

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Pseudouridine (Ψ) synthases are the enzymes responsible for the most abundant and phylogenetically conserved posttranscriptional modification of cellular RNAs. These enzymes catalyze an isomerization reaction of specific uridine residues within an RNA chain. Sequence and structure analyses have thus far demonstrated the existence of six Ψ synthase families that share a common core domain structure despite very low sequence identity. Ψ synthases display exquisite specificity in pinpointing the site of pseudouridylation within their RNA substrates, and each enzyme achieves this by structural elaborations of the conserved catalytic domain. The catalytic mechanism, presumably shared between all Ψ synthases, is still not well understood and remains a challenging problem in the enzymology of nucleic acid modifications.
Introduction and Nomenclature
Pseudouridine (Ψ) was discovered to be the most abundant posttranscriptionally modified nucleotide in cellular RNAs in the 1950s.1 Chemical characterization of what was called “the fifth nucleotide” showed Ψ to be the C5-glycoside isomer of uridine.2-6 Early biochemical analyses demonstrated that Ψ synthases produce the nucleotide by isomerizing uridines that already are part of RNAs; that is, they formally perform an internal transglycosylation reaction (Fig. 1). These enzymes function without cofactors and do not appear to release to bulk solvent the uracil base that they isomerize.7-10
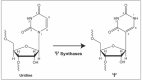
Figure 1
Pseudouridine (Ψ) and internal isomerization reaction catalyzed by Ψ synthases.
Molecular cloning and sequence analyses of Ψ synthases from Escherichia coli led to the definition of five protein families that share minimal sequence similarity with each other and have broad phylogenetic distribution.11,12 These were called RluA, RsuA, TruA, TruB and TruD, after the E. coli enzymes (Table 1). The names of the enzymes are derived from their substrate RNAs. Thus, RsuA is responsible for Ψ516 in the small-subunit rRNA in E. coli and TruB for Ψ55 in tRNA (RluA modifies position 746 in 23S rRNA, but also position 32 in some tRNAs). This historically evolved nomenclature results in, for instance, TruC being part of the RluA family (due to sequence similarity), rather than the TruA, TruB or TruD families. The nomenclature of budding yeast Ψ synthases, in which the enzymes have been numbered sequentially, developed independently of the bacterial system. Thus, Pus4p13 and Pus7p14,15 are the Saccharomyces cerevisiae orthologs of TruB and TruD from E. coli, respectively. Based on sequence,16,17 functional18 and structural19 considerations, it is possible to define a sixth family, named after the human Ψ synthase Pus10p.
Table 1
Pseudouridine synthase families.
Three-Dimensional Structure
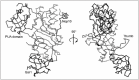
Site-directed mutagenesis experiments indicated that Ψ synthases of different families each have an aspartate residue that is critical for enzymatic activity.20-23 When their crystal structures were determined, it was found that Ψ synthases from all six families share a common catalytic domain structure despite having sometimes undetectably low sequence similarity.19,24-32 Nature appears to use this domain exclusively in Ψ synthases. The structure of the core domain comprises an extensive, saddle-shaped, predominantly anti-parallel β-sheet, which is decorated on one face by two groups α-helices and loops. These two groups flank the active site cleft. One side of the cleft is defined by a polypeptide stretch of irregular secondary structure that contains the catalytic aspartate residue (Fig. 2A).

The various Ψ synthases have three types of elaborations of the conserved core domain (Table 2). First, some Ψ synthases have extended loops that flank the active site cleft on either side. When the crystal structures of TruB25 and RluA33 bound to their respective substrate RNAs were determined, it was found that these loops “pinch” the RNA on the major and minor groove. These have been anthropomorphically named “thumb” and “forefinger” loops, respectively. Second, different Ψ synthases have N- or C-terminal extensions to the core domain. Some of these extensions are independently folded polypeptide units (that is, domains), which have been found to occur in proteins with other functions. Third, Ψ synthases of the Cbf5p/dyskerin class (a subset of the TruB family) have evolved to associate with additional polypeptides that are needed for enzymatic activity.
Table 2
Domain structure of enzymes of the six pseudouridine synthase families.
Figure 2 depicts the three-dimensional structures of some representative Ψ synthases. RluA (Fig. 2A) from E. coli is one of the most compact Ψ synthases and lacks either N- or C-terminal extensions. It does, however, employ both thumb and forefinger loops to associate with its substrate RNA.33 In terms of domains structure and evolutionary history, Ψ synthases of the TruD family (Fig. 2B) are remarkable because the linear arrangement of the secondary structure elements (the “topology”) of their core catalytic domain is a circular permutation of that found in all other structurally-characterized Ψ synthase families.32 Despite this, their core structure superimposes very closely on that of all other Ψ synthases. E. coli TruD does not have N- or C-terminal extensions or a forefinger loop but has a large domain in the location where Ψ synthases such as RluA have a simple thumb loop. Thus far, this TruD domain appears to be unique to this family of proteins.30-32 Enzymes of the Pus10p family (Fig. 2C) have both forefinger and thumb loops in their core domain and are also characterized by a large N-terminal extension called the THUMP domain. This domain has been found also to occur in some 4-thiouridine synthases and RNA methyltransferases.17,34-36
RNA Recognition
A remarkable feature of Ψ synthases is their substrate specificity. Many of these enzymes (Table 1) are capable of recognizing as their substrate a single nucleotide in one particular RNA among the myriad different RNAs in the cell. The fact that Ψ synthases employ the structural or sequence context of their target uridine residue for recognition of the isomerization locus in their substrate RNA has the biologically important corollary that these enzymes are incapable of isomerizing free uridine into Ψ. This is critical, because cells synthesize thymidine by methylation of uridine (and reduction of the 2′-OH of the sugar) and Ψ cannot be converted into T by methylation. Moreover, if the cell contained a pool of Ψ triphosphate, Ψ would be randomly incorporated instead of U into cellular RNAs, because the Watson-Crick base-pairing potential of Ψ is identical to that of U.
