preserving flexibility for future growth upon discovery of novel gene duplications or clade divisions. In order to maintain the gene name “coronin” with its changing usage and aliases during nomenclature development, a new symbol (e.g.,“CRN”) is needed to supplant the present symbol (CORO) and assume a dominant role for the immediate implementation and reliable dissemination of changes emanating from the HGNC via electronic database crosslinks to the worldwide research community. Its simple, strict format with contiguous number suffixes but without punctuation (i.e., no hyphens, spaces, periods, etc.) supplements other constant identifiers such sequence accessions, gene IDs or citation references to provide instant, unambiguous gene identification for all past and future scientific records, allowing the corresponding gene name to take a subordinate descriptive role. Lineage-specific gene duplicates would be designated with a standardized letter suffix (e.g., stick-leback CRN3a and CRN3b) and alternative DNA transcripts can also be specified (e.g., human CRN2v1, CRN2v2, CRN2v3).
It is prudent to bear in mind that a universal nomenclature scheme with an intuitively logical scientific basis offers the greatest assurance for easy learning by newcomers to the field, faithful adherence by established investigators and an effective, enduring knowledgebase for biomedical research. The proposed nomenclature revision has attempted to combine the simple elegance of the original scheme with the identification power of a unique gene symbol and to build on these features by embracing the entire family and inculcating rational order so as to place all relevant genetic information in proper context. Any additional refinements to the proposed scheme should resist the tendency to grasp familiar relics, especially the deficiencies of outdated systems for reasons of nostalgia or personal convenience as this would surely defeat the long-term objective of attaining a coherent nomenclature system that consolidates ongoing research. The current official records and future changes in coronin nomenclature can always be consulted at the websites of the Human Gene Nomenclature Committee (http://www.genenames.org/) and Mouse Genome Informatics (http://www.informatics.jax.org/). This chapter utilizes the proposed new nomenclature (Table 1) based on original data (Figs. 1A,B) and accompanied by these official symbols.
Domain Structures and Hidden Markov Models
The primary structure characterization of coronin homologs was a prerequisite for validating homolog authenticity in the input alignment for phylogenetic analysis. We observed that coronin protein sequences aligned well with many highly conserved sites and few insertions and deletions, at least up to the C-terminal coiled coil region. All were confirmed to have congruent domain organization with two unique “Domains of Unknown Function” designated DUF1899 and DUF1900 (PFAM entries PF08953 and PF08954, respectively), separated by three canonical WD40 domains (i.e., defined by significant match scores to the PFAM-HMM model) and terminating in a coiled coil region of variable composition and length (Fig. 2A). The DUF1899 and DUF1900 coronin domains served as effective “bait” for the detection of distant coronin homologs and their unique, highly conserved structures likely play key roles in regulating actin dynamics in the cytoskeleton. The 3 canonical WD40, two “pseudo WD40” (i.e., vestigial, eroded domains) and two flanking “atypical domains” form the characteristic 7-bladed propeller scaffold structure of coronins.8 One effective way to visualize the primary structure conservation or “molecular fingerprint” of these and other coronin domains is to use a multiple sequence alignment to generate a statistical “profle hidden Markov model” (pHMM) and transform it into a sequence logo (Fig. 2B). This composite logo outlines the amino acid distribution and over all conservation level at each site in a “typical” human coronin, based on the contribution of amino acids from all 8 human coronin monomers, including the component halves of coronin 7 separately in the alignment. Since the computation by HMMER9 and LogoMat10 also measures the relative probability that such a distribution of amino acid replacements would be found at random in nature, the total column height gives a measure of the entropy or information content that can be used to infer the functional importance of each site.
