

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

The seven members of the mammalian signal transducer and activator of transcription (STAT) family participate in a wide range of biological processes with impact both on the generation and the functional regulation of cells involved in antimicrobial immunity. Activation of STATs is a hallmark of innate as well as adaptive immune responses. Here we review stimuli and signals for STAT activation arising during immune responses to microbes. We describe the severe consequences of STAT deficiency. Moreover, we emphasize the dual nature of STATs as enhancers of immunity on one hand and as important mediators of immunosuppression and anti-inflammatory activity on the other.
Introduction
Identification of Stats and genetic proof of their dual function as signal transducers and activators of transcription in the labs of Jim Darnell and George Stark/Ian Kerr resulted from attempts to understand how interferons (IFN) act to rapidly change gene expression of a cell.1 The essential and nonredundant role of IFN as antiviral cytokines (type I and type III IFN, IFN-I and IFN-III) or as a macrophage activating cytokines (IFN–γ) strongly suggested that the STATs activated by IFN receptors, STAT1 and STAT2, would prominently determine the course of both viral and nonviral infections. Subsequent work in a number of labs identified five further members of the mammalian STAT family and unearthed their unique role in signal transduction and the reprogramming of gene expression downstream of class I and class II cytokine receptors.2 Moreover, an evolutionary origin of STATs in nonvertebrate organisms was revealed. Recent evidence suggests an important role of nonvertebrate STATs during development and for the innate immune response to virus.3,4
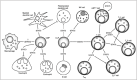
All mammalian STATs participate in both innate and adaptive immune responses to microbes. Compared to the immediate enhancement of immunocompetence by IFN/STAT1/STAT2, other STATs influence antimicrobial immunity in a more indirect way. For example, STATs 4, 5 and 6 exert a large impact on immune responses by regulating the generation of hematopoietic cells and/or their differentiation into effector populations (Fig. 1). Furthermore, while IFN are mostly linked to immune activation, other STATs like STAT3 and STAT6 participate in the suppression of immune and inflammatory responses by mediating effects of, respectively, IL-10 and IL-4. To add yet another level of complexity the same STAT can be part of stimulatory or suppressive pathways. As an example, STAT3 is activated by both the IL-6 cytokine family or by IL-22, which can promote inflammation in addition to IL-10, which suppresses it.5
Activation of STATs by tyrosine phosphorylation occurs during innate antimicrobial immune responses when signals emitted by pattern recognition receptors (PRR) stimulate the expression of cytokine genes (Fig. 2). Thus, ligands for class I or class II cytokine receptors are generated and cause JAK-STAT signal transduction. For example, signals emanating from RNA helicases after binding of viral RNA, or the TRIF signalling pathway downstream of toll-like receptors (TLR) 3 and 4 will stimulate transcription of type I IFN (IFN-I) genes. IFN-I, in turn, will signal through the IFN-I receptor and cause tyrosine phosphorylation of STATs 1 and 2.6 Other or additional pathways downstream of TLR target the IL-6 and/or IL-10 genes and cause activation of STAT3.7-9 A further possibility how PRR can influence the activity of STATs is through serine phosphorylation.10 Several STATs contain serine phosphorylation sites in their C-terminal transactivation domains. Addition of a phosphate changes their transcriptional activity. Mutation of this site in STAT1 or STAT4 reduces their immunostimulatory properties.11,12 Importantly, serine phosphorylation through PRR signals occurs also in absence of a cytokine deploying a canonical JAK-STAT pathway, i.e., in absence of tyrosine phosphorylation.13 It has long been speculated that STATs phosphorylated only at the C-terminal serine residue may regulate immunological and/or growth properties of cells, but compelling evidence for this hypothesis has not yet been provided.14
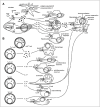
Figure 2
STAT activation in the innate and adaptive immune responses. Only the most prominent examples and their functional consequences are shown. For explanation see text.
In addition to their activation by the innate immune system, STAT-activating cytokines can be produced by activated lymphocytes during the adaptive antimicrobial immune response. Following exposure to cytokines produced during antigen presentation or in the early phase of T-cell activation, STATs are activated in T-cells and participate in their differentiation into distinct subsets. Each subset then produces a characteristic pattern of STAT activating cytokines. Examples of these are the IFN–γ produced by TH1 cells, or the IL-4 and IL-10 secreted by TH2 or TR1 T-cell subsets.
As a rule, a cytokine will activate the same STAT(s) in different cells. A well-documented exception to this rule, IFN-I, which usually target STATs 1 and 2, can stimulate STAT4 tyrosine phosphorylation in TH1 and NK cells.15 As will be described further below, this switch between STATs has important implications for the course of immune responses to pathogens.
It is clear from the abovementioned that there is a very limited number of generalizations that can be made for all STATs and their role in antimicrobial immunity. To account for this fact the following sections of our article will deal with members of the STAT family individually or group them into functional contexts.
STAT1 and STAT2
STAT1 and STAT2 are essential mediators of host responses to interferons (IFN), pleiotropic cytokines induced after infection with all classes of pathogens. Whereas STAT2 in general is only activated in response to type I and type III IFN (IFN-I and IFN-III, respectively), STAT1 plays an essential role in IFN-I, IFN–γ (type II) and IFN-III-stimulated signaling.1,16
IFN–γ, the only type II IFN species, is produced by a limited number of immune cells like activated TH1 or CD8+ T-lymphocytes and NK-cells. Its role in macrophage activation and protective immunity to intracellular bacteria such as Listeria, Mycobacteria, Salmonella or Chlamydia is well documented.17,18 Although not explicitly shown for each infection, it is safe to assume that STAT1 is essential wherever IFN–γ plays a major role. Production of IFN–γ is controlled by STAT4, a family member activated by IFN-I or IL-12 (see below).
Contrasting IFN–γ, most likely all cells in the body are able to produce IFN-I, a family of cytokines with about 20 members.19 Most important in the context of antimicrobial immunity are more than 10 IFN-α species and, in most mammals, a single IFN–β. IFN-I are best known as antiviral cytokines. However, IFN-I/STAT1/STAT2 have significant impact on innate immunity to nonviral pathogens as well.20,21 In some cases, best documented for Listeria monocytogenes infection of mice, type I IFN can have adverse effects on immunity to bacteria (reviewed in21). Induction of IFN-I is stimulated by the recognition of pathogen-associated molecular patterns (PAMPs) through pattern recognition receptors (PRRs) located either at the plasma membrane (e.g., toll-like receptors (TLR) 3 and 4 recognizing dsRNA or LPS, respectively, the endosome (TLRs 7 and 9 recognizing ssRNA or DNA) or in the cytoplasm (the RNA helicases RIG-I and MDA-5 sensing viral RNA, or the cytoplasmic DNA receptors). A number of distinct adapter molecules (TRIF, MAVS*, MyD88) and TRAF3/6 ubiquitin ligases relay the signal to serine threonine kinases (TBK1, IKKε, IRAK4, IKKα) which are directly or indirectly involved in phosphorylating two members of the family of interferon regulatory factors, IRF-3 and IRF-7.6,9,21,23 These in turn stimulate expression of IFN-I genes.24 A similar sequence of events most likely stimulates IFN-III synthesis (IFN-λ 1-3, also named, respectively, IL-29, IL-28A and IL-28B).25 IFN-III act via STAT1 and 2 and their biological activity is most likely very similar to that of IFN-I.16 Contrasting IFN-I, however, IFN-III target a very restricted number of cells. The benefit of having IFN-III in addition to IFN-I has not been clarified, but it may lie in reinforcing the first line of antiviral defense particularly in epithelia.26,27
IFN receptors are comprised of two subunits. These are IFNAR1 and IFNAR2, associated with TYK2 and JAK1 tyrosine kinases, for the IFN-I receptor and IFNGR1 and IFNGR2, associated with JAK1 and JAK2 tyrosine kinases, for the IFN–γ receptor. Binding of IFNs to the receptor triggers a series of phosphorylation events on the receptor chains and activation of the associated JAKs, leading to the formation of docking sites for STAT1 (in case of IFNGR) or of STAT1 and STAT2 (in case of IFNAR). STAT1 and STAT2 are phosphorylated on tyrosine residues 701 and 689, respectively, by the JAKs and can form heterodimers and homodimers via reciprocal binding of their SH2-domains. STAT1/STAT2 heterodimers, which are formed downstream of IFNAR stimulation, associate with a third protein, IFN regulatory factor 9 (IRF-9) to form the transcription factor interferon stimulated gene factor (ISGF) 3, which translocates to the nucleus and binds to interferon stimulated response element (ISRE) promoter sequences.1 In a highly similar fashion ISGF3 formation is brought about by the IFN-λ receptor consisting of IFN-λR1 and IL-10R2 chains.16 STAT1 homodimers, or γ-IFN-activated factor (GAF), which are formed downstream of IFNGR and to a lesser extent also downstream of IFNAR, bind to γ-IFN–activated-site (GAS) promoter sequences to stimulate ISG transcription.1
Classically, the role of IFN-I is to alert the immune system to fight against viral infections. Accordingly, many target genes with ISGF3 binding sites in their promoters have antiviral properties. In contrast, IFN–γ was mainly described as an important activator of macrophages in response to intracellular nonviral pathogens. So far, more than 1000 genes have been described to be stimulated by either IFN-I, IFN–γ or both and only for some of them has a clear microbicidal function been assigned.28,29
IFN/STAT Target Genes Involved in Antimicrobial Defense
A prominent group of IFN-target genes are members of the guanidine 5′ triphosphatase (GTPase) families including the genes encoding Mx proteins, the p65 Guanylate binding proteins (GBP) and the p47 GTPase family.
