

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Introduction
Almost 30 years ago, Kerr and co-workers proposed the existence of an intrinsic cell death program and introduced the term apoptosis for the execution of this program.1 Apoptosis is an active form of cell death enabling individual cells to commit suicide. In contrast, necrosis is a passive form of cell death induced by accidental damage of tissue and does not encompass activation of any specific cellular program during the death process. Initial classification of cell death into the apoptosis/necrosis dichotomy was mainly based on morphological criteria: hallmarks of apoptosis include membrane blebbing, cell shrinkage and chromatin condensation/fragmentation whereas necrosis typically is associated with early loss of plasma membrane integrity and swelling of the cell body. In recent years, significant progress has been made identifying key components of the apoptotic cell death machinery and deciphering the signaling pathways in which they are embedded. It is generally accepted that members of the caspase family of proteases are central executioners of apoptotic cell death (see other chapters of this book). For many years, apoptosis was thought to be a synonym for programmed cell death (PCD)*. However, an increasing number of studies substantiate the existence of caspase-independent forms of PCD. The initial model describing only one, stereotypical form of active cell death today is viewed as an oversimplification, because it is now generally accepted that multiple forms of PCD exist and that some forms of PCD do not require activation of caspases (Fig. 1). One single execution system, i.e., the caspases, could easily be overcome by viruses and transformed cells. Hence, alternative cell death pathways, acting as backup pathways, might have evolved during evolution.2 This chapter will focus on the current cognoscenti of caspase-independent forms of PCD. (* The term programmed cell death was initially reserved for developmental cell death. It will be used interchangeably with active cell death in this chapter.)
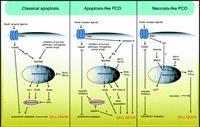
Multiple Forms of PCD
Although there is some controversy regarding the nomenclature of the different forms of PCD, in the majority of the current literature the term apoptosis in most cases is exclusively used for caspase-dependent cell death.3 A broad classification of different PCD forms has recently been proposed by Leist and Jäättelä.4 According to this classification which is based on both morphological and biochemical criteria, three different forms of PCD exist (Fig. 1) in addition to passive necrosis: 1) classical, caspase-dependent apoptosis associated with membrane blebbing, potent chromatin condensation/fragmentation, phosphatidylserine exposure, disruption of the cell into apoptotic bodies, activation of executioner caspases and internucleosomal DNA cleavage, 2) apoptosis-like PCD characterized by less compact chromatin condensation, phosphatidylserine exposure, but absence of executioner caspase activation, and 3) necrosis-like PCD which occurs in the absence of both chromatin condensation and caspase activation.
In addition to this tripartite classification, there are more specialized forms of PCD not fitting into any of the three above models. These additional forms which are restricted to distinct cell types include paraptosis and dark cell death.5,6 Yet another type of PCD is represented by autophagy, a process first described in yeast. In autophagy, which is characterized by prominent cytoplasmic vacuolization, cells are destroyed by degradation of cellular components via an autophagosomic-lysosomal pathway.7 In this Chapter, the biochemical features and genetic requirements for different forms of PCD will be discussed.
Caspase-Independent PCD Controlled by Bcl-2 Family Proteins
Activation of executioner caspases can occur after ligation of death receptors or via the release of proapoptotic factors from mitochondria. The latter pathway is controlled by Bcl-2 family proteins. In this pathway, activation of the initator caspase-9 occurs via binding of adaptor protein apoptotic protease activating factor-1 (Apaf-1) to the caspase recruitment domain (CARD). The association of caspase-9 and Apaf-1 and subsequent apoptosome formation is triggered by the proapoptotic factor cytochrome c. This factor resides in the mitochondrial intermembrane space where it participates in electron transport during respiration. During apoptosis, cytochrome c is released from the intermembrane space because of a significant increase in mitochondrial outer membrane permeability. This process is triggered and controlled by pro- and antiapoptotic Bcl-2 family proteins.8 However, mitochondria as central integrators of PCD signaling pathways are able to release multiple factors that may trigger a caspase-independent cell death. Moreover, cell death may be caused by altered mitochondrial energetics directly due to the loss of cytochrome c.