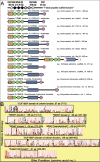
The 65-aa DUF1899 exhibits a prominent cluster of highly conserved (basic) amino acids at the amino terminus representing a coronin signature1 that has been previously suggested to harbor a putative actin binding site just downstream of a phosphoserine modification site.11,12 The three “true” WD40 domains that follow may vary in some coronin isoforms due to alternative exon splicing and two sequential “pseudo domains” exhibit much lower detectability by sequence search algorithms. While the typical WD40 domain (PFAM accession PF00400.23) generally has a conserved Gly-His early in the domain, internal and C-terminal Trp and several conserved Asp and Arg residues, other WD-like motifs in the center of DUF1899, a WL pair at the end of DUF1900 and atypical flanking domains may serve an analogous structural role together with the authentic WD40 domains.8,13 This cluster is followed by a 139-aa DUF1900 domain unique to coronins, with well-defined conservation of Pro residues (inducing structural turns), charged Asp and Arg residues (disposed to external interactions) and a bulky aromatic Trp near its C-terminus. Our attention was drawn to the presence of a highly conserved “KGD” motif spanning an exon splice site in phase 1 of the codon (i.e., between the first and second nucleotide) that is constant in all coronins. This feature is identical to that observed in actin-binding members of the annexin gene family and identifies a potential ligand for interaction with membrane-bound integrins and signalling complexes enriched with C2-domain containing enzymes.14 The C-terminal coiled coil region is most variable in composition and length, with additional insertions in coronin 5 and coronin 7 linker region and heterologous domain extensions (e.g., pleckstrin homology, gelsolin) in some yeast and amoeba monomeric coronins. The unique villidins of Dictyostelium (XP_636652) and Entamoeba (AF118397) are fascinating evolutionary anomalies resulting from gene fusion between the coronin and villin families of actin-binding proteins.
Structural Features of Coronin Subfamilies
The ability to decipher functionally relevant conserved sites or regions in the primary structure of the coronin family HMM (Fig. 2) can be obscured by subfamily divergence, so it is important to extend such analysis to ortholog groups of individual subfamilies to obtain distinctive molecular fngerprints for each. We therefore compiled pHMM models from multiple sequence alignments for each of the known subfamilies, including the invertebrate, fungal and protist monomeric coronins, to statistically validate new member classification and to conduct pairwise HMM-HMM comparisons.15 Another effective approach to detect “specifcity determining positions” in multiple alignments of subfamily ortholog groups was available in the SDPpred algorithm.16 The essential results of these analyses are presented in an alignment of pHMM consensus sequences for each of the coronin subfamilies (Fig. 3). Back-shaded residues within the top-line domain sequences denote sites which are conserved but different between subfamily groups, most evident in the multiple aa replacements between coronins 1-4 versus 5-6. Among members of the latter subfamilies, conserved Cys have been replaced by Thr-78 and Gly-152 in coronin 3 and Val-345 in coronin 1. The greatest divergence is seen between the two component halves of coronin 7 sequences, in agreement with the phylogenetic tree (Fig. 1B) and apparent binding and functional differences.17 Conserved characteristics within each of the three typical WD40 domains can be seen in the HMM logo of Figure 2 although variations between the individual domains and subfamilies can also be discerned, including the presence of a WD dipeptide in the fourth “pseudo” domain despite extensive loss of other WD40 sequence characteristics. Population allelic polymorphisms (e.g., human nsSNPs in Fig. 3) represent another confirmed source of coronin sequence variation.

Corroboration from Genetic Maps and Structures
Exon splicing patterns incorporated into Figure 3 confirm their conservation in species representatives of the individual subfamilies, but marked differences and similarities between subfamilies testify to their relatedness. Only two DUF1900 splice sites are universally conserved, including the split “KGD” motif, whereas marked differences between the monomeric halves of coronin 7 and their use of rare phase-2 introns (i.e., between codon bases 2 and 3) reflect their remote time of divergence. The (dis)similarity between splice sites of coronins 1-4 versus 5-6 reflects the extent of coevolutionary relatedness, with the exception of a coronin 3 WD40 cassette exon splice site, which instead coincides with coronins 5 and 6. It may be noted that total gene lengths, principally reflecting intron sizes, bear only minor resemblance to the evolutionary relatedness of coronins 1, 4 and 6 (all 5-7 kbp), coronins 2 and 3 (both 84 kbp), coronin 7 (62 kbp) and coronin 5 (149 kbp).