The promoter of Mx genes contains ISRE but no GAS and induction is thus strictly dependent on IFN-I signalling and the formation of ISGF3. The Mx familiy consists of relatively high molecular weight (70-80 kDa) proteins with a tendency for multimerization. It includes Mx1 and Mx2 in mice and MxA and MxB in humans.30 Mx1 is mostly localized in the nucleus and confers resistance to Orthomyxoviruses and Thogaviruses replicating in the nucleus, whereas the cytosolic Mx2 is directed against cytosolic viruses like Bunyaviruses. Oligomerization via the core GTPase domain initiates binding of the small ubiquitin-like modifier 1 (SUMO-1) protein system to trigger viral degradation. Human MxA has been shown to be active against a large variety of viruses like Orthomyxoviruses, Paramyxoviruses, Picornaviruses and Hepatitis B. It blocks the nuclear export of viral mRNA or sequesters viral capsid proteins from nuclear import.
Resistance to vesicular stomatitis virus (VSV) and Encephalomyocarditis viruses is accomplished by the expression of p65 GBP proteins, GBP1 and GBP2.31 The antiproliferative activity of GBP1 might limit viral spread, but otherwise their antiviral mechanism is poorly understood. p65 GBP proteins are induced by both IFN-I and IFN–γ and their promoters contain both ISRE and GAS. STAT1 phosphorylation at serine 727 enhances GBP expression because it increases the recruitment of p300/CBP histone acetylases.11,32
Beside the Mx proteins, dsRNA-dependent protein kinase R (PKR) and the 2′5′ oligoadenylate synthetase (OAS) are the antiviral ISGs, hence STAT1/2 target genes, that have been most extensively studied.33 PKR is activated by dsRNA produced during viral replication. Activated PKR undergoes dimerization and phosphorylates the α subunit of the eukaryotic translational initiation factor 2 (eIF2α), resulting in inhibition of translation. Additionally, PKR is involved in induction of apoptosis and cell cycle arrest.34 Similar to PKR, OAS is activated by dsRNA. It triggers oligomerization of ATP through unusual 2′-5′ phosphodiester linkages, which then bind and activate RNase L. RNase L in turn degrades cellular and viral RNAs. Recently, it has been shown that RNase L also has a function more upstream in the pathway, in the initiation of IFN-I production.35 In response to 2′-5′-linked oligoadenylate, it produces small RNA cleavage products from self-RNA which are recognized by RIG-I or MDA5 to induce type I IFN production.
Viruses have evolved an amazing capacity to inhibit IFN-I induction, signalling or response. Beside the attempts to minimize recognition by limiting the production of PAMPS (e.g., paramyxoviruses are capping the 5′-end of their mRNA to avoid recognition by RIG-I), the strategies involve for example the inhibition of PRRs (e.g., Influenza virus inhibiting RIG-I), targeting and degradation of adapters (e.g., NS3/4A protease of Hepatitis C Virus targeting MAVS), or interferon regulatory factors (e.g., Herpes simplex virus targeting IRF-3 and IRF-7). Neutralization of IFNs (e.g., Poxviruses) and degradation of STATs (e.g., the genus Rubulavirus), inhibition of IFN-induced enzymes with antiviral activity (e.g., downregulation of RNase L activity by HIV) or a combination of all45,46 are further weapons in the fight of viruses against the antiviral properties of IFN. Inhibition of STAT activation by nonviral intracellular pathogens has been observed.47,48 Compared to viruses, the underlying mechanisms are poorly understood.
The largest group of IFN-stimulated GTPases is the p47 family with more than 20 members in mice.36,37 However, only 6 members (IRG-47, LRG-47, TGTP/MG21, IGTP, LIGP1 and GTPI) have been characterized so far. Their expression is dramatically upregulated even at low concentrations of IFN–γ and can also be induced somewhat by IFN-I. As in the case of the p65 GTPases, expression requires STAT1 phosphorylated on serine 727. ISRE have been found upstream of some p47 gene promoters whereas GAS is present in all of them. The main antimicrobial mechanism elicited by p47 proteins is to alter the pathogen containing phagosome within infected macrophages. Thus, knock-out mice for members of this family are particularly susceptible to intracellular pathogens like Salmonella, Mycobacteria, Listeria, or Toxoplasma. In case of Mycobacterium tuberculosis or Toxoplasma gondii the activity of, respectively, the LRG-47 and IGTP proteins aids autophagic pathogen destruction by promoting the fusion of microbe-containing vacuoles with autophagic vesicles.38,40
Another antimicrobial mechanism of innate immune cells is the generation of NO and O2-radicals.18 IFNs and STAT1 enhance their synthesis by the upregulation of the inducible Nitric Oxide Synthase (iNOS) and the phagocyte NADPH oxidase, respectively.41 iNOS catalyzes the conversion of L-arginine and molecular oxygen to citrulline and NO, which further reacts to •NO. •NO and its derivatives directly lead to killing or reduced replication of infectious agents by mutation of DNA and inhibition of DNA repair and protein synthesis. Indirect effects of iNOS upregulation involve arginine depletion, since several infectious pathogens are dependent on the presence of exogenous arginine. The iNOS promoter contains binding sites for NFκB, AP-1 and STAT1 dimer (GAF). Full promoter activation is achieved by the combination of at least two stimuli, an IFN signal leading to GAF formation and a pathogen- associated molecular pattern (PAMP) leading to the activation of NFκB through signals from a PRR. iNOS-deficient mice are highly susceptible against Salmonella typhimurium, Mycobacterium tuberculosis, Leishmania major, Trypanosoma cruzi, Coxsackie virus and murine Cytomegalovirus, whereas the enzym is dispensable for the control of Shigella flexneri, Streptococcus pneumoniae, Trypanosoma brucei, Plasmodium chabaudi, mouse Hepatitis virus or Sendai virus. In certain cases like infection with Influenza virus or with Listeria monocytogenes, •NO can be detrimental to the host and exert cytotoxicity against cells and tissues.41,42 The NADPH or phagocyte oxidase (PHOX) catalyzes the conversion of molecular oxygen to O2-radicals, one of several reactive oxygen species (ROS) with antimicrobial activity. The active enzyme is a multi-subunit complex assembled in response to the phagocytosis of microbes. IFN–γ/STAT1 increase PHOX activity because they stimulate expression of at least two subunits of the enzyme (gp91phox, p67phox;43,44).