Studies in Yeast
Caspase-independent cell death controlled by Bcl-2 family proteins and mitochondria has been initially studied in the fission yeast Schizosaccharomyces pombe, since the yeast genome does not contain any caspase genes. Nonetheless, overexpression of proapoptotic Bcl-2 family members Bax and Bak has been shown to cause cell death in S. pombe.9 Bax- and Bak-induced cell death in yeast is associated with prominent cytosolic vacuolization and nuclear chromatin condensation. Furthermore, it can be inhibited by the antiapoptotic counteraction of Bcl-XL. This observation might indicate that the emergence of caspase-independent cell death pathways has occurred much earlier than classical caspase-dependent apoptosis during evolution. As a matter of fact, PCD has even been observed in bacteria.10
Studies in Genetically Altered Background
In concordance to the yeast model, overexpressed Bax and Bak are capable of inducing mitochondrial dysfunction-triggered death in cells lacking apoptosome-mediated caspase activation due to deficiency of caspase-3, caspase-9 or Apaf-1.11 This mitochondrial step requires the activation of Bax and/or Bak, since Bax/Bak double knockouts are completely deficient to trigger the intrinsic apoptosis pathway.11 Since it is well established that the major executioner caspase is caspase-3,12 cells which lack expression of caspase-3 represent an often used experimental system to study the dependence of morphological and biochemical alterations on this particular caspase during cell death.13,14 However, execution of cell death does not necessarily occur in caspase-independent fashion in cells devoid of caspase-3, since other executioner caspases such as caspase-6 and -7 might in part substitute for caspase-3 activity.15,16
Studies Using Caspase Inhibitors
Inhibition of the enzymatic activity is another widely used experimental approach to study the caspase dependence of cell death. The employed caspase inhibitors are of either biological (viral or cellular caspase inhibitors) or chemical-origin (synthetic peptide inhibitors). A myriad of experiments has been performed with various peptide inhibitors, either directed against individual caspases, or the zVAD-fmk broad-spectrum inhibitor. zVAD-fmk, the compound used in most studies, binds irreversibly to the catalytic site of caspases, forming a covalent inhibitor/enzyme complex. Generally, execution of cell death can be decelerated, but not prevented by abrogation of caspase activity.17 It has been proposed that no single experimental system exists in which zVAD-fmk can save cells from dying.18 This has been verified for multiple apoptotic stimuli18 and both major cell death pathways, the death receptor pathway (see below) and the mitochondrial pathway.17,19–21 These observations imply that in an individual cell receiving any given apoptotic death signal both caspase-dependent and caspase-independent cell death pathways are activated in parallel. In cells with inhibited caspase activity caspase-independent cell death mechanisms suffice to eventually cause cell death, albeit in a slower, less efficient manner.17 Although active caspases are not a prerequisite for execution of cell death, the time frame and caspase dependence of individual events during PCD might be stimulus- and cell type-specific. For example, conflicting data exist on the dependence of membrane blebbing on caspase activity. Two recent reports describe caspase-dependent activation of serine/threonine kinase ROCK as critical step in initiation of membrane blebbing.22,23 On the other hand, application of caspase inhibitors revealed that some of the early, cytoplasmic changes in apoptotic cell death do not depend on activation of caspases. In a seminal study by McCarthy and coworkers, cell shrinkage and membrane blebbing was not inhibited by abrogation of caspase activity with pancaspases inhibitors after induction of cell death by a variety of stimuli including overexpression of oncogenes and induction of DNA damage.17 In an alternative cell death pathway, members of the death-associated protein (DAP) kinase family might trigger membrane blebbing in caspase-independent fashion.24 The catalytic domain of these kinases shares a high sequence homology to the myosin light chain kinase (MLCK). Of note, phosphorylation of myosin light chain (MLC) has been implicated in caspase-independent membrane blebbing.25,26
Although caspase inhibitors can dramatically alter the death response of cells, the observed effects of these inhibitors must be discussed critically. Especially, it remains to be established if so called “pan-caspase inhibitors”, such as the zVAD-fmk, really completely inhibit all proapoptotic caspases in the cell. The biggest drawbacks of the chemical caspase inhibitors are their limited stability and the gross differences in binding affinity to the individual caspase family members. In addition, other cysteine proteases such as calpains and cathepsins may also be inhibited by these compounds, thus aggravating interpretation of experimental data.
Mitochondrial Dysfunction Due to Loss of Cytochrome c
While many studies have focused on the role of cytochrome c release as the trigger of apoptosome activation,27,28 loss of cytochrome c may also directly affect mitochondrial free radical and ATP production. Confocal time-lapse imaging experiments using cytochrome-c-GFP-expressing cells suggested that the release of cytochrome c during apoptosis is rapid and complete.29,30 Cytochrome c normally transports electrons between mitochondrial complexes III and IV. A disruption of the mitochondrial electron flow caused by a significant loss of cytochrome c will maintain complex I and the ubiquinone at complex II in their reduced state. This condition may favor 1-electron reduction of molecular oxygen, presumably due to autooxidation processes.31,32 This is also a potential mechanism for the known effect of complex III and IV inhibitors to increase the mitochondrial production of superoxide. Inhibition of mitochondrial electron flow and increased mitochondrial superoxide production secondary to cytochrome c release have been observed during Fas and staurosporine-mediated apoptosis of Jurkat and HL60 cells.33,34 Interestingly, cytochrome c release and superoxide production also occur at similar time points in the death cascade during trophic factor withdrawal- or staurosporine-induced apoptosis in neurons.35–39 Moreover, in cell lines deficient in mitochondrial respiration (ρ– cells), cytochrome c release and activation of apoptosis are preserved, while an increased superoxide production can not be detected.39,40 Therefore, cytochrome c release may occur upstream of mitochondria-derived ROS production. The protective effects of antioxidants, SOD-mimetics, and superoxide dismutase overexpression in several apoptosis models suggest that the production of superoxide due to the loss of cytochrome c may play an important role in the execution of cell death, in particular in nontransformed cells.35,37,39,41,42 In contrast, inhibition of executioner caspases reduces the biochemical and morphological signs of apoptosis, but does not necessarily inhibit cell death.37,43,44 Therefore, mitochondrial superoxide production may significantly contribute to cell death during apoptosis, particularly in cell types that are sensitive to oxidant stress.