The first structure of a Ψ synthase bound to a substrate RNA and indeed, of any RNA nucleobase-modifying enzyme bound to substrate, was that of E. coli TruB cocrystallized with a TΨC stem-loop minihelix.25 TruB is responsible for the universally conserved Ψ at position 55 of elongator tRNAs.37 This one enzyme is capable of recognizing and modifying all the different elongator tRNAs of the cell. Biochemical analyses demonstrated that TruB has equivalent catalytic activity with a minihelix comprising the TΨC stem-loop or full-length tRNA.38 This indicates that the TΨC stem-loop contains all the recognition determinants for the enzyme. The structure (Fig. 3) showed how the enzyme uses a combination of shape complementarity to the characteristic39 three-dimensional structure of T-loops and also direct readout of nucleotides that are invariant specifically in the T-loops of elongator tRNAs (e.g., the C residue at position 56 in the sequence TΨC). This structure defined the thumb loop as an RNA-recognition element and provided the first high-resolution view of the thumb loop interacting with tRNA. Subsequent studies have shown that the TruB thumb undergoes a folding transition concomitant with binding of substrate RNA.40-42 The TruB cocrystal structure was also the first to show how the PUA domain can interact with RNA.25,43 This domain was discovered on the basis of sequence analyses of Ψ synthases and a different class of RNA nucleobase modifying enzymes, the archaeosine-guanine transglycosylases. The PUA domain is a compact mixed α/β fold that has since been found in many types of proteins involved in RNA metabolism.44,45 TruB uses this domain to associate with the A-form double helical portion of the acceptor stem of its substrate tRNA (Fig. 3). Interestingly other structurally characterized instances of PUA domains in RNA-protein complexes show that the PUA domain can interact with RNA using different surfaces.46,47 Thus, while presence of this domain in a protein correlates with its RNA binding, its exact mode of association with the nucleic acid appears to be evolutionarily variable.48

While TruB recognizes one nucleotide in the same structural context in many different substrate tRNAs, other Ψ synthases recognize multiple RNAs, or multiple adjacent sites in one or a few closely related RNAs. For instance, Pus7p from budding yeast recognizes both tRNAs and the U2 spliceosomal snRNA.14 RluA from E. coli was the first multi-substrate Ψ synthase for which a structure in complex with substrate was determined. This Ψ synthase modifies position 32 in four tRNAs and position 746 in 23S rRNA. All its substrate uridines lie in a similar sequence context (ΨUXXAAA, where X is any nucleotide) in a terminal loop, so it was expected that the enzyme would be carrying out direct readout of the conserved sequence elements. The crystal structure revealed, instead, that RluA melts and refolds its substrates and imposes on them a new pairing scheme. The structure and site-directed mutagenesis experiments suggest that the enzyme selects as substrates those RNAs that can adopt this protein binding-induced structure.33
Unlike most single-polypeptide Ψ synthases, which are monomeric, TruA functions as a dimer.24 In E. coli, this enzyme modifies multiple neighboring positions in the anticodon stem-loop of some tRNAs.49 Crystal structures of this enzyme in complex with various tRNAs have been reported.50 However, none of these appear to represent productive complexes because a uridine is not bound in the active site of the enzyme in any of them. Thus, a structural understanding of how a single enzyme modifies multiple adjacent positions in an RNA will have to await further studies.
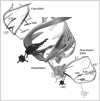
The foregoing discussion on the interaction of Ψ synthases with RNA concerns the majority of these enzymes, which are comprised of a single polypeptide chain (or a dimer of the same chain, in the case of TruA) that directly recognizes its substrate(s). The yeast Ψ synthase Cbf5p and its orthologs from archaea and eukarya (called dyskerin in human) have evolved a completely different mechanism for substrate recognition. Cbf5p binds to a guide RNA that has sequence complementarity to nucleotides flanking the site of modification of its substrate RNAs. Base pairing between the guide RNA and the substrate RNA brings the Cbf5p active site into proximity with its substrate. This has been proven formally by devising artificial guide RNAs that target pseudouridylation in vivo into previously unmodified positions of cellular RNAs. The guide RNAs have a conserved secondary structure and sequence motifs. The latter are named “box H” and “box ACA” and hence, the RNAs are known as the box H/ACA RNAs (reviewed in ref. 51).
Cbf5p also associates with three additional polypeptides. Two of these, Nop10p and Gar1p, bind directly to Cbf5p (Fig. 4). The other, Nhp2p (or its archaeal homolog L7Ae), binds to the guide RNA through a K-turn52 or K-loop53,54 motif. The first structural information on these Ψ synthases came from a crystal structure of the archaeal Cbf5p-Nop10p complex.55,56 This structure demonstrated that Cbf5p is a close homolog of TruB and that Nop10p indirectly stabilizes the active site of Cbf5p by buttressing a critical segment of the enzyme structure, called Motif I.57 Subsequently, structures of the triple Cbf5p-Nop10p-Gar1p complex,58 as well as of the complex of the four proteins with a guide RNA47 and of the Cbf5p-Nop10p-Gar1p complex with a guide RNA and a substrate RNA have been reported.59 These structures demonstrated how the complex recognizes the conserved elements of the box H/ACA RNAs. However, at the time of writing, there is still no structure of a full complex in a catalytically competent conformation, that is, with the substrate uridine closer than 11 Å from the active site aspartate. (For a more extensive discussion, see chapter by Grozdanov and Meier in this volume).