Impact of STAT1/2 Deficiency on Infections
STAT1-deficient mice have been generated more than ten years ago by two independent labs.49,50 Cells derived from these mice are nonresponsive to both IFN-α/β and IFN–γ and do not induce classical ISGs like IRF-1, GBP1, iNOS or MHC II. STAT1-defient mice are born at a Mendelian ratio and show no abnormal growth defects, however, when hosted under conventional conditions, die readily due to infections with opportunistic pathogens. Similar to IFNAR1-deficient mice, STAT1-deficient mice succumb to a sublethal dose of VSV with extremely high titers of virus replication, whereas mice heterozygous for the disrupted STAT1 allele survive like wild-type mice. Likewise, STAT1-deficient mice are 100-fold more sensitive to Influenza virus infection than their wild-type counterparts.51 Additionally, they are highly susceptible to infection with the intracellular bacterium Listeria monocytogenes and die with similar kinetics as mice treated with an anti-IFN–γ antibody. STAT1 also enhances the systemic inflammation resulting from LPS administration. Consistently, STAT1-/- mice survive moderate LPS quantities better than wt mice.11,52
More recent studies employing microarray analysis showed that a subset of genes is induced by IFN–γ through IFNGR and JAK1 without a requirement for STAT1.53,54 Other genes are repressed by STAT1 and therefore upregulated by IFNs in STAT1 null, but not in wild-type cells. This may explain why STAT1-deficient mice are 100 times more resistant to murine cytomegalovirus (MCMV) and Sindbis virus (SV) than mice double-deficient for type I and type II IFN receptors.53
Human STAT1 deficiencies or mutations confirm the pivotal importance of STAT1 for innate immunity to intracellular pathogens deduced from studies with knock-out mice. In two cases infants carrying homozygous mutated STAT1 alleles suffered from mycobacterial disease but died of viral disease.57 Other patients with mycobacterial disease were found to carry a STAT1 mutation which was dominant in one cell type but recessive for the other. Nuclear accumulation of GAF but not of ISGF3 was impaired in heterozygous cells. Consequently, these patients did not suffer from increased susceptibility to virus infection.58
Similar to STAT1 deficiency the absence of STAT2 produces an almost complete loss of protection against RNA viruses such as VSV.55 Mouse Cytomegalovirus has evolved a mechanism to selectively inhibit STAT2 without an effect on STAT1 activation and signalling by coding for a 79 kD protein named M27. M27 expression confers resistance to IFN. Surprisingly, the absence of M27 increases the susceptibility of this virus to IFN–γ much more dramatically then to type I IFNs. The study explains that this is in part due to STAT2 activation directly by IFN–γ.56 The general implications of these findings for the role of STAT2 in antiviral immunity need to be further explored.
Regulation of Adaptive Immunity by STAT1/2
Besides its role in innate immune response to pathogens, STAT1 also has an important function in the modulation of adaptive immunity. STAT1-deficient mice show an exacerbated, proinflammatory pathological process with a strong bias towards a TH2 response in response to Influenza infection, which was mainly assigned to a lack of IFN-I responsiveness.59 STAT1-deficient lymphocytes display hyperproliferation after TCR stimulation and reduced apoptosis with lower levels of caspases 1 and 11, an effect, which is only partly due to the abrogated response to IFN–γ or IFN-I.60 Despite the evidence for an antiproliferative and proapoptotic effect of IFN on T-cells in vitro, others have shown that IFN–γ stimulation increases the abundance of CD8+ T-cells during viral infection of mice.61
Intriguingly, the antiproliferative and proapoptotic effects of IFN-I on wild-type T-cells change to antiapoptotic and mitogenic effects in T-cells lacking STAT1 or STAT2.62,63 The growth-promoting effect may result from STAT5 activation by the IFNAR in absence of STAT1.62 It is independent of STAT3 because type I IFN treatment of STAT1/STAT3 double-knock-out T-cells still stimulates growth.63
Expansion of antigen-specific CD8+ T-cells after Lymphocytic Choriomeningitis virus infection in vivo and the ability to generate memory cells is critically dependent on type I IFN signalling, shown by adoptive transfer experiments with IFNAR1-deficient CD8+ T-cells.64 In line with this, Vaccinia virus infection of mice demonstrated the importance of STAT1 for the generation of virus-specific CD8+ T-cells and for CD8+ cell memory.65 A critical role for dendritic cell STAT1 for the induction of TH1 responses was shown by infection studies with the intracellular parasite Leishmania major.66 Adoptive transfer experiments in this model suggest that IFNGR and STAT1 are dispensable for the differentiation of protective TH1 cells, whereas the lack of STAT1 in DCs results in impaired upregulation of MHC and costimulatory molecules and, consequently, reduced antigen presentation and T-cell priming.
Cross-priming of CD8+ T-cells after infection with Lymphocytic Choriomeningitis virus is enhanced by type I IFN.67 The data suggest that this is probably not due to increased antigen presentation after IFN-α stimulation, but rather to the upregulation of costimulatory signals. However, for full induction of cross-priming a direct effect of IFN-α on CD8+ T-cells is needed.68,69
STAT3
STAT3 activation is caused by many cytokine receptors. Most relevant for immunity to infection are the IL-6 family signalling through the common gp130 receptor subunit (including IL-6, IL-11, IL-27 and IL-3170), IL-21,71 IL-22,72 the antiinflammatory IL-10,5 but also cytokines with predominantly hematopoietic activity like G-CSF73 and Flt3L.74 Deletion of the STAT3 gene in the germ line causes embryonic lethality.2 Therefore most available genetic information about nonredundant roles of STAT3 in the immune system has been obtained by tissue-specific disruption of the STAT3 gene in mice. Moreover, two STAT3 isoforms, STAT3α and STAT3β, differ in their C-termini due to alternative splicing and induce partially distinct sets of target genes. Mice expressing only one of the isoforms have delivered additional information about STAT3 functions.75,76 While these alterations of the STAT3 locus by gene targeting have yielded a considerable amount of information about pro- and antiinflammatory activities of the transcription factor, their consequences have not been explored extensively with regard to infection. Therefore, available knowledge how the complex activity of STAT3 in immune responses translates into antimicrobial action is rather limited.
The spectrum of STAT3-activating cytokines predicts its pleiotropic impact on antimicrobial immunity. The protein negatively regulates granulopoiesis73 and contributes to the Flt3L-dependent expansion of DC progenitors.74 Moreover, it is involved in the IL-6 and IL-23-dependent differentiation of TH17 cells and their IL-21-mediated production of IL-17 and IL-22.77,79 Consistent with the important role of TH17 cytokines IL-17 and IL-22 in neutrophil recruitment and inflammation, they provide defense against infection with extracellular bacteria such as Klebsiella pneumoniae.80 Although not directly shown, STAT3 in this context is clearly linked to proinflammatory and bactericidal activity through its regulation of TH17 development and function and, possibly, by mediating the effects of IL-22. When mice with myeloid cell ablation of STAT3 were tested in a cecal ligation and puncture model of septic peritonitis, the outcome was strikingly different.81 Such mice showed strongly increased inflammation, but nevertheless failed to clear bacteria more efficiently than wild-type controls. This study thus suggests a predominant role of myeloid cell STAT3 in limiting inflammation and providing effector function. Taken together, the data point to a proinflammatory and microbicidal effect of T-cell STAT3 and an inflammation-limiting, yet microbicidal role in myeloid cells. The antiinflammatory properties of myeloid cell STAT3 were further revealed by its important role in controlling colitis as a response to gut commensals.82,84
The antiinflammatory and immunesuppressive activities of STAT3 result to a large extent from its critical role in IL-10 signalling. More precisely, STAT3′s suppressive effect on the LPS-induced septic shock or on the development of chronic enterocolitis is attributed to the lack of a myeloid cell response to IL-10. As a downside of its immunesuppressive activity, STAT3 may mediate the IL-10-dependent reactivation of tuberculosis in murine infection models.85 IL-10-independent suppression of proinflammatory cytokine production by STAT3 was reported for macrophages infected with Toxoplasma gondii86 and for endothelial cells.87
IL-10 signalling is an exclusive property of the STAT3α isoform.75 By contrast, both STAT3α and STAT3β contribute to the regulation of inflammation and infection through transcriptional control of acute phase gene expression in the liver. STAT3α and STAT3β stimulated by IL-6 family cytokines elicit an acute phase response that balances the LPS-induced septic shock. Whereas STAT3β improves recovery, but not so much survival, STAT3α has a much more profound effect on the resistance to LPS.75,76 Of note, the liver acute phase response counteracts the LPS-induced septic shock, but is part of systemic inflammation in response to other noxious agents such as turpentine.88 This further emphasizes the yin and yang of STAT3 in the regulation of inflammation.