Mitochondria that have released their cytochrome c are likewise less capable of producing ATP. Mitochondria are able to maintain a mitochondrial membrane potential after the release of cytochrome c.29,37,45 Evidence has been provided that this is caused by a reversal of the F0F1-ATPase operating in the reverse mode, hence even consuming ATP.46,47 Readdition of cytochrome c to isolated mitochondria that underwent an outer membrane permeability increase likewise restores membrane potential and ATP production.47,48 Evidence has also been provided that mitochondria are able to maintain their membrane potential in intact cells by diffusion of cytosolic released cytochrome c back to the mitochondrial inner membrane.29,48 However, mitochondria will eventually depolarize after the release of cytochrome c, a process that is caspase-dependent in some systems.45 Mitochondrial depolarization will lead to ATP depletion, followed by a disturbance of ion homeostasis, cellular Ca2+ overloading, and finally cellular necrosis. In cultured rat sympathetic neurons deprived of NGF in the presence of caspase inhibitors, cells can be rescued from cell death until the time point of mitochondrial depolarization.49 Recent studies have shown that the opening of the permeability transition pore is involved in this final depolarization.50
Role of Other Proapoptotic Factors Released from Mitochondria in a Bcl-2-Dependent Manner
There are alternative signaling pathways leading to PCD-associated apoptotic events, such as degradation of chromosomal DNA. One of them is mediated by the apoptosis-inducing factor (AIF), a mitochondrial protein, that is released into the cytosol during execution of PCD (Table 1).51 AIF, most likely the best studied gene product involved in caspase-independent cell death to date belongs to the gene family of oxidoreductases. However, the enzymatic activity of AIF is not required for its cell death-inducing properties. Apparently, upon activation of the intrinsic cell death pathway, both caspase-dependent (apoptosome/caspase-3/CAD/DFF-40) and caspase-independent (AIF) execution pathways can be triggered simultaneously, both leading to distinct nuclear events during PCD. In contrast to CAD/DFF-40, AIF induces large-scale DNA fragmentation, thus leading to chromatin condensation. Peripheral condensation of chromatin is an early nuclear event in classical apoptosis.52,53 The DNA-degrading activity of AIF is caspase-independent as partial chromatinolysis and cell death caused by nuclear AIF is not inhibited by the presence of zVAD-fmk. In contrast, Bcl-2 overexpression inhibits AIF translocation from the mitochondria to the cytosol, thus abrogating AIF-triggered PCD.52 Knockout studies revealed that AIF might control early morphogenesis during embryonal development.54 The AIF gene of Dictyostelium discoideum, which has been recently identified,55 is capable of inducing cell death. This suggests that AIF-based death pathway might be evolutionary older than the caspase-dependent death cascade.
Table 1
Genes implicated in caspase-independent cell death pathways.
Not only large-scale fragmentation, but internucleosomal DNA cleavage might also occur in caspase-independent fashion under certain circumstances. In CAD/DFF-40 knockout cells, DNA laddering, indicative of internucleosomal DNA cleavage, can be observed after induction of cell death, although to a lesser extent than in wild-type cells. Recently, a novel apoptotic DNase, the endonuclease G, capable of internucleosomal DNA processing was characterized. Just like AIF, endonuclease G is released from the mitochondria and translocates to the nucleus during PCD.56
Caspase-Independent Cell Death in Response to Death Receptor Signaling
Activation of the death receptor by binding of tumor necrosis factor-α (TNF) and Fas ligand (FasL) to their respective receptors can induce both classical apoptosis and necrosis-like PCD upon certain experimental conditions.57,58 Furthermore, knockout studies revealed that necrosis-like PCD triggered by the extrinsic cell death pathway depends on both Fas-associated death domain (FADD)-mediated activation of the protein kinase receptor interacting protein (RIP). Interestingly, this type of cell death was shown to require the enzymatic activty of RIP which is dispensable for RIP-mediated activation of nuclear factor kB (NF-κB).57 Although the molecular mechanisms of death receptor-mediated necrosis are poorly characterized, mitochondrial dysfunction59,60 and non-caspase-proteases61 seem to be critically involved in this process. In the presence of zVAD-fmk, death receptor-mediated necrosis requires a mitochondrial step, although neither Bid cleavage, nor cytochrome c release are observed.59,60 Instead, this type of necrosis-like PCD is associated with increased production of ROS by the mitochondria.57–59 ROS are released from the mitochondria during TNF-induced PCD, and antioxidants inhibit this form of cell death.59,62
Necrosis Controlled by Bcl-2 Family Members
BNIP3, a member of the Bcl-2 family and direct interaction partner of Bcl-2, induces necrosis-like cell death through mitochondrial permeability transition. Cell death triggered by BNIP3 is associated with translocation of BNIP3 to the outer mitochondrial membrane, loss of mitochondrial membrane potential and increased production of reactive oxygen species (ROS). However, BNIP3-mediated cell death is independent of Apaf-1, caspase activation and cytochrome c release.63 It has been suggested that Bcl-2 exerts its anti-necrotic effect by complex formation with BNIP3.60
Involvement of Non-Caspase Proteases in PCD