The pattern of coronin subfamily expansion near the dawn of vertebrates (Fig. 1A) suggests that it was a consequence of the whole genome duplications believed to have taken place during the chordate to vertebrate transition.18 Since coronin genes are generally dispersed in vertebrate genomes (see ref. 2) their genetic loci may coincide with the emerging picture of chromosomal duplications being deduced from ongoing genome assemblies. One of the better documented examples of these events comes from the large human “paralogon” groups located on chromo-some 9 near band q22.3 (coronin 5 locus) and chromosome 15 in band q23 (coronin 6). We have observed homologous relationships of these same chromosomal regions and the phylogenetic branching between annexins A1 on human chromosome 9q21 and its direct descendent annexin A2 on 15q22.19 Partial sequences from lamprey and elephant shark strongly suggest their presence in all vertebrate coronin subfamilies, hence their origin from duplication events in the chordate common ancestor. The precise timing of this putative chromosome duplication event involving pairs of coronins, annexins and many other paralogous gene pairs awaits final genome assembly maps for lamprey, hagfish and their chordate predecessors.
3D Modelling of Evolutionary Information
The evaluation of evolutionarily conserved sites for their functional relevance needs to be considered in a realistic spatial context such as 3-dimensional models to distinguish, for example, whether their importance lies in preserving intramolecular protein structure and conformation or in directing intermolecular interactions via external charged residues or surface contours. CONSURF is one of several webservers that employs protein family alignments together with 3D crystal or NMR structural homolog models deposited in the Protein DataBank (http://www. rcsb.org/pdb/) to calculate and display site-specific information about evolutionary conservation.20 Figure 4 portrays such a model, rendered by MolMol (http://www.mol.biol.ethz.ch/wuthrich/ sofware/molmol/) as a space-filling model of human coronin 4 limited to the backbone atoms of DUF1899, WD40 and DUF1900 domains. The structure is based on the pdb:2aq5 model of mouse coronin 4 (i.e., CORO1A in ref. 7) and the graded shading indicates the level of amino acid conservation at each site based on the entire protein alignment utilized for phylogenetic analysis (Fig. 3). Analytical tools accompanying protein modelling programs assist in molecular inspection by supplying helpful information about solvent accessibility and bond angles and the 3D view offers rotational movement and enhanced visual perspective. The static 2D image in Fig. 4 does however provide some orientative information by localizing the N-terminal basic cluster in DUF1899 on the top surface, the WD dipeptides in WD40 domains comprising part of the propeller scaffold structure at the bottom-right, the highly conserved “KGD” motif in a central, externally exposed region of DUF1900 and part of the C-terminal coiled coil region hidden at the back of the image.

Conclusion
The phylogenetic analysis of known coronins presents a comprehensive overview of this gene family and lays a foundation to advance studies on their distribution, structure and functional diversity in other species. The knowledge of evolutionary relationships should facilitate comparative genomic studies that will help to elucidate the fundamental cellular roles of coronins and rationalize their apparent absence from plants, which are also distinguished by their exceptionally high ratio of monomeric to filamentous actin. The diversity of coronins in unicellular organisms is likely to be much more expansive than is presently known and may help to define basic aspects of their multifunctionality in relation to cellular morphology, vesicular trafficking, locomotion and cytokinesis. Although coronin 7 exhibits the greatest evolutionary success in terms of species distribution, hence functional adaptation, the divergence of the six monomeric subfamilies within vertebrates may be more closely associated with vertebrate-specific functions. The precise timing and mechanisms of the duplicative expansion of these six vertebrate coronin subfamilies will be clarified upon completion of the genome assemblies for early chordates, jawless and cartilaginous fishes. The available data provide perspective and rationale for developing a universal nomenclature for the entire coronin family based on a simple, ordered numbering scheme. Vertebrate coronin subfamilies could be enumerated in reverse evolutionary order of their appearance as coronins 1-7, followed by the incorporation of new coronin 8 and 9 subfamilies descended from their invertebrate common ancestors and terminating with three distant subfamilies based on their species distribution in alveolata (coronin 10), fungi and yeasts (coronin 11) and euglenozoa or amoebae (coronin 12).