It is not entirely clear how STAT3 regulates both pro- and anti-inflammatory pathways, sometimes in the same cell type. An important factor appears to be the feedback inhibitor of cytokine signalling, SOCS3. The SOCS3 gene is a STAT3 target and provides feedback inhibition on IL-6 family receptors. However, IL-10 receptors are not inhibited by SOCS3 and STAT3 signalling can continue in presence of SOCS3.77,89 Studies in SOCS3-deficient cells suggest this difference to be part of the explanation for the pro- and antiinflammatory action of STAT3 in IL-6 and IL-10 responses, respectively.90 SOCS3 inhibition of the G-CSF receptor is most likely also underlying the increased granulopoiesis observed in absence of STAT3.73
STAT4 and STAT6
STATs 4 and 6 can be regarded as immunological antagonists based on their supportive role for the development of type I and type II immune responses, respectively. It results mainly from their nonredundant function in IL-12 and IL-4/13 pathways. Gene-deficient mice clearly support the notion that STAT4 is required for TH1 development and IFN–γ production by TH1 as well as NK cells, whereas STAT6 mediates TH2 differentiation and the synthesis of the TH2 signature cytokines IL-4, IL-5 and IL-13.91,95 Consistent with this, STAT4 strengthens immunity against pathogens cleared by predominantly cellular mechanisms, most prominently intracellular viral, bacterial and protozoan pathogens. The STAT4 requirement for IFN–γ production in response to signalling by the IL-12 receptor links immunity against all pathogens requiring IFN–γ/STAT1 for clearance with those requiring STAT4.93,96,100 Conversely, STAT6 enhances antibody-mediated effector mechanisms that arise from IL-4-mediated isotype switching, particularly IgE. This may contribute to immunity against helminths such as gastrointestinal nematodes.101 It should be noted, however, that the undisputed importance of IL-4/IL-13/STAT6 for immunity against gastrointestinal nematodes does not primarily derive from their regulation of the humoral immune response. For example, STAT6 appears to influence mast cells and smooth muscle cells in the process of Trichinella spiralis expulsion from the intestinal tract.102,103 IL-4 signalling by non-bone marrow-derived cells was shown to protect from infection with Strongyloides venezuelensis.104,105 Studies with the larval stage of Taenia crassiceps show that IL-4/IL-13/STAT6 do not generally provide protective immunity against helminth infections. In case of this parasite, STAT6 correlated with susceptibility and STAT4 with increased control of infection.106,107
Deficiency for STAT4 or STAT6 skews TH differentiation towards TH2 and TH1 subsets, respectively. In some cases such as Ectromelia virus, cutaneous leishmaniasis, T. cruzi or mycobacterial infection, the increased propensity to develop type I immunity in STAT6-deficient mice produces better clearance of infections.108,110 This results from an increase in IL-12-driven, cell-mediated effector mechanisms on the one hand and a decreased activity of the immunesuppressive TH2 cytokines on the other. While the importance of STAT4 and STAT6 for type I and type II immunity against infection has been documented mostly by work with gene-targeted mice, it appears that at least some results apply to humans as well. For example, a defect in IL-12-mediated STAT4 nuclear translocation was reported for T-cells of a patient with recurrent mycobacterial infections.111 In case of STAT6, polymorphisms in the STAT6 gene were linked with increased resistance to nematode infections. The same haplotypes predisposed to increased IgE and allergic responses.112,113
Besides TH development, STAT4 and STAT6 also produce opposing effects on the inflammatory response that accompanies infection. IL-12/STAT4 are positively and IL-4/IL-13/STAT6 are negatively correlated with inflammation. The enhancement by IL-12 derives from its importance for IFN–γ production, from p38MAPK signalling and most likely additional mechanisms. The antiinflammatory activity of IL-4/IL-13/STAT6 results from its supportive role in the development of IL-10 and TGF–β- producing TH2 cells. Moreover, STAT6 was recently shown to limit the life span of T effector cells.114 A further important mechanism accounting for the immunesuppressive and antiinflammatory activity of IL-4 is alternative macrophage activation. M2 macrophages arise upon treatment with IL-4 and have a phenotypical appearance opposed to that of classically activated, microbicidal M1 macrophages.115 Alternatively activated macrophages contribute to resistance against nematodes.116 Besides a characteristic profile of chemokine production and response, a hallmark of alternatively activated macrophages is the production of arginase, a STAT6 target gene.117 This enzyme counteracts the proinflammatory and microbicidal effect of nitric oxide. Pro- and antiinflammatory activities as part of a complex immune response produce the seemingly paradox finding that both STAT4 and STAT6-deficient mice display an increased resistance to septic peritonitis.118 This is explained with an increased local clearance of bacteria in STAT6-/- animals and reduced systemic inflammation upon STAT4 deficiency. A different situation was observed upon administration of LPS. In this case STAT6 deficiency increased the endotoxin shock as expected by the absence of antiinflammatory potential. Surprisingly, STAT4 deficiency also increased the susceptibility to the septic shock syndrome, owing to increased levels of IL-12 in LPS-treated STAT4-/- mice.119
Besides IL-12, IFN-I are capable of activating STAT4 in TH1 and NK cells.15 This was shown to contribute to type I immunity against Salmonella typhimurium120 and also to the protective effect of exogenous IFN-I on the course of Leishmania major infection.121 The STAT4/STAT1 expression ratio in TH1 and NK cells was shown to determine association with the IFN-I receptor and preferential activation of one or the other STAT by IFN-I. Elegant studies with Lymphotropic Choriomengitis virus (LCMV) demonstrated an altered expression ratio and a concomitant switch from preferential IFN-I activation of STAT4 in the early immune response to a preferential activation of STAT1 at later stages.122 Since STAT1 cannot substitute for STAT4 in regulating IFN–γ transcription, preferential STAT1 activation limits excessive IFN–γ production during viral infection.
STAT5
Two STAT5 isoforms, STAT5a and STAT5b are encoded by distinct, but highly related genes. Notwithstanding their relatedness, the functional redundancy of the two transcription factors is incomplete, due to distinct expression patterns and unique sets of target genes. Disruption of the STAT5 genes in mice either individually or together revealed essential roles, among others, in hematopoiesis, mammary gland development and function, the control of body size and metabolic homeostasis. Mammary gland development appears to be mediated predominantly by STAT5a, whereas body size control and metabolic homeostasis are STAT5b mediated (reviewed in123). The interaction and/or complementarity of STAT5a and b is evident from the fact that disruption of both genes causes more severe hematopoietic defects compared to STAT5a alone.123 STAT5 is activated by single chain hematopoietin receptors (e.g., the EpoR) as well as by receptors using the common β and γ chains, including receptors for GM-CSF, IL-2, IL-3 and IL-7. Consistently STAT5a/b-deficiency or disregulation of STAT5 activity causes defects in erythroid, myeloid and lymphoid lineages.124,127 STAT5 not only influences the hematopoietic generation of these cells but also contributes to the expansion of peripheral T-cells during immune responses,128 the production of IL-4 by TH2 cells and Eosinophils,129,130 the activation of mast cells by IgE131 and the M2 programming of macrophages.132 GM-CSF and its target transcription factor STAT5 determine the fate of dendritic cell progenitors. The transcription factor suppresses the promoter of the IRF8 gene. Since IRF8 is critically required for the differentiation of plasmacytoid DC, STAT5 instructs a myeloid DC fate at the expense of plasmacytoid DC.133
The crucial role of IL-2 in the generation of regulatory T-cells (Treg) is reflected by the fact that absence of STAT5 leads to a severe defect in Treg development, hence a loss of immune tolerance.134,135 STAT5 is suggested to regulate the Treg commitment factor FoxP3.126,135,136 The critical role of STAT5 isoforms particularly for the normal development of Treg as well as other T-cell or NK populations is also demonstrated by patients with STAT5b mutations which display various immune defects besides showing small body size (reviewed in123).
Despite numerous reports about the important role of the STAT5 isoforms in the generation and functional integrity of an antimicrobial immune system, there are hardly any studies testing the impact of STAT5 deficiency on immune responses to infection. This is explained by the perinatal lethality of STAT5a/b -/- mice. The scarcity of data on the role of STAT5 in infection will eventually be overcome by tissue restricted ablation. A link has been established, however, between STAT5 and SIV or HIV infection. On the one hand STAT5 was shown to contribute to HIV transcription and its productive replication in CD4+ T-cells.137 On the other hand STAT5 expression/activation appears to be suppressed by HIV. This leads to defects in T-cell effector functions and in hematopoiesis.138,139 A recent study shows that both HIV and SIV suppress STAT5 expression in hematopoietic progenitors and that there is a corresponding loss of the multipotency of these cells that requires the SIV-encoded Nef protein. This defect could be rescued by forced expression of STAT5b.139
Drosophila STAT
The JAK/STAT pathway is highly conserved during evolution and thought to be a landmark of the single cell: metazoan boundary.140 In Drosophila, it was originally discovered through its role in embryonic development. The main components of this pathway are unpaired (Upd—the ligand), domeless (Dome—the receptor), hopscotch (Hop—the JAK) and STAT92E (Marelle—the STAT).3 STAT92E is most similar to human STAT5.3
Drosophila immunity to bacterial pathogens and fungi relies on activation of the toll-pathway, which is specific for Gram-positive bacteria and fungi and the Imd-pathway, which is specific for Gram-negative bacteria and is mainly accomplished by NFκB-mediated induction of antimicrobial peptides. Although some genes expressed in response to bacterial infection require STAT92E, mutants for Hop control bacterial infections much like wild-type insects. In contrast, the response against viruses relies on two distinct mechanisms: The activation of the JAK/STAT pathway141 and RNA interference employing Dicer-2 to control virus replication.142 Infection of flies with drosophila C virus triggers activation of STAT92E and upregulation of a set of genes distinct from those regulated by the toll or the Imd pathways. Furthermore, flies with a loss-of-function mutation for Hop are more sensitive to infection with Drosophila C virus with increased viral load and enhanced mortality.141 RNAi-mediated screening of Drosophila cells confirmed the importance of the JAK-STAT pathway to defend this organism against viral pathogens.4
Acknowledgements
Work in our lab is supported by the Austrian Science Foundation (FWF) through grant P20522-B05 and through SFB F28.