In addition to caspases, other proteases such as serine proteases, cathepsins and calpains might be involved in PCD as well.18 The general serine protease inhibitor AEBSF has been shown to inhibit oncogene-driven PCD in rat fibroblasts.64 Granzymes A and B have been implicated in caspase-independent cell death pathways triggered by granule-mediated cytotoxicity of T lymphocytes. Granzyme B triggers the intrinsic cell death pathway via truncation of Bid in a caspase-independent cleavage event upstream of mitochondria.65 However, granzyme B-induced cell death is significantly delayed by abrogation of caspase activity.66 In contrast, granzyme A triggers caspase-independent cell death by activating the endonuclease granzyme A-activated DNase (GAAD), leading to single-strand DNA nicking and chromatin condensation.67 Another recently identified serine protease, HtrA2, which is released from the mitochondria during PCD, activates both caspase-dependent and caspase-independent cell death pathways. Caspase-independent cell death triggered by HtrA2 depends on its enzymatic activity.68 Two members of the cathepsin family, cathepsin B and D, lysosomal proteins, have been suggested to translocate to the cytoplasm during PCD.69 Under certain conditions cathepsin B can become the dominant execution protease in death receptor-induced PCD.61 The PCD-related role of an another cathepsin family member, cathepsin D, has also been recently described.70 Similiar to granzyme B, cathepsins have been implicated in cleavage and activation of Bid.71
Elevated cellular Ca2+ concentration during apoptosis, e.g., following mitochondrial dysfunction, may lead to the activation of PCD-related, Ca2+-dependent enzymes, such as calpains or death associated protein (DAP) kinase. Like caspases, calpains are a family of cytosolic cysteine proteases, but require Ca2+ for their activity. Activation of calpains can be amplified by caspase cleavage of the endogenous calpain inhibitor calpastatin.72 Calpains have been suggested to be involved in the regulation of caspase activity during apoptosis. The cleavage of upstream caspases-9 and -8, as well as executioner caspases-3 and -7 by calpains have been described. Calpain-cleaved procaspases-3 and -9 could still be activated by granzyme B.73 In a study by Ruiz-Vela and co-workers,74 calpain I-mediated proteolysis of procaspase-7 led to its activation. ER stress-induced apoptosis mediated via murine caspase-12 has also been shown to require calpain activation.75 On the other hand, several reports support the role of calpains as negative regulators of caspase activity.76,77 Calpain-generated fragments of caspases-7, -8 and -9 were inactive and/or unable to activate downstream executioner caspases, and calpain potently inhibited the ability of cytochrome c to activate executioner caspases. A recent study also demonstrated calpain-dependent cleavage of the cytochrome cbinding protein Apaf-1.78 It is therefore conceivable that the upstream or concomitant activation of calpains exerts a negative feedback signal on caspase activation.
A significant number of studies revealed that specific calpain inhibitors can inhibit PCD in many cases.69 Interestingly, calpains promote apoptosis-like events during platelet activation and excitotoxic neuron death,73,76 including chromatin condensation, phosphatidylserine exposure, caspase substrate cleavage and cell shrinkage, thus mimicking aspects of caspase-mediated apoptosis. Hence, calpains are among candidates for the execution of apoptosis-like PCD.
Oncogenic Transformation: Escape from PCD
One of the fundamental functions of PCD is to protect higher organisms from cancer. A number of oncogenes, including c-Myc, E2F and Ras have been shown to induce PCD upon overexpression in nontransformed cells, although the mechanism of oncogene-driven PCD remains elusive.79 Importantly, both caspase-dependent and caspase-independent cell death mechanisms must be evaded by tumor cells during malignant transformation. A number of oncogene-driven caspase-independent forms of PCD have been described. Oncogenic Ras induces a caspase-independent and Bcl-2-insensitive form of PCD in human cancer cells.80 In contrast, c-Myc triggers both caspase-dependent and caspase-independent cell death pathways.81 Inhibition of PCD by tumor cells is achieved by either enhancement of antiapoptotic signaling pathways or inactivation of tumor suppressor genes. Survivin has been shown to protect tumor cells from both classical apoptosis and caspase-independent PCD.82 A constitutively active mutant of Akt/protein kinase B(PKB) has been recently implicated in suppression of caspase-independent PCD.83 In this study, ceramide-triggered cell death occurred in the presence of both zVAD-fmk and overexpressed Bcl-XL in glioma cells. However, this type of PCD could be counteracted by the dominant-active Akt/PKB mutant.83 Loss of function of tumor suppressors PML and Bin-1 might be implicated in abrogation of caspase-independent cell death.84,85 The tumor suppressor PML is involved in cell death induced by a wide variety of stimuli known to activate classical caspase-dependent apoptosis.86 In addition, a p53-coactivator function of PML has been recently established.87 However, PML-triggered cell death does not require activation of caspases as zVAD-fmk may even enhance cell death induced by PML.88 Caspase-independent signaling by c-Myc seems to require Bin-1.85 Similar to mutant Ras, PCD induced by Bin-1-overexpression cannot be rescued by zVAD-fmk or Bcl-2. However, Bin-1-triggered DNA degradation is abrogated by inhibition of serine proteases.85 Execution of oncogene-driven caspase-independent cell death likely involves other noncaspase proteases, such as cathepsins and calpains.
Outlook
The discovery of alternative, caspase-independent cell death pathways increases our understanding of the evolution of PCD mechanisms, but also demands the search for new strategies for the treatment of disorders associated with a deregulation of PCD, such as cancer, ischemic and degenerative diseases. Apart from agents that inhibit the activity of caspases potential targets for future drug development are the Bcl-2 family proteins. New anticancer drugs that facilitate mitochondrial outer membrane permeability may help to modulate death pathways within the cell. Several novel cancer drugs activating caspase-independent death programs in tumor cells has already been described.4 On the other hand, inhibitors of caspases, calpains, and cathepsins, but also antioxidants may prove beneficial for the treatment of ischemic and degenerative disorders involving a PCD component. Further experiments and clinical trials will reveal the effectiveness of these innovative therapies.
References
- 1.
- Kerr J F, Wyllie A H, Currie A R. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–57. [PMC free article: PMC2008650] [PubMed: 4561027]
- 2.