Substrate Nucleobase Flipping and Active Site Conservation
The cocrystal structure of TruB bound to its substrate RNA was also the first to show that an RNA base-modifying enzyme accesses its substrate through base flipping.25 This mechanism was first discovered in DNA methyltransferases and has been extensively discussed elsewhere (see chapters by Priyakumar and MacKerell and by Klimasauskas and Liutkeviciute in this volume). TruB binding results in extrusion from the TΨC-loop of Ψ55, C56 and X57 (X is any nucleotide). As indicated above, the extruded C56, a conserved sequence element of elongator tRNAs, is recognized by the enzyme through direct hydrogen bond interactions. The extruded nucleotide at position 55 is buried deep in the active site pocket of the enzyme. Because it was known that some Ψ synthases are strongly inhibited by RNAs that contain 5-fluorouridine (f5U) in place of their substrate uridine, TruB was cocrystallized with an RNA containing this unnatural base at position 55. The structure (Fig. 5A) showed that TruB had in fact isomerized f5U, producing a nonplanar nucleotide. Subsequently, the cocrystal structure of a point mutant of TruB in which the catalytic aspartate was replaced with asparagine was determined.42 To facilitate unambiguous orientation of the base of residue 55 during crystallographic model building, f5U instead of U was used in the substrate RNA (fluorine scatters electrons more strongly than hydrogen, but the radii of the two atoms are similar). Confirming previous mutagenesis studies, this structure showed that the mutation resulted in complete inactivation of the enzyme, as the precursor, rather than the isomerized product was bound in the active site (Fig. 5B). Subsequently, the structure of RluA bound to an f5U containing substrate RNA was also determined and this enzyme too was found to flip its substrate uridine into the active site. At present, nothing is known about the trajectory of the flipping reaction. However, on steric grounds, it is more plausible that the flipping occurs on the major groove side (Fig. 6).
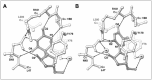
Structural superposition of the two available Ψ synthase-substrate RNA complex structures (those of TruB and RluA) as well as the numerous free enzyme structures (Table 1) demonstrates the conservation of three amino acids within the active site cleft throughout enzymes belonging to the six Ψ synthase families. First, a deeply buried aspartic acid (D48 in Fig. 5A) is absolutely conserved. Second, a basic residue, either arginine or lysine, is also buried in the active site and positioned to make a salt bridge with the conserved active site aspartate (R181 in Fig. 5A). Third, an aromatic residue (Y179 in Fig. 5A) packs close to the ribose of the isomerized uridine. While the identities of these residues are invariant across Ψ synthases, enzymes of the TruD family are thus far unique because the position of the basic residue within the conserved structural scaffold of the enzymes is different and because instead of the aromatic residue being a tyrosine, as in all other Ψ synthases, it is a phenylalanine. Beyond the conserved aspartate, basic residue and aromatic residue, very little sequence conservation is present in the active sites of Ψ synthases. These have a predominantly hydrophobic character, so a leucine (L200 in Fig. 5) is often, but not invariably, present.
The functional basis for the conservation of some of the characteristic sequence elements of the active site consensus (Table 2) for TruB and RluA family proteins has been clarified by the cocrystal structures. TruB employs the conserved histidine five residues N-terminal to the catalytic aspartate to occupy the location within the RNA helical stack vacated by flipping out of residue 55 from the TΨC-loop of its tRNA substrate. Similarly, the arginine two residues N-terminal from the catalytic aspartate in RluA occupies the position vacated by residue 32 from the anticodon stem of tRNA. The “intercalating” histidine of TruB is conserved in all members of the TruB family, including Cbf5p, so that a similar mode of interaction with RNA for all these enzymes is expected.25 An arginine two residues N-terminal to the catalytic aspartate is present in members of TruA and RsuA families, in addition to RluA Ψ synthases, so that those other enzymes probably also use the arginine to occupy the space vacated by their flipped-out substrate uridines.33 Comparison of the TruB and RluA complexes demonstrated that even though these two enzymes bind to different substrate RNAs employing different recognition mechanisms and their substrate RNAs approach their respective active sites with pronouncedly different angles, the disposition of the flipped-out nucleotide and the conserved active-site amino acid residues is very nearly indistinguishable.33 This underscores the versatility of the evolutionarily conserved Ψ synthase fold.
Catalytic Mechanism
The conserved fold and constellation of active site residues argue that the Ψ synthases share a common mechanism. The abolition of detectable activity upon mutation of the active site aspartic acid residue in all Ψ synthase families20-22,60 suggests that it serves a role beyond a simple active site acid/base group since substitutions of such groups typically result in catalytic impairments of 2-3 orders of magnitude rather than the loss of all activity. Two alternative mechanisms in which the aspartic acid residue (in its aspartate form) serves as a nucleophile have been proposed (Fig. 7). The first49,61 involves attack of the aspartate at C6 of the uracil ring (in organic parlance, a Michael addition) based on the precedent of the pyrimidine methylases, including thymidylate synthase.62 With that enzyme, an active site cysteine residue is the nucleophile and the attack generates carbanionic character at C5 for its subsequent attack on a methyl group donor. With the uracil ring safely linked to the Ψ synthase (intermediate I, Fig. 7A), the glycosidic bond breaks and the tethered uracil rotates about the ester bond with the active site aspartic acid residue to reposition C5 near C1′ and the new C–C bond forms (to yield intermediate II). The aspartate then departs as a leaving group to generate the penultimate intermediate (III), which is deprotonated at C5 and protonated at N1 to generate Ψ. The second mechanism21 rests on the precedent of the retaining glycosidases63 and involves the nucleophilic attack of the active site aspartate at C1′ to liberate the anion of uracil and generate an acylal intermediate (Fig. 7B). The tightly bound uracilate anion then rotates about an axis perpendicular to the ring or flips about an axis running through C6 and N3 to position C5 adjacent to C1′ for C–C bond formation with the aspartate as the leaving group (to form III). The subsequent deprotonation of C5 and protonation of N1 yield Ψ.