References
- 1.
- Darnell JE Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signalling proteins. Science. 1994;264:1415–421. [PubMed: 8197455]
- 2.
- Levy DE, Darnell JE Jr. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–62. [PubMed: 12209125]
- 3.
- Agaisse H, Perrimon N. The roles of JAK/STAT signalling in drosophila immune responses. Immunol revs. 2004;198:72–82. [PubMed: 15199955]
- 4.
- Arbouzova NI, Zeidler MP. JAK/STAT signalling in drosophila: insights into conserved regulatory and cellular functions. Development. 2006;133:2605–616. [PubMed: 16794031]
- 5.
- Murray PJ. Understanding and exploiting the endogenous interleukin-10/STAT3-mediated anti-inflammatory response. Curr opin Pharmacol. 2006;6:379–86. [PubMed: 16713356]
- 6.
- Akira S. TLR signalling. Curr Top Microbiol Immunol. 2006;311:1–16. [PubMed: 17048703]
- 7.
- Hirotani T, Yamamoto M, Kumagai Y. et al. Regulation of lipopolysaccharide-inducible genes by MyD88 and toll/IL-1 domain containing adaptor inducing IFN-beta. Biochem Biophys Res Commun. 2005;328:383–92. [PubMed: 15694359]
- 8.
- Chang EY, Guo B, Doyle SE. et al. Cutting edge: involvement of the type I IFN production and signalling pathway in lipopolysaccharide-induced IL-10 production. J Immunol. 2007;178:6705–6709. [PubMed: 17513714]
- 9.
- ONeill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in toll-like receptor signalling. Nat rev Immunol. 2007;7:353–364. [PubMed: 17457343]
- 10.
- Decker T, Kovarik P. Serine phosphorylation of STATs. Oncogene. 2000;19:2628–637. [PubMed: 10851062]
- 11.
- Varinou L, Ramsauer K, Karaghiosoff M. et al. Phosphorylation of the stat1 transactivation domain is required for full-fledged IFN-gamma-dependent innate immunity. Immunity. 2003;19:793–802. [PubMed: 14670297]
- 12.
- Morinobu A, Gadina M, Strober W. et al. STAT4 serine phosphorylation is critical for IL-12-induced IFN-gamma production but not for cell proliferation. Proc Natl Acad Sci USA. 2002;99:12281–86. [PMC free article: PMC129436] [PubMed: 12213961]
- 13.
- Kovarik P, Stoiber D, Eyers PA. et al. Stress-induced phosphorylation of STAT1 at ser727 requires p38 mitogen- activated protein kinase whereas IFN-gamma uses a different signalling pathway. Proc Natl Acad Sci USA. 1999;96:13956–61. [PMC free article: PMC24172] [PubMed: 10570180]
- 14.
- Ramana CV, Chatterjee-Kishore M, Nguyen H. et al. Complex roles of stat1 in regulating gene expression. Oncogene. 2000;19:2619–27. [PubMed: 10851061]
- 15.
- Nguyen KB, Watford WT, Salomon R. et al. Critical role for STAT4 activation by type 1 interferons in the interferon-gamma response to viral infection. Science. 2002;297:2063–66. [PubMed: 12242445]
- 16.
- Ank N, West H, Paludan SR. IFN-lambda: novel antiviral cytokines. J Interferon Cytokine Res. 2006;26:373–79. [PubMed: 16734557]
- 17.
- Schroder K, Hertzog PJ, Ravasi T. et al. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–89. [PubMed: 14525967]
- 18.
- Decker T, Stockinger S, Karaghiosoff M. et al. IFNs and STATs in innate immunity to microorganisms. J Clin Invest. 2002;109:1271–77. [PMC free article: PMC150987] [PubMed: 12021240]
- 19.
- Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines and their receptors. Immunol revs. 2004;202:8–32. [PubMed: 15546383]
- 20.
- Bogdan C, Mattner J, Schleicher U. The role of type I interferons in nonviral infections. Immunol. 2004;202:33–48. [PubMed: 15546384]
- 21.
- Decker T, Muller M, Stockinger S. The yin and yang of type I interferon activity in bacterial infection. Nat revs. 2005;5:675–87. [PubMed: 16110316]
- 22.
- Meylan E, Tschopp J, Karin M. Intracellular pattern recognition receptors in the host response. Nature. 2006;442:39–44. [PubMed: 16823444]
- 23.
- He JQ, Oganesyan G, Saha SK. et al. TRAF3 and its biological function. Adv Exp Med Biol. 2007;597:48–59. [PubMed: 17633016]
- 24.
- Honda K, Taniguchi T. IRFs: master regulators of signalling by toll-like receptors and cytosolic pattern-recognition receptors. Nat revs Immunol. 2006;6:644–58. [PubMed: 16932750]
- 25.
- Osterlund PI, Pietila TE, Veckman V. et al. IFN regulatory factor family members differentially regulate the expression of type III IFN (IFN-lambda) genes. J Immunol. 2007;179:3434–42. [PubMed: 17785777]
- 26.
- Ank N, Iversen MB, Bartholdy C. et al. An important role for type III interferon (IFN-lambda/IL-28) in TLR-induced antiviral activity. J Immunol. 2008;180:2474–85. [PubMed: 18250457]
- 27.
- Sommereyns C, Paul S, Staeheli P. et al. IFN-lambda (IFN-lambda) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 2008;4:e1000017. [PMC free article: PMC2265414] [PubMed: 18369468]
- 28.
- Ehrt S, Schnappinger D, Bekiranov S. et al. Reprogramming of the macrophage transcriptome in response to interferon-gamma and mycobacterium tuberculosis: signalling roles of nitric oxide synthase-2 and phagocyte oxidase. J Exp Med. 2001;194:1123–40. [PMC free article: PMC2193509] [PubMed: 11602641]
- 29.
- Der SD, Zhou A, Williams BR. et al. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc Natl Acad Sci USA. 1998;95:15623–28. [PMC free article: PMC28094] [PubMed: 9861020]
- 30.
- Haller O, Stertz S, Kochs G. The Mx GTPase family of interferon-induced antiviral proteins. Microbes Infect. 2007;9:1636–43. [PubMed: 18062906]
- 31.
- Martens S, Howard J. The interferon-inducible GTPases. Ann rev Cell Dev Biol. 2006;22:559–89. [PubMed: 16824009]
- 32.
- Ramsauer K, Farlik M, Zupkovitz G. et al. Distinct modes of action applied by transcription factors STAT1 and IRF1 to initiate transcription of the IFN-gamma-inducible gbp2 gene. Proc Natl Acad Sci USA. 2007;104:2849–2854. [PMC free article: PMC1815270] [PubMed: 17293456]
- 33.
- Stark GR, Kerr IM, Williams BR. et al. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–64. [PubMed: 9759489]
- 34.
- Sadler AJ, Williams BR. Structure and function of the protein kinase R. Curr Top Microbiol Immunol. 2007;316:253–92. [PubMed: 17969452]
- 35.
- Malathi K, Dong B, Gale M Jr. et al. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature. 2007;448:816–19. [PMC free article: PMC3638316] [PubMed: 17653195]
- 36.
- MacMicking JD. IFN-inducible GTPases and immunity to intracellular pathogens. Trends Immunol. 2004;25:601–09. [PubMed: 15489189]
- 37.
- Taylor GA. IRG proteins: key mediators of interferon-regulated host resistance to intracellular pathogens. Cell Microbiol. 2007;9:1099–107. [PubMed: 17359233]
- 38.
- Singh SB, Davis AS, Taylor GA. et al. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science. 2006;313:1438–41. [PubMed: 16888103]
- 39.
- Deretic V, Singh S, Master S. et al. Mycobacterium tuberculosis inhibition of phagolysosome biogenesis and autophagy as a host defence mechanism. Cell Microbiol. 2006;8:719–27. [PubMed: 16611222]
- 40.