- Nicotera P, Leist M, Single B. et al. Execution of apoptosis: converging or diverging pathways? Biol Chem. 1999;380:1035–40. [PubMed: 10543440]
- 3.
- Blagosklonny M V. Cell death beyond apoptosis. Leukemia. 2000;14:1502–8. [PubMed: 10942250]
- 4.
- Leist M, Jaattela M. Four deaths and a funeral: from caspases to alternative mechanisms. Nat Rev Mol Cell Biol. 2001;2:589–98. [PubMed: 11483992]
- 5.
- Sperandio S, de Belle I, Bredesen D E. An alternative, nonapoptotic form of programmed cell death. Proc Natl Acad Sci USA. 2000;97:14376–81. [PMC free article: PMC18926] [PubMed: 11121041]
- 6.
- Turmaine M, Raza A, Mahal A. et al. Nonapoptotic neurodegeneration in a transgenic mouse model of Huntington's disease. Proc Natl Acad Sci USA. 2000;97:8093–7. [PMC free article: PMC16675] [PubMed: 10869421]
- 7.
- Lee C Y, Baehrecke E H. Steroid regulation of autophagic programmed cell death during development. Development. 2001;128:1443–55. [PubMed: 11262243]
- 8.
- Desagher S, Martinou J C. Mitochondria as the central control point of apoptosis. Trends Cell Biol. 2000;10:369–77. [PubMed: 10932094]
- 9.
- Jurgensmeier J M, Krajewski S, Armstrong R C. et al. Bax- and Bak-induced cell death in the fission yeast Schizosaccharomyces pombe. Mol Biol Cell. 1997;8:325–39. [PMC free article: PMC276083] [PubMed: 9190211]
- 10.
- Yarmolinsky M B. Programmed cell death in bacterial populations. Science. 1995;267:836–7. [PubMed: 7846528]
- 11.
- Cheng E H, Wei M C, Weiler S. et al. Bcl-2, bclx(l) sequester bh3 domainonly molecules preventing bax and bakmediated mitochondrial apoptosis. Mol Cell. 2001;8:705–11. [PubMed: 11583631]
- 12.
- Slee E A, Adrain C, Martin S J. Executioner caspase-3, 6, and 7 perform distinct, nonredundant roles during the demolition phase of apoptosis. J Biol Chem. 2001;276:7320–6. [PubMed: 11058599]
- 13.
- Janicke R U, Sprengart M L, Wati M R. et al. caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J Biol Chem. 1998;273:9357–60. [PubMed: 9545256]
- 14.
- Fiers W, Beyaert R, Declercq W. et al. More than one way to die: apoptosis, necrosis and reactive oxygen damage. Oncogene. 1999; 18:7719–30. [PubMed: 10618712]
- 15.
- Liang Y, Yan C, Schor N F. Apoptosis in the absence of caspase 3. Oncogene. 2001;20:6570–8. [PubMed: 11641782]
- 16.
- Zheng T S, Hunot S, Kuida K. et al. Deficiency in caspase-9 or caspase-3 induces compensatory caspase activation. Nat Med. 2000;6:1241–7. [PubMed: 11062535]
- 17.
- McCarthy N J, Whyte M K, Gilbert C S. et al. Inhibition of Ced3/ICErelated proteases does not prevent cell death induced by oncogenes, DNA damage, or the Bcl-2 homologue Bak. J Cell Biol. 1997;136:215–27. [PMC free article: PMC2132458] [PubMed: 9008715]
- 18.
- Borner C, Monney L. Apoptosis without caspases: an inefficient molecular guillotine? Cell Death Differ. 1999;6:497–507. [PubMed: 10381652]
- 19.
- Deas O, Dumont C, MacFarlane M. et al. caspase-independent cell death induced by antiCD2 or staurosporine in activated human peripheral T lymphocytes. J Immunol. 1998;161:3375–83. [PubMed: 9759854]
- 20.
- Miller T M, Moulder K L, Knudson C M. et al. Bax deletion further orders the cell death pathway in cerebellar granule cells and suggests a caspase-independent pathway to cell death. J Cell Biol. 1997;139:205–17. [PMC free article: PMC2139809] [PubMed: 9314540]
- 21.
- Xiang J, Chao D T, Korsmeyer S J. BAXinduced cell death may not require interleukin 1 betaconverting enzymelike proteases. Proc Natl Acad Sci USA. 1996;93:14559–63. [PMC free article: PMC26172] [PubMed: 8962091]
- 22.
- Coleman M L, Sahai E A, Yeo M. et al. Membrane blebbing during apoptosis results from caspasemediated activation of ROCK I. Nat Cell Biol. 2001;3:339–45. [PubMed: 11283606]
- 23.
- Sebbagh M, Renvoize C, Hamelin J. et al. caspase-3mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat Cell Biol. 2001;3:346–52. [PubMed: 11283607]
- 24.
- Kogel D, Prehn J H, Scheidtmann K H. The DAP kinase family of proapoptotic proteins: novel players in the apoptotic game. Bioessays. 2001;23:352–8. [PubMed: 11268041]
- 25.
- Mills J C, Stone N L, Erhardt J. et al. Apoptotic membrane blebbing is regulated by myosin light chain phosphorylation. J Cell Biol. 1998;140:627–36. [PMC free article: PMC2140178] [PubMed: 9456322]
- 26.
- Mills J C, Stone N L, Pittman R N. Extranuclear apoptosis. The role of the cytoplasm in the execution phase. J Cell Biol. 1999;146:703–8. [PMC free article: PMC2156138] [PubMed: 10459006]
- 27.