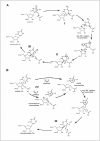
To test the first mechanism, RNA containing f5U was used to probe the formation of the adduct between the active site aspartate and the uracil ring (I, Fig. 7A).61 The fluoro group at C5 should inductively stabilize such an adduct, similar to the behavior seen with 5-fluorouridine and thymidylate synthase.62 Incubation of TruA with the in vitro transcript of tRNAPhe prepared using f5UTP instead of UTP resulted in irreversible inhibition of TruA, and a shifted band containing both RNA and protein was detected by both SDS-PAGE and urea-PAGE.61 The formation of a covalent adduct between the pyrimidine ring and the active site aspartate would account for these observations. After heat disruption, a hydrated product of f5U was recovered, and this product was reasonably ascribed to ester hydrolysis of the adduct (I, Fig. 7A) once the protein was denatured.61 The results with f5U, then, seemed to indicate that the first mechanism was followed.
The cocrystal structure of TruB with TΨC stem-loop containing f5U, however, showed not a covalent adduct but instead a hydrated and rearranged product: the f5U had been converted to a C-glycoside as uridine is converted to Ψ.25 The reasonable conclusion was that the reaction proceeded along the first mechanism until formation of the C-glycoside (II, Fig. 7A) rather than stop at the first covalent intermediate; the intermediate then underwent slow ester hydrolysis in the crystal. Gel shift analysis, however, gave no indication of a covalent adduct between TruB and RNA containing f5U.64 Instead, TruB converted the f5U into two hydrated products on a time scale comparable to the natural reaction64, and both hydrated products are rearranged to C-glycosides (E. Miracco, unpublished observations). RluA behaved similarly to TruA in its irreversible inhibition by RNA containing f5U and a band shift in denaturing gels.64 To test the assumption that the hydration occurred by ester hydrolysis of the putative covalent intermediates, the Ψ synthases were incubated with their cognate RNA containing f5U in buffer containing [18O]water. Analysis of both the protein and the RNA products revealed that the label was in the products of f5U rather than the active site aspartic acid residue for TruB,65 RluA66 and TruA (M. McDonald, unpublished observations). The hydrated products, therefore, cannot result from ester hydrolysis but must arise from the direct hydration of the f5U. The interpretation of a hydrated product of f5U in support of the first mechanism rested on the generation of that product by ester hydrolysis, which is not the case. Furthermore, since both proposed mechanisms can account for these results by the reaction of water (whether enzyme-catalyzed or free in solution) with the rearranged f5U intermediate (III, Fig. 7, both A and B), the appearance of hydrated products cannot be used to distinguish the mechanistic course followed by the Ψ synthases.65,66 Current information, then, does not decisively favor either mechanism although the position of the aspartic acid residue relative to the f5U products in the active sites in the cocrystals of TruB25 and RluA33 seem to make attack at the pyrimidine ring (and hence the first mechanism) more plausible than attack at C1′ since the former would require less structural reorganization than the latter. Other mechanistic probes and methods will be required to establish the reaction mechanism with confidence and then to elucidate details specific to that one (for example, how the oxocarbenium ion is stabilized if an acylal intermediate does not form).
The identities of the active site residues that serve as general acids/bases remain uncertain. A general base is required for the deprotonation of the penultimate intermediate, and two different residues have been proposed to play that role.67,68 Two residues lie in the general vicinity of the proton on C5 in the cocrystal structures, the essential aspartate and a tyrosine that is conserved in the TruA, TruB, RluA, RsuA and Pus10p families. The absolute conservation of the aspartate and a descending limb in the TruB pH activity profile with a pKa of ∼10 led to the assignment of aspartate as the general base,67 as shown in Figure 7. If the descending limb reflects the protonation state of the tyrosine, then the protonated form is required for activity. However, the descending limb might instead reflect the deprotonation of bound uridine substrate, which would dramatically slow the reaction by requiring a dianionic leaving group.67 The hydrogen bond between the phenolic group (protonated) of the tyrosine and the fluoro group of rearranged and hydrated f5U in the TruB and RluA cocrystals has been interpreted to indicate that the Tyr serves as the base to deprotonate C5 in the penultimate intermediate.68 The mismatch between the protonation state required to form the observed hydrogen bond and that required to perform the deprotonation of C5 and the ability of the TruD family to function with phenylalanine in place of this tyrosine argue against its assignment as the general base. The conserved aromatic side chain (tyrosine or phenylalanine) may serve to position the uracil ring during the reaction rather than serve an acid base role. The relatively low pKa of Ψ (9.0)69 and lack of an appropriately positioned acid group (other than, perhaps, the essential aspartate if it is sufficiently mobile) near N1 led to the assumption of the specific acid protonation of N1 depicted in Figure 7, which could occur either in solution or at the active site.67 More work will be required to resolve the role of active site general acid/base groups in catalysis.