- Ling YM, Shaw MH, Ayala C. et al. Vacuolar and plasma membrane stripping and autophagic elimination of toxoplasma gondii in primed effector macrophages. J Exp Med. 2006;203:2063–71. [PMC free article: PMC2118399] [PubMed: 16940170]
- 41.
- Bogdan C, Rollinghoff M, Diefenbach A. The role of nitric oxide in innate immunity. Immunol revs. 2000;173:17–26. [PubMed: 10719664]
- 42.
- Zwaferink H, Stockinger S, Reipert S. et al. Stimulation of inducible nitric oxide synthase expression by interferon beta increases necrotic death of macrophages upon listeria monocytogenes infection. Infect Immun. 2008;76:1649–56. [PMC free article: PMC2292882] [PubMed: 18268032]
- 43.
- Eklund EA, Kakar R. Recruitment of CREB-binding protein by PU.1, IFN-regulatory factor-1 and the IFN consensus sequence-binding protein is necessary for IFN-gamma-induced p67phox and gp91phox expression. J Immunol. 1999;163:6095–105. [PubMed: 10570299]
- 44.
- Kumatori A, Yang D, Suzuki S. et al. Cooperation of STAT-1 and IRF-1 in interferon-gamma-induced transcription of the gp91(phox) gene. J Biol Chem. 2002;277:9103–111. [PubMed: 11781315]
- 45.
- Weber F, Haller O. Viral suppression of the interferon system. Biochimie. 2007;89:836–42. [PMC free article: PMC7126635] [PubMed: 17336442]
- 46.
- Randall RE, Goodbourn S. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol. 2008;89:1–47. [PubMed: 18089727]
- 47.
- Nandan D, Reiner NE. Attenuation of gamma interferon-induced tyrosine phosphorylation in mononuclear phagocytes infected with leishmania donovani: selective inhibition of signalling through janus kinases and stat1. Infect Immun. 1995;63:4495–500. [PMC free article: PMC173640] [PubMed: 7591091]
- 48.
- Prabhakar S, Qiao Y, Hoshino Y. et al. Inhibition of response to alpha interferon by mycobacterium tuberculosis. Infect Immun. 2003;71:2487–97. [PMC free article: PMC153238] [PubMed: 12704120]
- 49.
- Durbin JE, Hackenmiller R, Simon MC. et al. Targeted disruption of the mouse stat1 gene results in compromised innate immunity to viral disease. Cell. 1996;84:443–50. [PubMed: 8608598]
- 50.
- Meraz MA, White JM, Sheehan KC. et al. Targeted disruption of the stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signalling pathway. Cell. 1996;84:431–42. [PubMed: 8608597]
- 51.
- Garcia-Sastre A, Durbin RK, Zheng H. et al. The role of interferon in influenza virus tissue tropism. J Virol. 1998;72:8550–58. [PMC free article: PMC110265] [PubMed: 9765393]
- 52.
- Karaghiosoff M, Steinborn R, Kovarik P. et al. Central role for type I interferons and Tyk2 in lipopolysaccharide-induced endotoxin shock. Nat Immunol. 2003;4:471–77. [PubMed: 12679810]
- 53.
- Gil MP, Bohn E, O'Guin AK. et al. Biologic consequences of stat1-independent IFN signalling. Proc Natl Acad Sci USA. 2001;98:6680–85. [PMC free article: PMC34412] [PubMed: 11390995]
- 54.
- Ramana CV, Gil MP, Han Y. et al. Stat1-independent regulation of gene expression in response to IFN-gamma. Proc Natl Acad Sci USA. 2001;98:6674–79. [PMC free article: PMC34411] [PubMed: 11390994]
- 55.
- Park C, Li S, Cha E. et al. Immune response in stat2 knockout mice. Immunity. 2000;13:795–804. [PubMed: 11163195]
- 56.
- Zimmermann A, Trilling M, Wagner M. et al. A cytomegaloviral protein reveals a dual role for STAT2 in IFN-{gamma} signalling and antiviral responses. J Exp Med. 2005;201:1543–53. [PMC free article: PMC2212917] [PubMed: 15883169]
- 57.
- Dupuis S, Jouanguy E, Al-Hajjar S. et al. Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat Genet. 2003;33:388–91. [PubMed: 12590259]
- 58.
- Dupuis S, Dargemont C, Fieschi C. et al. Impairment of mycobacterial but not viral immunity by a germline human STAT1 mutation. Science. 2001;293:300–303. [PubMed: 11452125]
- 59.
- Durbin JE, Fernandez-Sesma A, Lee CK. et al. Type I IFN modulates innate and specific antiviral immunity. J Immunol. 2000;164:4220–28. [PubMed: 10754318]
- 60.
- Lee CK, Smith E, Gimeno R. et al. STAT1 affects lymphocyte survival and proliferation partially independent of its role downstream of IFN-gamma. J Immunol. 2000;164:1286–92. [PubMed: 10640742]
- 61.
- Whitmire JK, Tan JT, Whitton JL. Interferon-gamma acts directly on CD8+ T-cells to increase their abundance during virus infection. J Exp Med. 2005;201:1053–59. [PMC free article: PMC2213135] [PubMed: 15809350]
- 62.
- Tanabe Y, Nishibori T, Su L. et al. Cutting edge: role of STAT1, STAT3 and STAT5 in IFN-alpha beta responses in T-lymphocytes. J Immunol. 2005;174:609–13. [PubMed: 15634877]
- 63.
- Gimeno R, Lee CK, Schindler C. et al. Stat1 and stat2 but not stat3 arbitrate contradictory growth signals elicited by alpha/beta interferon in T-lymphocytes. Mol Cell Biol. 2005;25:5456–65. [PMC free article: PMC1156979] [PubMed: 15964802]
- 64.
- Kolumam GA, Thomas S, Thompson LJ. et al. Type I interferons act directly on CD8 T-cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med. 2005;202:637–50. [PMC free article: PMC2212878] [PubMed: 16129706]
- 65.
- Quigley M, Huang X, Yang Y. STAT1 signalling in CD8 T-cells is required for their clonal expansion and memory formation following viral infection in vivo. J Immunol. 2008;180:2158–64. [PubMed: 18250422]
- 66.
- Johnson LM, Scott P. STAT1 expression in dendritic cells, but not T-cells, is required for immunity to leishmania major. J Immunol. 2007;178:7259–66. [PubMed: 17513775]
- 67.
- Le Bon A, Etchart N, Rossmann C. et al. Cross-priming of CD8+ T-cells stimulated by virus-induced type I interferon. Nat Immunol. 2003;4:1009–15. [PubMed: 14502286]
- 68.
- Le Bon A, Durand V, Kamphuis E. et al. Direct stimulation of T-cells by type I IFN enhances the CD8+ T-cell response during cross-priming. J Immunol. 2006;176:4682–89. [PubMed: 16585561]
- 69.
- Le Bon A, Tough DF. Type I interferon as a stimulus for cross-priming. Cytokine Growth Factor Rev. 2008;19:33–40. [PubMed: 18068417]
- 70.
- Heinrich PC, Behrmann I, Muller-Newen G. et al. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J. 1998;334:297–314. [PMC free article: PMC1219691] [PubMed: 9716487]
- 71.
- Spolski R, Leonard WJ. Interleukin-21: basic biology and implications for cancer and autoimmunity. Ann rev Immunol. 2008;26:57–79. [PubMed: 17953510]
- 72.
- Lejeune D, Dumoutier L, Constantinescu S. et al. Interleukin-22 (IL-22) activates the JAK/STAT ERK, JNK and p38 MAP kinase pathways in a rat hepatoma cell line. Pathways that are shared with and distinct from IL-10. J Biol Chem. 2002;277:33676–82. [PubMed: 12087100]
- 73.
- Lee CK, Raz R, Gimeno R. et al. STAT3 is a negative regulator of granulopoiesis but is not required for G-CSF-dependent differentiation. Immunity. 2002;17:63–72. [PubMed: 12150892]
- 74.
- Laouar Y, Welte T, Fu XY. et al. STAT3 is required for Flt3L-dependent dendritic cell differentiation. Immunity. 2003;19:903–12. [PubMed: 14670306]
- 75.
- Maritano D, Sugrue ML, Tininini S. et al. The STAT3 isoforms alpha and beta have unique and specific functions. Nat Immunol. 2004;5:401–09. [PubMed: 15021879]
- 76.