- Liu X, Kim C N, Yang J. et al. Induction of apoptotic program in cellfree extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–57. [PubMed: 8689682]
- 28.
- Li P, Nijhawan D, Budihardjo I. et al. Cytochrome c and dATPdependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–89. [PubMed: 9390557]
- 29.
- Goldstein J C, Waterhouse N J, Juin P. et al. The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant [see comments] Nat Cell Biol. 2000;2:156–62. [PubMed: 10707086]
- 30.
- Luetjens C M, Kogel D, Reimertz C. et al. Multiple kinetics of mitochondrial cytochrome c release in drug-induced apoptosis. Mol Pharmacol. 2001;60:1008–19. [PubMed: 11641429]
- 31.
- Turrens J F, Boveris A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem J. 1980;191:421–7. [PMC free article: PMC1162232] [PubMed: 6263247]
- 32.
- Boveris A, Cadenas E, Stoppani A O. Role of ubiquinone in the mitochondrial generation of hydrogen peroxide. Biochem J. 1976; 156:435–44. [PMC free article: PMC1163765] [PubMed: 182149]
- 33.
- Krippner A, MatsunoYagi A, Gottlieb R A. et al. Loss of function of cytochrome c in Jurkat cells undergoing fasmediated apoptosis. J Biol Chem. 1996;271:21629–36. [PubMed: 8702951]
- 34.
- Cai J, Jones D P. Superoxide in apoptosis. Mitochondrial generation triggered by cytochrome c loss. J Biol Chem. 1998; 273:11401–4. [PubMed: 9565547]
- 35.
- Greenlund L J, Deckwerth T L, Johnson E M Jr. Superoxide dismutase delays neuronal apoptosis: a role for reactive oxygen species in programmed neuronal death. Neuron. 1995;14:303–15. [PubMed: 7857640]
- 36.
- Deshmukh M, Johnson E M Jr. Evidence of a novel event during neuronal death: development of competence-to-die in response to cytoplasmic cytochrome c. Neuron. 1998;21:695–705. [PubMed: 9808457]
- 37.
- Krohn A J, Preis E, Prehn J H. Staurosporine-induced apoptosis of cultured rat hippocampal neurons involves caspase-1-like proteases as upstream initiators and increased production of superoxide as a main downstream effector. J Neurosci. 1998;18:8186–97. [PMC free article: PMC6792864] [PubMed: 9763465]
- 38.
- Martinou I, Desagher S, Eskes R. et al. The release of cytochrome c from mitochondria during apoptosis of NGFdeprived sympathetic neurons is a reversible event. J Cell Biol. 1999;144:883–9. [PMC free article: PMC2148194] [PubMed: 10085288]
- 39.
- Luetjens C M, Bui N T, Sengpiel B. et al. Delayed mitochondrial dysfunction in excitotoxic neuron death: cytochrome c release and a secondary increase in superoxide production. J Neurosci. 2000;20:5715–23. [PMC free article: PMC6772544] [PubMed: 10908611]
- 40.
- Jiang S, Cai J, Wallace D C. et al. Cytochrome c-mediated apoptosis in cells lacking mitochondrial DNA. Signaling pathway involving release and caspase 3 activation is conserved. J Biol Chem. 1999;274:29905–11. [PubMed: 10514472]
- 41.
- Jordan J, Ghadge G D, Prehn J H. et al. Expression of human copper/zincsuperoxide dismutase inhibits the death of rat sympathetic neurons caused by withdrawal of nerve growth factor. Mol Pharmacol. 1995;47:1095–1100. [PubMed: 7603446]
- 42.
- Schulz J B, Weller M, Klockgether T. Potassium deprivation-induced apoptosis of cerebellar granule neurons: a sequential requirement for new mRNA and protein synthesis, ICE-like protease activity, and reactive oxygen species. J Neurosci. 1996;16:4696–706. [PMC free article: PMC6579033] [PubMed: 8764657]
- 43.
- Stefanis L, Park D S, Yan C Y. et al. Induction of CPP32-like activity in PC12 cells by withdrawal of trophic support. Dissociation from apoptosis. J Biol Chem. 1996;271:30663–71. [PubMed: 8940042]
- 44.
- Taylor J, Gatchalian C L, Keen G. et al. Apoptosis in cerebellar granule neurones: involvement of interleukin1 beta converting enzyme-like proteases. J Neurochem. 1997;68:1598–605. [PubMed: 9084431]
- 45.
- Bossy-Wetzel E, Newmeyer D D, Green D R. Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO J. 1998;17:37–49. [PMC free article: PMC1170356] [PubMed: 9427739]
- 46.
- Rego A C, Vesce S, Nicholls D G. The mechanism of mitochondrial membrane potential retention following release of cytochrome c in apoptotic GT17 neural cells. Cell Death Differ. 2001;8:995–1003. [PubMed: 11598797]
- 47.
- Polster B M, Kinnally K W, Fiskum G. Bh3 death domain peptide induces cell type-selective mitochondrial outer membrane permeability. J Biol Chem. 2001;276:37887–94. [PubMed: 11483608]
- 48.
- Waterhouse N J, Goldstein J C, von Ahsen O. et al. Cytochrome c maintains mitochondrial transmembrane potential and ATP generation after outer mitochondrial membrane permeabilization during the apoptotic process. J Cell Biol. 2001;153:319–28. [PMC free article: PMC2169468] [PubMed: 11309413]
- 49.