The development of pHMM statistical models for each of the individual coronin subfamilies is a powerful tool to classify new homologs and to make pairwise comparisons of molecular profiles with the aim of defining the structural basis of functional specificity for each subfamily. The observation that the domain structures of monomeric coronins are congruent and contain unique, conserved sequence domains (largely of unknown function) will facilitate their structural characterization and comparison with other family members by sequence threading of available 3D structures. These approaches will help to define key functional domains and the basis of sub-family differences. For closely related subfamilies, it is equally important to define the functional determinants of gene and protein regulation, which are also amenable to evolutionary analysis by phylogenetic footprinting.
Computational biology and in silico modelling strategies that utilize evolutionary information from comparative genomic studies can facilitate the prediction, validation and inference of concepts and hypotheses useful in the design and conduct of molecular research. The possibilities go far beyond the present study but they should realistically include molecular docking studies to test the feasibility of putative functional interactions of coronins with, for example, actin and components of known regulatory complexes. Experimental verification of the key functional structures in the unique coronin DUF domains would make a particularly valuable contribution to understanding their subcellular localization, physiological roles and mechanisms. Finally, clinical genetic knowledge about phenotype changes involving coronin single nucleotide polymorphisms (SNPs, see Fig. 3) affecting protein expression, structure and function could be validated by population studies such as HapMap (http://www.hapmap.org/) and combined with molecular modelling studies to help elucidate coronin physiological roles and mechanisms and to identify pathological alleles of medical relevance.
Acknowledgements
This work was supported by research grant BFU2007-67876 from the Ministry of Science and Technology of Spain, research grant IB05-128, personnel support from the “Fundación para el Fomento en Asturias de la Investigación Científica Aplicada y la Tecnología” (FICYT) and the “Programa Ramón y Cajal” jointly funded by the European Union, Spanish Ministry of Education and Science and University of Oviedo.
References
- 1.
- Rybakin V, Clemen CS. Coronin proteins as multifunctional regulators of the cytoskeleton and membrane trafficking. Bioessays. 2005;27:625–632. [PubMed: 15892111]
- 2.
- Uetrecht AC, Bear JE. Coronins: the return of the crown. Trends Cell Biol. 2006;16:421–426. [PubMed: 16806932]
- 3.
- de Hostos EL. The coronin family of actin-associated proteins. Trends Cell Biol. 1999;9:345–350. [PubMed: 10461187]
- 4.
- Hussey PJ, Allwood EG, Smertenko AP. Actin-binding proteins in the Arabidopsis genome database: properties of functionally distinct plant actin-depolymerizing factors/cofilins. Phil Trans R Soc Lond B. 2002;357:791–798. [PMC free article: PMC1692981] [PubMed: 12079674]
- 5.
- Tamura K, Dudley J, Nei M. et al. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. [PubMed: 17488738]
- 6.
- Ronquist F, Huelsenbeck JP. MRBAYES 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. [PubMed: 12912839]
- 7.
- Vinh LS, von Haeseler A. IQPNNI: Moving fast through tree space and stopping in time. Mol Biol Evol. 2004;21:1565–1571. [PubMed: 15163768]
- 8.
- Appleton BA, Wu P, Wiesmann C. The crystal structure of murine coronin-1: a regulator of actin cytoskeletal dynamics in lymphocytes. Structure. 2006;14:87–96. [PubMed: 16407068]
- 9.
- Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. [PubMed: 9918945]
- 10.