Conclusion: Outstanding Questions
Several questions regarding the Ψ synthases remain largely unexplored. Do the bacterial Ψ synthases and the eukaryotic and archaeal enzymes that act alone in vitro form a complex in vivo with other Ψ synthases or RNA-modifying enzymes? What is the metal ion dependence of the Ψ synthases? The effects of magnesium and monovalent ions on the kinetic parameters of Ψ formation have not been widely explored, but substantial effects can be reasonably anticipated due to the impact on the structure and flexibility of the substrate RNA, which could affect its binding to the enzymes or the rate of reaction once bound (or both). Intriguingly, three Ψ synthases, yeast Pus1p70 and Pus10p from human19 and Pyrococcus abyssi (L. Droogmans, J. Armengaud and H. Grosjean, unpublished observations) bind zinc ions. Given that the tightly bound and solvent-inaccessible zinc ion in human Pus10p sits in a domain other than the one containing the active site for uridine isomerization and the same binding motif is present in yeast Pus1p, the zinc ion almost certainly plays no direct role in Ψ generation but instead provides for the organization and stability of the domain that contains it,19 as originally concluded for yeast Pus1p.70 This structural role for the zinc ion is analogous to that in the zinc ribbon domain of the archaeal (but not the eukaryal) Nop10p protein that is part of the H/ACA RNP.55 Structural characterization of Pus1p and Pus10p from other species should more fully elucidate the role of zinc ions as well as resolve other questions regarding potential differences between Ψ synthases from the three domains of life.
Perhaps the most striking universal feature of the Ψ synthases is their ability to carry out conformational rearrangements both gross and subtle, from the disruption of the tertiary structure and sometimes the secondary structure of the RNA substrate to the fine positioning of the detached uracil ring relative to the ribose ring. Although simple to envision in principle, the determination of the course of events in practice may prove difficult and will require a host of techniques. The grosser structural changes can perhaps be probed by stopped flow spectroscopy, including CD, fluorescence and perhaps UV. FRET and EPR may also prove a useful means to monitor the structural changes, assuming that appropriate fluorophores and spin labels can be installed in the substrate or the enzyme, which seems likely. The more subtle changes that facilitate the disruption of the glyosidic bond and the means by which the enzyme so finely positions C5 relative to C1′ to form the C-glycoside will likely prove harder to probe. Sorting out these issues will require a more thorough integration of the structural and mechanistic lines of investigation described in this chapter than has been the case to date, and the design and interpretation of the experiments to probe the conformational changes will rely heavily on the knowledge obtained from each of the current lines of investigation, even to the point of determining which particular Ψ synthase/ RNA substrate pairs are most likely to be amenable to a given line of experiments. With the firm foundation provided by the work to date, however, one can reasonably be optimistic that we will eventually understand in great detail the fascinating and deceptively simple reaction that the families of Ψ synthases carry out in so many RNA contexts.
Acknowledgements
The work summarized here was funded in part by grants from the National Institutes of Health (GM59636 to E.G.M. and GM63576 to A.R.F.), the Rita Allen Foundation (to A.R.F.) and the W.M. Keck Foundation (to A.R.F.) A.R.F. is a Distinguished Young Scholar in Medical Research of the W.M. Keck Foundation and an Investigator of the Howard Hughes Medical Institute. The authors gratefully acknowledge the efforts of their past and current research group members.
References
- 1.
- Cohn WE, Volkin E. Nucleoside-5′-phosphates from ribonucleic acid. Nature. 1951;167:483–484.
- 2.
- Davis FF, Allen FW. Ribonucleic acids from yeast which contain a fifth nucleotide. J Biol Chem. 1957;227:907–915. [PubMed: 13463012]
- 3.
- Cohn WE. 5-Ribosyl uracil, a carbon-carbon ribofuranosyl nucleoside in ribonucleic acids. Biochim Biophys Acta. 1959;32:569–571. [PubMed: 13811055]
- 4.
- Cohn WE. Pseudouridine, a carbon-carbon linked ribonucleoside in ribonucleic acids: isolation, structure and chemical characteristics. J Biol Chem. 1960;235:1488–1498. [PubMed: 13811056]
- 5.
- Yu CT, Allen FW. Studies on an isomer of uridine isolated from ribonucleic acids. Biochim Biophys Acta. 1959;32:393–406. [PubMed: 13846687]
- 6.
- Scannell JP, Crestfield AM, Allen FW. Methylation studies on various uracil derivatives and on an isomer of uridine isolated from ribonucleic acids. Biochim Biophys Acta. 1959;32:406–412. [PubMed: 14442211]
- 7.
- Johnson L, Söll D. In vitro biosynthesis of pseudouridine at the polynucleotide level by an enzyme extract from Escherichia coli. Proc Natl Acad Sci USA. 1970;67:943–950. [PMC free article: PMC283296] [PubMed: 4943184]
- 8.
- Cortese R, Kammen HO, Spengler SJ. et al. Biosynthesis of pseudouridine in transfer ribonucleic acid. J Biol Chem. 1974;249:1103–1108. [PubMed: 4592259]
- 9.
- Arena F, Ciliberto G, Ciampi S. et al. Purification of pseudouridylate synthetase I from Salmonella typhimurium. Nucleic Acids Res. 1978;5:4523–4536. [PMC free article: PMC342770] [PubMed: 370771]
- 10.
- Samuelsson T, Olsson M. Transfer RNA pseudouridine synthases in Saccharomyces cerevisiae. J Biol Chem. 1990;265:8782–8787. [PubMed: 2187870]
- 11.
- Koonin EV. Pseudouridine synthases: four families of enzymes containing a putative uridine-binding motif also conserved in dUTPases and dCTP deaminases. Nucleic Acids Res. 1996;24:2411–2415. [PMC free article: PMC145960] [PubMed: 8710514]
- 12.