- Deshmukh M, Kuida K, Johnson E M Jr. Caspase inhibition extends the commitment to neuronal death beyond cytochrome c release to the point of mitochondrial depolarization. J Cell Biol. 2000;150:131–43. [PMC free article: PMC2185568] [PubMed: 10893262]
- 50.
- Chang L K, Johnson E M. Cyclosporin A prevents NGF deprivation-induced death of rat sympathetic neurons only in the presence of a caspase inhibitor. Society for Neuroscience Meeting. 2001;595:2–4.
- 51.
- Daugas E, Susin S A, Zamzami N. et al. Mitochondrionuclear translocation of AIF in apoptosis and necrosis. FASEB J. 2000;14:729–39. [PubMed: 10744629]
- 52.
- Daugas E, Nochy D, Ravagnan L. et al. Apoptosis-inducing factor (AIF): a ubiquitous mitochondrial oxidoreductase involved in apoptosis. FEBS Lett. 2000;476:118–23. [PubMed: 10913597]
- 53.
- Susin S A, Daugas E, Ravagnan L. et al. Two distinct pathways leading to nuclear apoptosis. J Exp Med. 2000;192:571–80. [PMC free article: PMC2193229] [PubMed: 10952727]
- 54.
- Joza N, Susin S A, Daugas E. et al. Essential role of the mitochondrial apoptosisinducing factor in programmed cell death. Nature. 2001;410:549–54. [PubMed: 11279485]
- 55.
- Arnoult D, Tatischeff I, Estaquier J. et al. On the evolutionary conservation of the cell death pathway: mitochondrial release of an apoptosis-inducing factor during Dictyostelium discoideum cell death. Mol Biol Cell. 2001;12:3016–30. [PMC free article: PMC60152] [PubMed: 11598188]
- 56.
- Li L Y, Luo X, Wang X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature. 2001;412:95–9. [PubMed: 11452314]
- 57.
- Holler N, Zaru R, Micheau O. et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1:489–95. [PubMed: 11101870]
- 58.
- Los M, Mozoluk M, Ferrari D. et al. Activation and caspase-mediated inhibition of PARP: a molecular switch between fibroblast necrosis and apoptosis in death receptor signaling. Mol Biol Cell. 2002;13:978–88. [PMC free article: PMC99613] [PubMed: 11907276]
- 59.
- Vercammen D, Brouckaert G, Denecker G. et al. Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J Exp Med. 1998;188:919–30. [PMC free article: PMC2213397] [PubMed: 9730893]
- 60.
- Denecker G, Vercammen D, Steemans M. et al. Death receptor-induced apoptotic and necrotic cell death: differential role of caspases and mitochondria. Cell Death Differ. 2001;8:829–40. [PubMed: 11526436]
- 61.
- Foghsgaard L, Wissing D, Mauch D. et al. Cathepsin B acts as a dominant execution protease in tumor cell apoptosis induced by tumor necrosis factor. J Cell Biol. 2001;153:999–1010. [PMC free article: PMC2174340] [PubMed: 11381085]
- 62.
- Schulze-Osthoff K, Bakker A C, Vanhaesebroeck B. et al. Cytotoxic activity of tumor necrosis factor is mediated by early damage of mitochondrial functions. Evidence for the involvement of mitochondrial radical generation. J Biol Chem. 1992;267:5317–23. [PubMed: 1312087]
- 63.
- Vande Velde C, Cizeau J, Dubik D. et al. BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol Cell Biol. 2000;20:5454–68. [PMC free article: PMC85997] [PubMed: 10891486]
- 64.
- Kagaya S, Kitanaka C, Noguchi K. et al. A functional role for death proteases in s-Myc and c-Myc-mediated apoptosis. Mol Cell Biol. 1997;17:6736–45. [PMC free article: PMC232528] [PubMed: 9343438]
- 65.
- Wang G Q, Wieckowski E, Goldstein L A. et al. Resistance to granzyme B-mediated cytochrome c release in Bak-deficient cells. J Exp Med. 2001;194:1325–1337. [PMC free article: PMC2195982] [PubMed: 11696597]
- 66.
- Darmon A J, Ley T J, Nicholson D W. et al. Cleavage of CPP32 by granzyme B represents a critical role for granzyme B in the induction of target cell DNA fragmentation. J Biol Chem. 1996; 271:21709–12. [PubMed: 8702964]
- 67.
- Beresford P J, Zhang D, Oh D Y. et al. Granzyme A activates an endoplasmic reticulum-associated caspase-independent nuclease to induce single-stranded DNA nicks. J Biol Chem. 2001;276:43285–43293. [PubMed: 11555662]
- 68.
- Suzuki Y, Imai Y, Nakayama H. et al. A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol Cell. 2001;8:613–21. [PubMed: 11583623]
- 69.
- Johnson D E. Noncaspase proteases in apoptosis. Leukemia. 2000;14:1695–703. [PubMed: 10995018]
- 70.
- Deiss L P, Galinka H, Berissi H. et al. Cathepsin D protease mediates programmed cell death induced by interferon-γ, Fas/APO1 and TNF-α EMBO J. 1996;15:3861–70. [PMC free article: PMC452079] [PubMed: 8670891]
- 71.
- Stoka V, Turk B, Schendel S L. et al. Lysosomal protease pathways to apoptosis. Cleavage of bid, not procaspases, is the most likely route. J Biol Chem. 2001;276:3149–57. [PubMed: 11073962]
- 72.