- Schuster-Bockler B, Schultz J, Rahmann S. HMM logos for visualization of protein families. BMC Bioinformatics. 2004;5(7) [PMC free article: PMC341448] [PubMed: 14736340]
- 11.
- Oku T, Itoh S, Okano M. et al. Two regions responsible for the actin binding of p57, a mammalian coronin family actin-binding protein. Biol Pharm Bull. 2003;26:409–416. [PubMed: 12673016]
- 12.
- Cai L, Holoweckyj N, Schaller MD. et al. Phosphorylation of coronin 1b by protein kinase C regulates interaction with Arp2/3 and cell motility. J Biol Chem. 2005;280:31913–31923. [PubMed: 16027158]
- 13.
- Rosentreter A, Hofmann A, Xavier CP. et al. Coronin 3 involvement in F-actin-dependent processes at the cell cortex. Exp Cell Res. 2007;313:878–895. [PubMed: 17274980]
- 14.
- Morgan RO, Martin-Almedina S, Garcia M. et al. Deciphering function and mechanism of calcium-binding proteins from their evolutionary imprints. Biochim Biophys Acta—MCR. 2006;1763:1238–1249. [PubMed: 17092580]
- 15.
- Söding J. Protein homolog y detection by HMM-HMM comparison. Bioinformatics. 2005;21:951–960. [PubMed: 15531603]
- 16.
- Kalinina OV, Mironov AA, Gelfand MS. et al. Automated selection of positions determining functional specificity of proteins by comparative analysis of orthologous groups in protein families. Protein Sci. 2004;13:443–456. [PMC free article: PMC2286703] [PubMed: 14739328]
- 17.
- Rybakin V, Gounko NV, Spate K. et al. Crn7 interacts with AP-1 and is required for the maintenance of Golgi morphology and protein export from the Golgi. J Biol Chem. 2006;281:31070–31078. [PubMed: 16905771]
- 18.
- Panopoulou G, Poustka AJ. Timing and mechanism of ancient vertebrate genome duplications—the adventure of a hypothesis. Trends Genetics. 2005;21:559–567. [PubMed: 16099069]
- 19.
- Fernandez MP, Morgan RO. Structure, function and evolution of the annexin gene superfamily. In: Bandorowicz-Pikula J Ed. Annexins: biological importance and annexin-related pathologies, Landes Bioscience/Kluwer Academic/Plenum. 2003:21–37.
- 20.
- Glaser F, Pupko T, Paz I. et al. ConSurf: identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics. 2003;19:163–164. [PubMed: 12499312]
- Introduction
- Homolog Search and Assembly
- Phylogenetic Analysis
- Nomenclature Proposal for the Coronin Gene Family
- Domain Structures and Hidden Markov Models
- Structural Features of Coronin Subfamilies
- Corroboration from Genetic Maps and Structures
- 3D Modelling of Evolutionary Information
- Conclusion
- Acknowledgements
- References
- Molecular Phylogeny and Evolution of the Coronin Gene Family - Madame Curie Bios...Molecular Phylogeny and Evolution of the Coronin Gene Family - Madame Curie Bioscience Database
- Cell-Cell Fusion: Transient Channels Leading to Plasma Membrane Merger - Madame ...Cell-Cell Fusion: Transient Channels Leading to Plasma Membrane Merger - Madame Curie Bioscience Database
- Replacement of Specific Neuronal Populations in the Spinal Cord - Madame Curie B...Replacement of Specific Neuronal Populations in the Spinal Cord - Madame Curie Bioscience Database
- The End in Sight: Poly(A), Translation and mRNA Stability in Eukaryotes - Madame...The End in Sight: Poly(A), Translation and mRNA Stability in Eukaryotes - Madame Curie Bioscience Database
- Origin and Evolution of DNA and DNA Replication Machineries - Madame Curie Biosc...Origin and Evolution of DNA and DNA Replication Machineries - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...