- Yoo JY, Huso DL, Nathans D. et al. Specific ablation of stat3beta distorts the pattern of stat3-responsive gene expression and impairs recovery from endotoxic shock. Cell. 2002;108:331–44. [PubMed: 11853668]
- 77.
- O'Shea JJ, Murray PJ. Cytokine signalling modules in inflammatory responses. Immunity. 2008;28:477–87. [PMC free article: PMC2782488] [PubMed: 18400190]
- 78.
- Nurieva R, Yang XO, Martinez G. et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T-cells. Nature. 2007;448:480–83. [PubMed: 17581589]
- 79.
- Yang XO, Panopoulos AD, Nurieva R. et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T-cells. J Biol Chem. 2007;282:9358–63. [PubMed: 17277312]
- 80.
- Aujla SJ, Chan YR, Zheng M. et al. IL-22 mediates mucosal host defense against gram-negative bacterial pneumonia. Nat Med. 2008;14:275–81. [PMC free article: PMC2901867] [PubMed: 18264110]
- 81.
- Matsukawa A, Takeda K, Kudo S. et al. Aberrant inflammation and lethality to septic peritonitis in mice lacking STAT3 in macrophages and neutrophils. J Immunol. 2003;171:6198–6205. [PubMed: 14634136]
- 82.
- Takeda K, Clausen BE, Kaisho T. et al. Enhanced TH1 activity and development of chronic enterocolitis in mice devoid of stat3 in macrophages and neutrophils. Immunity. 199;10:39–49. [PubMed: 10023769]
- 83.
- Kobayashi M, Kweon MN, Kuwata H. et al. toll-like receptor-dependent production of IL-12p40 causes chronic enterocolitis in myeloid cell-specific Stat3-deficient mice. J Clin Invest. 2003;111:1297–308. [PMC free article: PMC154445] [PubMed: 12727921]
- 84.
- Alonzi T, Newton IP, Bryce PJ. et al. Induced somatic inactivation of STAT3 in mice triggers the development of a fulminant form of enterocolitis. Cytokine. 2004;26:45–56. [PubMed: 15050604]
- 85.
- Turner J, Gonzalez-Juarrero M, Ellis DL. et al. In vivo IL-10 production reactivates chronic pulmonary tuberculosis in C57BL/6 mice. J Immunol. 2002;169:6343–51. [PubMed: 12444141]
- 86.
- Butcher BA, Kim L, Panopoulos AD. et al. IL-10-independent STAT3 activation by toxoplasma gondii mediates suppression of IL-12 and TNF-alpha in host macrophages. J Immunol. 1999;174:3148–52. [PubMed: 15749841]
- 87.
- Kano A, Wolfgang MJ, Gao Q. et al. Endothelial cells require STAT3 for protection against endotoxin-induced inflammation. J Exp Med. 2003;198:1517–25. [PMC free article: PMC2194113] [PubMed: 14623907]
- 88.
- Fattori E, Cappelletti M, Costa P. et al. Defective inflammatory response in interleukin 6-deficient mice. J Exp Med. 1994;180:1243–50. [PMC free article: PMC2191674] [PubMed: 7931061]
- 89.
- Murray PJ. The JAK-STAT signalling pathway: input and output integration. J Immunol. 2007;178:2623–29. [PubMed: 17312100]
- 90.
- Yasukawa H, Ohishi M, Mori H. et al. IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages. Nat Immunol. 2003;4:551–56. [PubMed: 12754507]
- 91.
- Thierfelder WE, van Deursen JM, Yamamoto K. et al. Requirement for stat4 in interleukin-12-mediated responses of natural killer and T-cells. Nature. 1996;382:171–74. [PubMed: 8700208]
- 92.
- Kaplan MH, Schindler U, Smiley ST. et al. Stat6 is required for mediating responses to IL-4 and for development of TH2 cells. Immunity. 1996;4:313–19. [PubMed: 8624821]
- 93.
- Kaplan MH, Sun YL, Hoey T. et al. Impaired IL-12 responses and enhanced development of TH2 cells in stat4-deficient mice. Nature. 1996;382:174–77. [PubMed: 8700209]
- 94.
- Shimoda K, van Deursen J, Sangster MY. et al. Lack of IL-4-induced TH2 response and IgE class switching in mice with disrupted stat6 gene. Nature. 1996;380:630–33. [PubMed: 8602264]
- 95.
- Takeda K, Tanaka T, Shi W. et al. Essential role of stat6 in IL-4 signalling. Nature. 1996;380:627–30. [PubMed: 8602263]
- 96.
- Elvin SJ, Williamson ED. Stat 4 but not stat 6 mediated immune mechanisms are essential in protection against plague. Microb Pathog. 2004;37:177–84. [PubMed: 15458778]
- 97.
- Sugawara I, Yamada H, Mizuno S. Relative importance of STAT4 in murine tuberculosis. J Med Microbiol. 2003;52:29–34. [PubMed: 12488562]
- 98.
- Deng JC, Zeng X, Newstead M. et al. STAT4 is a critical mediator of early innate immune responses against pulmonary klebsiella infection. J Immunol. 2004;173:4075–83. [PMC free article: PMC3001230] [PubMed: 15356157]
- 99.
- Cai G, Radzanowski T, Villegas EN. et al. Identification of STAT4-dependent and independent mechanisms of resistance to toxoplasma gondii. J Immunol. 2000;165:2619–27. [PubMed: 10946290]
- 100.
- Stamm LM, Satoskar AA, Ghosh SK. et al. STAT-4 mediated IL-12 signalling pathway is critical for the development of protective immunity in cutaneous leishmaniasis. Eur J Immunol. 2000;29:2524–29. [PubMed: 10458767]
- 101.
- Finkelman FD, Shea-Donohue T, Morris SC. et al. Interleukin-4- and interleukin-13-mediated host protection against intestinal nematode parasites. Immunol revs. 2004;201:139–55. [PubMed: 15361238]
- 102.
- Urban JF Jr, Schopf L, Morris SC. et al. Stat6 signalling promotes protective immunity against trichinella spiralis through a mast cell- and T-cell-dependent mechanism. J Immunol. 2000;164:2046–52. [PubMed: 10657657]
- 103.
- Khan WI, Vallance BA, Blennerhassett PA. et al. Critical role for signal transducer and activator of transcription factor 6 in mediating intestinal muscle hypercontractility and worm expulsion in trichinella spiralis-infected mice. Infect Immun. 2001;69:838–44. [PMC free article: PMC97960] [PubMed: 11159976]
- 104.
- Urban JF Jr, Noben-Trauth N, Donaldson DD. et al. IL-13, IL-4ralpha and stat6 are required for the expulsion of the gastrointestinal nematode parasite nippostrongylus brasiliensis. Immunity. 1998;8:255–64. [PubMed: 9492006]
- 105.
- Negrao-Correa D, Pinho V, Souza DG. et al. Expression of IL-4 receptor on nonbone marrow-derived cells is necessary for the timely elimination of strongyloides venezuelensis in mice, but not for intestinal IL-4 production. Int J Parasitol. 2006;36:1185–95. [PubMed: 16793046]
- 106.
- Rodriguez-Sosa M, David JR, Bojalil R. et al. Cutting edge: susceptibility to the larval stage of the helminth parasite taenia crassiceps is mediated by TH2 response induced via STAT6 signalling. J Immunol. 2002;168:3135–39. [PubMed: 11907063]
- 107.
- Rodriguez-Sosa M, Saavedra R, Tenorio EP. et al. A STAT4-dependent TH1 response is required for resistance to the helminth parasite taenia crassiceps. Infect Immun. 2004;72:4552–60. [PMC free article: PMC470677] [PubMed: 15271915]
- 108.
- Mahalingam S, Karupiah G, Takeda K. et al. Enhanced resistance in STAT6-deficient mice to infection with ectromelia virus. Proc Natl Acad Sci USA. 2001;98:6812–17. [PMC free article: PMC34435] [PubMed: 11371617]
- 109.
- Stamm LM, Raisanen-Sokolowski A, Okano M. et al. Mice with STAT6-targeted gene disruption develop a TH1 response and control cutaneous leishmaniasis. J Immunol. 1998;161:6180–88. [PubMed: 9834104]
- 110.
- Tarleton RL, Grusby MJ, Zhang L. Increased susceptibility of Stat4-deficient and enhanced resistance in stat6-deficient mice to infection with trypanosoma cruzi. J Immunol. 2000;165:1520–25. [PubMed: 10903759]
- 111.
- Toyoda H, Ido M, Hayashi T. et al. Impairment of IL-12-dependent STAT4 nuclear translocation in a patient with recurrent mycobacterium avium infection. J Immunol. 2004;172:3905–12. [PubMed: 15004198]
- 112.