- Kaya Y, Ofengand J. A novel unanticipated type of pseudouridine synthase with homologs in bacteria archaea and eukarya. RNA. 2003;9:711–721. [PMC free article: PMC1370438] [PubMed: 12756329]
- 13.
- Becker HF, Motorin Y, Planta RJ. et al. The yeast gene YNL292w encodes a pseudouridine synthase (Pus4) catalyzing the formation of Ψ55 in both mitochondrial and cytoplasmic tRNAs. Nucleic Acids Res. 1997;25:4493–4499. [PMC free article: PMC147073] [PubMed: 9358157]
- 14.
- Ma X, Zhao X, Yu Y-T. Pseudouridylation of U2 snRNA in S. cerevisiae is catalyzed by an RNA-independent mechanism. EMBO J. 2003;22:1889–1897. [PMC free article: PMC154481] [PubMed: 12682021]
- 15.
- Behm-Ansmant I, Urban A, Ma X. et al. The Saccharomyces cerevisiae U2 snRNA:pseudouridine-synthase Pus7p is a novel multisite-multisubstrate RNA:Ψ-synthase also acting on tRNAs. RNA. 2003;9:1371–1382. [PMC free article: PMC1287059] [PubMed: 14561887]
- 16.
- Watanabe Y, Gray MW. Evolutionary appearance of genes encoding proteins associated with box H/ACA snoRNAs: Cbf5p in Euglena gracilis, an early diverging eukaryote and candidate Gar1p and Nop10p homologs in archaebacteria. Nucleic Acids Res. 2000;28:2342–2352. [PMC free article: PMC102724] [PubMed: 10871366]
- 17.
- Aravind L, Koonin EV. THUMP—a predicted RNA-binding domain shared by 4-thiouridine, pseudouridine synthases and RNA methylases. Trends Biochem Sci. 2001;26:215–217. [PubMed: 11295541]
- 18.
- Roovers M, Hale C, Tricot C. et al. Formation of the conserved pseudouridine at position 55 in archaeal tRNA. Nucleic Acids Res. 2006;34:4293–4301. [PMC free article: PMC1616971] [PubMed: 16920741]
- 19.
- McCleverty C, Hornsby M, Spraggon G. et al. Crystal structure of human Pus10, a novel pseudouridine synthase. J Mol Biol. 2007;373:1243–1254. [PubMed: 17900615]
- 20.
- Raychaudhuri S, Niu L, Conrad J. et al. Functional effect of deletion and mutation of the Escherichia coli ribosomal RNA and tRNA pseudouridine synthase RluA. J Biol Chem. 1999;274:18880–18886. [PubMed: 10383384]
- 21.
- Huang L, Pookanjanatavip M, Gu X. et al. A conserved aspartate of tRNA pseudouridine synthase is essential for activity and a probable nucleophilic catalyst. Biochemistry. 1998;37:344–351. [PubMed: 9425056]
- 22.
- Ramamurthy V, Swann SL, Paulson JL. et al. Critical aspartic acid residues in pseudouridine synthases. J Biol Chem. 1999;274:22225–22230. [PubMed: 10428788]
- 23.
- Del Campo M, Kaya Y, Ofengand J. Identification and site of action of the remaining four putative pseudouridine synthases in Escherichia coli. RNA. 2001;7:1603–1615. [PMC free article: PMC1370202] [PubMed: 11720289]
- 24.
- Foster PG, Huang L, Santi D. et al. The structural basis for tRNA recognition and pseudouridine formation by pseudouridine synthase I. Nat Struct Biol. 2000;7:23–27. [PubMed: 10625422]
- 25.
- Hoang C, Ferré-D'Amaré AR. Cocrystal structure of a tRNA Ψ55 pseudouridine synthase: nucleotide flipping by an RNA-modifying enzyme. Cell. 2001;107:929–939. [PubMed: 11779468]
- 26.
- Sivaraman J, Sauve V, Larocque R. et al. Structure of the 16S rRNA pseudouridine synthase RsuA bound to uracil and UMP. Nat Struct Biol. 2002;9:353–358. [PubMed: 11953756]
- 27.
- Sivaraman J, Iannuzzi P, Cygler M. et al. Crystal structure of the RluD pseudouridine synthase catalytic module, an enzyme that modifies 23S rRNA and is essential for normal cell growth of Escherichia coli. J Mol Biol. 2004;335:87–101. [PubMed: 14659742]
- 28.
- Del Campo M, Ofengand J, Malhotra A. Crystal structure of the catalytic domain of RluD, the only rRNA pseudouridine synthase required for normal growth of Escherichia coli. RNA. 2004;10:231–239. [PMC free article: PMC1370535] [PubMed: 14730022]
- 29.
- Mizutani K, Machida Y, Unzai S. et al. Crystal structures of the catalytic domains of pseudouridine synthases RluC and RluD from Escherichia coli. Biochemistry. 2004;43:4454–4463. [PubMed: 15078091]
- 30.
- Ericsson UB, Nordlund P, Hallberg BM. X-ray structure of tRNA pseudouridine synthase TruD reveals an inserted domain with a novel fold. FEBS Letters. 2004;565:59–64. [PubMed: 15135053]
- 31.
- Kaya Y, Del Campo M, Ofengand J. et al. Crystal structure of TruD, a novel pseudouridine synthase with a new protein fold. J Biol Chem. 2004;279:18107–18110. [PubMed: 14999002]
- 32.
- Hoang C, Ferré-D'Amaré AR. Crystal structure of the highly divergent pseudouridine synthase TruD reveals a circular permutation of a conserved fold. RNA. 2004;10:1026–1033. [PMC free article: PMC1370594] [PubMed: 15208439]
- 33.