- Porn-Ares M I, Samali A, Orrenius S. Cleavage of the calpain inhibitor, calpastatin, during apoptosis. Cell Death Differ. 1998;5:1028–33. [PubMed: 9894609]
- 73.
- Wolf B B, Goldstein J C, Stennicke H R. et al. Calpain functions in a caspase-independent manner to promote apoptosis-like events during platelet activation. Blood. 1999;94:1683–92. [PubMed: 10477693]
- 74.
- RuizVela A, Gonzalez de Buitrago G, Martinez A C. Implication of calpain in caspase activation during B cell clonal deletion. EMBO J. 1999;18:4988–98. [PMC free article: PMC1171570] [PubMed: 10487751]
- 75.
- Nakagawa T, Yuan J. Crosstalk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J Cell Biol. 2000;150:887–94. [PMC free article: PMC2175271] [PubMed: 10953012]
- 76.
- Lankiewicz S, Marc Luetjens C, Truc Bui N. et al. Activation of calpain I converts excitotoxic neuron death into a caspase-independent cell death. J Biol Chem. 2000;275:17064–71. [PubMed: 10828077]
- 77.
- Chua B T, Guo K, Li P. Direct cleavage by the calcium-activated protease calpain can lead to inactivation of caspases. J Biol Chem. 2000;275:5131–5. [PubMed: 10671558]
- 78.
- Reimertz C, Kogel D, Lankiewicz S. et al. Ca(2+)-induced inhibition of apoptosis in human SHSY5Y neuroblastoma cells: degradation of apoptotic protease activating factor-1 (Apaf-1). J Neurochem. 2001;78:1256–66. [PubMed: 11579134]
- 79.
- Zornig M, Hueber A, Baum W. et al. Apoptosis regulators and their role in tumorigenesis. Biochim Biophys Acta. 2001;1551:F1–F37. [PubMed: 11591448]
- 80.
- Chi S, Kitanaka C, Noguchi K. et al. Oncogenic Ras triggers cell suicide through the activation of a caspase-independent cell death program in human cancer cells. Oncogene. 1999;18:2281–90. [PubMed: 10327074]
- 81.
- DuHadaway J B, Sakamuro D, Ewert D L. et al. Bin1 mediates apoptosis by c-Myc in transformed primary cells. Cancer Res. 2001;61:3151–6. [PubMed: 11306501]
- 82.
- Shankar S L, Mani S, O'Guin K N. et al. Survivin inhibition induces human neural tumor cell death through caspase-independent and dependent pathways. J Neurochem. 2001;79:426–36. [PubMed: 11677271]
- 83.
- Mochizuki T, Asai A, Saito N. et al. Akt protein kinase inhibits non-apoptotic programmed cell death induced by ceramide. J Biol Chem. 2002;277:2790–7. [PubMed: 11706021]
- 84.
- Rego E M, Wang Z G, Peruzzi D. et al. Role of promyelocytic leukemia (PML) protein in tumor suppression. J Exp Med. 2001; 193:521–29. [PMC free article: PMC2195907] [PubMed: 11181703]
- 85.
- Elliott K, Ge K, Du W. et al. The c-Myc-interacting adaptor protein Bin1 activates a caspase-independent cell death program. Oncogene. 2000;19:4669–84. [PubMed: 11032017]
- 86.
- Wang Z G, Ruggero D, Ronchetti S. et al. PML is essential for multiple apoptotic pathways. Nat Genet. 1998;20:266–72. [PubMed: 9806545]
- 87.
- Gottifredi V, Prives C. P53 and PML: new partners in tumor suppression. Trends Cell Biol. 2001;11:184–7. [PubMed: 11316590]
- 88.
- Quignon F, De Bels F, Koken M. et al. PML induces a novel caspase-independent death process. Nat Genet. 1998;20:259–65. [PubMed: 9806544]
- Introduction
- Multiple Forms of PCD
- Caspase-Independent PCD Controlled by Bcl-2 Family Proteins
- Studies Using Caspase Inhibitors
- Mitochondrial Dysfunction Due to Loss of Cytochrome c
- Role of Other Proapoptotic Factors Released from Mitochondria in a Bcl-2-Dependent Manner
- Caspase-Independent Cell Death in Response to Death Receptor Signaling
- Necrosis Controlled by Bcl-2 Family Members
- Involvement of Non-Caspase Proteases in PCD
- Oncogenic Transformation: Escape from PCD
- Outlook
- References
- Caspase-Independent Cell Death Mechanisms - Madame Curie Bioscience DatabaseCaspase-Independent Cell Death Mechanisms - Madame Curie Bioscience Database
- Neurotrophins - Madame Curie Bioscience DatabaseNeurotrophins - Madame Curie Bioscience Database
- In Vitro Endothelialization: Its Contribution Towards an Ideal Vascular Replacem...In Vitro Endothelialization: Its Contribution Towards an Ideal Vascular Replacement - Madame Curie Bioscience Database
- Cryptosporidium parvum isolate kurnool small subunit ribosomal RNA gene, partial...Cryptosporidium parvum isolate kurnool small subunit ribosomal RNA gene, partial sequencegi|2806264502|gb|PQ345813.1|Nucleotide
- PREDICTED: Megachile rotundata uncharacterized LOC100879616 (LOC100879616), tran...PREDICTED: Megachile rotundata uncharacterized LOC100879616 (LOC100879616), transcript variant X4, mRNAgi|805827549|ref|XM_012298214.1|Nucleotide
Your browsing activity is empty.
Activity recording is turned off.
See more...