- Peisong G, Yamasaki A, Mao XQ. et al. An asthma-associated genetic variant of STAT6 predicts low burden of ascaris worm infestation. Genes Immun. 2004;5:58–62. [PubMed: 14735150]
- 113.
- Moller M, Gravenor MB, Roberts SE. et al. Genetic haplotypes of Th-2 immune signalling link allergy to enhanced protection to parasitic worms. Hum Mol Genet. 2007;16:1828–36. [PubMed: 17519224]
- 114.
- King SB, Knorn AM, Ohnmacht C. et al. Accumulation of effector CD4 T-cells during type 2 immune responses is negatively regulated by Stat6. J Immunol. 2008;180:754–63. [PubMed: 18178813]
- 115.
- Gordon S. Alternative activation of macrophages. Nat Revs Immunol. 2003;3:23–35. [PubMed: 12511873]
- 116.
- Anthony RM, Urban JF Jr, Alem F. et al. Memory T(H)2 cells induce alternatively activated macrophages to mediate protection against nematode parasites. Nat Med. 2006;12:955–60. [PMC free article: PMC1955764] [PubMed: 16892038]
- 117.
- Gray MJ, Poljakovic M, Kepka-Lenhart D. et al. Induction of arginase I transcription by IL-4 requires a composite DNA response element for STAT6 and C/EBPbeta. Gene. 2005;353:98–106. [PubMed: 15922518]
- 118.
- Matsukawa A, Kaplan MH, Hogaboam CM. et al. Pivotal role of signal transducer and activator of transcription (stat)4 and stat6 in the innate immune response during sepsis. J Exp Med. 2001;193:679–88. [PMC free article: PMC2193416] [PubMed: 11257135]
- 119.
- Lentsch AB, Kato A, Davis B. et al. STAT4 and Stat6 regulate systemic inflammation and protect against lethal endotoxemia. J Clin Invest. 2001;108:1475–82. [PMC free article: PMC209422] [PubMed: 11714739]
- 120.
- Freudenberg MA, Merlin T, Kalis C. et al. Cutting edge: a murine, IL-12-independent pathway of IFN-gamma induction by gram-negative bacteria based on STAT4 activation by type I IFN and IL-18 signalling. J Immunol. 2002;169:1665–68. [PubMed: 12165484]
- 121.
- Mattner J, Wandersee-Steinhauser A, Pahl A. et al. Protection against progressive leishmaniasis by IFN-beta. J Immunol. 2004;172:7574–82. [PubMed: 15187137]
- 122.
- Miyagi T, Gil MP, Wang X. et al. High basal STAT4 balanced by STAT1 induction to control type 1 interferon effects in natural killer cells. J Exp Med. 2007;204:2383–96. [PMC free article: PMC2118450] [PubMed: 17846149]
- 123.
- Hennighausen L, Robinson GW. Interpretation of cytokine signalling through the transcription factors STAT5A and STAT5B. Genes Dev. 2008;22:711–21. [PMC free article: PMC2394721] [PubMed: 18347089]
- 124.
- Kieslinger M, Woldman I, Moriggl R. et al. Antiapoptotic activity of stat5 required during terminal stages of myeloid differentiation. Genes Dev. 2000;14:232–44. [PMC free article: PMC316353] [PubMed: 10652277]
- 125.
- Hoelbl A, Kovacic B, Kerenyi MA. et al. Clarifying the role of stat5 in lymphoid development and abelson-induced transformation. Blood. 2006;107:4898–06. [PMC free article: PMC2875852] [PubMed: 16493008]
- 126.
- Yao Z, Cui Y, Watford WT. et al. Stat5a/b are essential for normal lymphoid development and differentiation. Proc Natl Acad Sci USA. 2006;103:1000–05. [PMC free article: PMC1327727] [PubMed: 16418296]
- 127.
- Grebien F, Kerenyi MA, Kovacic B. et al. Stat5 activation enables erythropoiesis in the absence of EpoR and jak2. Blood. 2008;111:4511–22. [PMC free article: PMC2976848] [PubMed: 18239084]
- 128.
- Moriggl R, Topham DJ, Teglund S. et al. Stat5 is required for IL-2 induced cell cycle progression of peripheral T-cells. Immunity. 1999;10:249–59. [PubMed: 10072077]
- 129.
- Zhu J, Cote-Sierra J, Guo L. et al. Stat5 activation plays a critical role in TH2 differentiation. Immunity. 2003;19:739–48. [PubMed: 14614860]
- 130.
- Zhu Y, Chen L, Huang Z. et al. Cutting edge: IL-5 primes TH2 cytokine-producing capacity in eosinophils through a STAT5-dependent mechanism. J Immunol. 2004;173:2918–22. [PubMed: 15322148]
- 131.
- Barnstein BO, Li G, Wang Z. et al. Stat5 expression is required for IgE-mediated mast cell function. J Immunol. 2006;177:3421–26. [PubMed: 16920984]
- 132.
- Xiao W, Hong H, Kawakami Y. et al. Regulation of myeloproliferation and M2 macrophage programming in mice by Lyn/Hck, SHIP and stat5. J Clin Invest. 2008;118:924–34. [PMC free article: PMC2214849] [PubMed: 18246197]
- 133.
- Esashi E, Wang YH, Perng O. et al. The signal transducer STAT5 inhibits plasmacytoid dendritic cell development by suppressing transcription factor IRF8. Immunity. 2008;28:509–20. [PMC free article: PMC2864148] [PubMed: 18342552]
- 134.
- Yao Z, Kanno Y, Kerenyi M. et al. Nonredundant roles for stat5a/b in directly regulating foxp3. Blood. 2007;109:4368–75. [PMC free article: PMC1885496] [PubMed: 17227828]
- 135.
- Burchill MA, Yang J, Vogtenhuber C. et al. IL-2 receptor beta-dependent STAT5 activation is required for the development of foxp3+ regulatory T-cells. J Immunol. 2007;178:280–90. [PubMed: 17182565]
- 136.
- Zorn E, Nelson EA, Mohseni M. et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T-cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood. 2006;108:1571–79. [PMC free article: PMC1895505] [PubMed: 16645171]
- 137.
- Selliah N, Zhang M, DeSimone D. et al. The gammac-cytokine regulated transcription factor, STAT5, increases HIV-1 production in primary CD4 T-cells. Virology. 2006;344:283–91. [PubMed: 16289657]
- 138.
- Zheng CF, Jones GJ, Shi M. et al. Late expression of granulysin by microbicidal CD4+ T-cells requires PI3K- and STAT5-dependent expression of IL-2R{beta} that is defective in HIV-infected patients. J Immunol. 2008;180:7221–29. [PMC free article: PMC2661617] [PubMed: 18490721]
- 139.
- Prost S, Le Dantec M, Auge S. et al. Human and simian immunodeficiency viruses deregulate early hematopoiesis through a Nef/PPARgamma/STAT5 signalling pathway in macaques. J Clin Invest. 2008;118:1765–75. [PMC free article: PMC2323187] [PubMed: 18431514]
- 140.
- Darnell JE Jr. Phosphotyrosine signalling and the single cell: metazoan boundary. Proc Natl Acad Sci USA. 1997;94:11767–69. [PMC free article: PMC33778] [PubMed: 9342310]
- 141.
- Dostert C, Jouanguy E, Irving P. et al. The jak-STAT signalling pathway is required but not sufficient for the antiviral response of drosophila. Nat Immunol. 2005;6:946–53. [PubMed: 16086017]
- 142.
- Galiana-Arnoux D, Dostert C, Schneemann A. et al. Essential function in vivo for dicer-2 in host defense against RNA viruses in drosophila. Nat Immunol. 2006;7:590–97. [PubMed: 16554838]
- STATs and Infection - Madame Curie Bioscience DatabaseSTATs and Infection - Madame Curie Bioscience Database
- Regulation of Medical Devices - Madame Curie Bioscience DatabaseRegulation of Medical Devices - Madame Curie Bioscience Database
- Calreticulin in Cytotoxic Lymphocyte-Mediated Cytotoxicity - Madame Curie Biosci...Calreticulin in Cytotoxic Lymphocyte-Mediated Cytotoxicity - Madame Curie Bioscience Database
- Predicting the Spread of a Transgene in African Malaria Vector Populations: Curr...Predicting the Spread of a Transgene in African Malaria Vector Populations: Current Knowledge and Limitations - Madame Curie Bioscience Database
- Coronin 1 in Innate Immunity - Madame Curie Bioscience DatabaseCoronin 1 in Innate Immunity - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...