- Hoang C, Chen J, Vizthum CA. et al. Crystal structure of pseudouridine synthase RluA: indirect sequence readout through protein-induced RNA structure. Mol Cell. 2006;24:535–545. [PubMed: 17188032]
- 34.
- Armengaud J, Urbonavicius J, Fernandez B. et al. N2-Methylation of guanosine at position 10 in tRNA is catalyzed by a THUMP domain-containing, S-adenosylmethionine-dependent methyltransferase, conserved in archaea and eukaryota. J Biol Chem. 2004;279:37142–37152. [PubMed: 15210688]
- 35.
- Waterman D, Ortizlombardia M, Fogg M. et al. Crystal structure of bacillus anthracis ThiI, a tRNA-modifying enzyme containing the predicted RNA-binding THUMP domain. J Mol Biol. 2006;356:97–110. [PubMed: 16343540]
- 36.
- Gabant G, Auxilien S, Tuszynska I. et al. THUMP from archaeal tRNA:m2 2G10 methyltransferase, a genuine autonomously folding domain. Nucleic Acids Res. 2006;34:2483–2494. [PMC free article: PMC1459410] [PubMed: 16687654]
- 37.
- Nurse K, Wrzesinski J, Bakin A. et al. Purification, cloning and properties of the tRNA Ψ55 synthase from Escherichia coli. RNA. 1995;1:102–112. [PMC free article: PMC1369054] [PubMed: 7489483]
- 38.
- Gu X, Yu M, Ivanetich KM. et al. Molecular recognition of tRNA by tRNA pseudouridine 55 synthase. Biochemistry. 1998;37:339–343. [PubMed: 9425055]
- 39.
- Krasilnikov AS, Mondragón A. On the occurrence of the T-loop RNA folding motif in large RNA molecules. RNA. 2003;9:640–643. [PMC free article: PMC1370430] [PubMed: 12756321]
- 40.
- Pan H, Agarwalla S, Moustakas DT. et al. Structure of tRNA pseudouridine synthase TruB and its RNA complex: RNA recognition through a combination of rigid docking and induced fit. Proc Natl Acad Sci USA. 2003;100:12648–12653. [PMC free article: PMC240672] [PubMed: 14566049]
- 41.
- Phannachet K, Huang RH. Conformational change of pseudouridine 55 synthase upon its association with RNA substrate. Nucleic Acids Res. 2004;32:1422–1429. [PMC free article: PMC390278] [PubMed: 14990747]
- 42.
- Hoang C, Hamilton CS, Mueller EG. et al. Precursor complex structure of pseudouridine synthase TruB suggests coupling of active site perturbations to an RNA-sequestering peripheral protein domain. Protein Sci. 2005;14:2201–2206. [PMC free article: PMC2279332] [PubMed: 15987897]
- 43.
- Ferré-D'Amaré AR. RNA-modifying enzymes. Curr Opin Struct Biol. 2003;13:49–55. [PubMed: 12581659]
- 44.
- Aravind L, Koonin EV. Novel predicted RNA-binding domains associated with the translation machinery. J Molec Evol. 1999;48:291–302. [PubMed: 10093218]
- 45.
- Anantharaman V, Koonin EV, Aravind L. Comparative genomics and evolution of proteins involved in RNA metabolism. Nucleic Acids Res. 2002;30:1427–1464. [PMC free article: PMC101826] [PubMed: 11917006]
- 46.
- Ishitani R, Nureki O, Nameki N. et al. Alternative tertiary structure of tRNA for recognition by a posttranscriptional modification enzyme. Cell. 2003;113:383–394. [PubMed: 12732145]
- 47.
- Li L, Ye K. Crystal structure of an H/ACA box ribonucleoprotein particle. Nature. 2006;443:302–307. [PubMed: 16943774]
- 48.
- Pérez-Arellano I, Gallego J, Cervera J. The PUA domain a structural and functional overview. FEBS J. 2007;274:4972–4984. [PubMed: 17803682]
- 49.
- Kammen HO, Marvel CC, Hardy L. et al. Purification, structure and properties of Escherichia coli tRNA pseudouridine synthase I. J Biol Chem. 1988;263:2255–2263. [PubMed: 3276686]
- 50.
- Hur S, Stroud R. How U38, 39 and 40 of many tRNAs become the targets for pseudouridylation by TruA. Mol Cell. 2007;26:189–203. [PMC free article: PMC3562137] [PubMed: 17466622]
- 51.
- Reichow SL, Hamma T, Ferré-D'Amaré AR. et al. The structure and function of small nucleolar ribonucleoproteins. Nucleic Acids Res. 2007;35:1452–1464. [PMC free article: PMC1865073] [PubMed: 17284456]
- 52.
- Klein DJ, Schmeing TM, Moore PB. et al. The kink-turn: a new RNA secondary structure motif. EMBO J. 2001;20:4214–4221. [PMC free article: PMC149158] [PubMed: 11483524]
- 53.
- Nolivos S, Carpousis AJ, Clouet-d'Orval B. The K-loop, a general feature of the Pyrococcus C/D guide RNAs, is an RNA structural motif related to the K-turn. Nucleic Acids Res. 2005;33:6507–6514. [PMC free article: PMC1289080] [PubMed: 16293637]
- 54.
- Hamma T, Ferré-D'Amaré AR. Structure of protein L7Ae bound to a K-turn derived from an archaeal box H/ACA sRNA at 1.8 Å resolution. Structure. 2004;12:893–903. [PubMed: 15130481]
- 55.
- Hamma T, Reichow SL, Varani G. et al. The Cbf5-Nop10 complex is a molecular bracket that organizes box H/ACA RNPs. Nat Struct Mol Biol. 2005;12:1101–1107. [PubMed: 16286935]
- 56.
- Manival X, Charron C, Fourmann JB. et al. Crystal structure determination and site-directed mutagenesis of the Pyrococcus abyssi aCBF5-aNOP10 complex reveal crucial roles of the C-terminal domains of both proteins in H/ACA sRNP activity. Nucleic Acids Res. 2006;34:826–839. [PMC free article: PMC1361308] [PubMed: 16456033]
- 57.
- Spedaliere CJ, Hamilton CS, Mueller EG. Functional importance of motif I of pseudouridine synthases: mutagenesis of aligned lysine and proline residues. Biochemistry. 2000;39:9459–9465. [PubMed: 10924141]
- 58.
- Rashid R, Liang B, Baker DL. et al. Crystal structure of a Cbf5-Nop10-Gar1 complex and implications in RNA-guided pseudouridylation and dyskeratosis congenita. Mol Cell. 2006;21:249–260. [PubMed: 16427014]
- 59.
- Liang B, Xue S, Terns RM. et al. Substrate RNA positioning in the archaeal H/ACA ribonucleoprotein complex. Nat Struct Mol Biol. 2007;14:1189–1195. [PubMed: 18059286]
- 60.
- Conrad J, Niu L, Rudd K. et al. 16S ribosomal RNA pseudouridine synthase RsuA of Escherichia coli: deletion, mutation of the conserved Asp102 residue and sequence comparison among all other pseudouridine synthases. RNA. 1999;5:751–763. [PMC free article: PMC1369802] [PubMed: 10376875]
- 61.
- Gu X, Liu Y, Santi DV. The mechanism of pseudouridine synthase I as deduced from its interaction with 5-fluorouracil-tRNA. Proc Natl Acad Sci USA. 1999;96:14270–14275. [PMC free article: PMC24426] [PubMed: 10588695]
- 62.
- Santi DV, McHenry CS, Sommer H. Mechanism of interaction of thymidylate synthetase with 5-fluorodeoxyuridylate. Biochemistry. 1974;13:471–481. [PubMed: 4203910]
- 63.
- Zechel DL, Withers SG. Glycosidase mechanisms: anatomy of a finely tuned catalyst. Accounts Chem Res. 2000;33:11–18. [PubMed: 10639071]
- 64.
- Spedaliere CJ, Mueller EG. Not all pseudouridine synthases are potently inhibited by RNA containing 5-fluorouridine. RNA. 2004;10:192–199. [PMC free article: PMC1370531] [PubMed: 14730018]
- 65.
- Spedaliere CJ, Ginter JM, Johnston MV. et al. The pseudouridine synthases: revisiting a mechanism that seemed settled. J Am Chem Soc. 2004;126:12758–12759. [PubMed: 15469254]
- 66.
- Hamilton CS, Greco TM, Vizthum CA. et al. Mechanistic investigations of pseudouridine synthase RluA using RNA containing 5-fluorouridine. Biochemistry. 2006;45:12029–12039. [PMC free article: PMC2580076] [PubMed: 17002302]
- 67.
- Hamilton CS, Spedaliere CJ, Ginter JM. et al. The roles of the essential Asp-48 and highly conserved His-43 elucidated by the pH dependence of the pseudouridine synthase TruB. Arch Biochem Biophys. 2005;433:322–334. [PubMed: 15581587]
- 68.
- Phannachet K, Elias Y, Huang RH. Dissecting the roles of a strictly conserved tyrosine in substrate recognition and catalysis by pseudouridine 55 synthase. Biochemistry. 2005;44:15488–15494. [PubMed: 16300397]
- 69.
- Dawson RMC, Elliot DC, Elliot WH. et al. Data for biochemical research (Oxford Science Publications, 1986)
- 70.
- Arluison V, Hountondji C, Robert B. et al. Transfer RNA-pseudouridine synthetase Pus1 of Saccharomyces cerevisiae contains one atom of zinc essential for its native conformation and tRNA recognition. Biochemistry. 1998;37:7268–7276. [PubMed: 9585540]
- 71.
- Carson M. Ribbons. Meth Enzymol. 1997;277:493–505. [PubMed: 18488321]
- 72.
- Shi H, Moore PB. The crystal structure of yeast phenylalanine tRNA at Å 1.93 resolution: a classic structure revisited. RNA. 2000;6:1091–1105. [PMC free article: PMC1369984] [PubMed: 10943889]
- 73.
- Krebs WG, Gerstein M. The morph server: a standarized system for analyzing and visualizing macromolecular motions in a database framework. Nucleic Acids Res. 2000;28:1665–1675. [PMC free article: PMC102811] [PubMed: 10734184]
- Pseudouridine Formation, the Most Common Transglycosylation in RNA - Madame Curi...Pseudouridine Formation, the Most Common Transglycosylation in RNA - Madame Curie Bioscience Database
- Modeling Metastasis In Vivo - Madame Curie Bioscience DatabaseModeling Metastasis In Vivo - Madame Curie Bioscience Database
- The Immune-Suppressive Effects of Pain - Madame Curie Bioscience DatabaseThe Immune-Suppressive Effects of Pain - Madame Curie Bioscience Database
- β-Secretase: Progress and Open Questions - Madame Curie Bioscience Databaseβ-Secretase: Progress and Open Questions - Madame Curie Bioscience Database
- The Sarcoglycans - Madame Curie Bioscience DatabaseThe Sarcoglycans - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...