

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Controlled cell proliferation, differentiation, activation and cell removal are the key events during the development and existence of multicellular organisms. Proliferating mammalian cells undertake a repeated sequence of DNA synthesis, mitosis, and cell division, a series of complicated processes that when going astray, may become deleterious not only to the particular cell, but also to the whole organism. Regulation and proper control of the cell cycle and of the programmed cell death (PCD, apoptosis) is therefore essential for mammalian development and tissue homeostasis. The molecular networks that regulate these processes are critical targets for drug development, gene therapy, and metabolic engineering. In this chapter we will focus on apoptotic pathways converging on caspase family proteases, summarizing “under development” pharmacological attempts towards genes, proteins, and intermolecular interactions presently known to control apoptosis. We also propose new potential molecular targets that may prove to be effective in controlling cell death in vivo.
Introduction
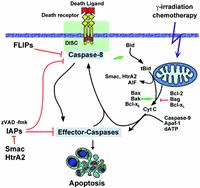
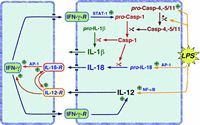
Programmed cell death is involved in almost every existence aspect of higher multicellular organisms. Multicellular animals developed a controlled way of selective removal and replacement of their building blocks, a process tightly surveyed by the neighbor cells and intrinsic mechanisms.1,2 Caspases, the key effector molecules in apoptosis, together with a battery of triggers and regulators of their activity are among the most promising targets for pharmacological modulation of cell death. The search for caspase inhibitors was undertaken way before the discovery of these proteases as a key-effectors in apoptosis. The target of interest has been the interleukin-1β-converting enzyme (ICE, now caspase-1). Caspase-1, -4 and -5 are crucial regulators of secretion of inflammatory cytokines like IL-1ß, IL-16, IL-18 and indirectly IFN-γ.3,4 Therefore search programs focusing on single-caspase, or caspase-subfamily-specific inhibitors are pursued by a number of pharmaceutical companies (Table 1). In addition to caspases, modulators of their activity are also increasingly gaining the interest as potential targets for drug development. Among them the pro- and antiapoptotic Bcl-2 family members, especially the Bcl2 death inhibitor itself, are amid the most frequent targets. In recent years a family of caspase inhibitors called IAPs that bind and inactivate already active caspases attracted attention of the pharmaceutical industry. The interest in IAPs increased with the discovery of IAP inhibitors, Smac/DIABLO and HtrA2, that allow an additional level of apoptosis modulation. Depending on the part of IAP which would become occupied by a designed inhibitor, the net outcome could be either caspase activation and apoptosis if the interaction with caspase is disrupted, or downregulation of caspase activity and apoptosis inhibition, if the interaction with Smac/DIABLO becomes disrupted.5 Yet, another mechanism for apoptosis control can be applied. A number of cells express so called death receptors on the surface. They are able to activate caspases and induce apoptosis, when bound by appropriate ligand. A subfamily of caspases, termed apical/initiator caspases become activated upon enrollment to death inducing signaling complex (DISC), a multiprotein conglomerate recruited to death receptor within seconds, or minutes after its triggering.6 Once activated, the initiator caspases trigger downstream/effector caspases and other components of the apoptotic machinery.7,8 Modulation of interaction among DISC components, or triggering death receptors by naturally-occurring, or artificial ligand provides another mean of control of apoptotic process for clinical applications. Below we discuss in more details the progress, as well as positive and negative aspects of mentioned targets for drug development.
Table 1
Novel anticancer approaches, based on recent development of apoptosis research.
Modulation of Caspase Activity, Implications for Apoptosis and Cytokine Maturation
In lower eucaryotes, such as the worm C. elegans, caspases (ced-3) seems to be involved only in apoptosis. In higher eucaryotes, including mammals caspases form a large family comprising at least 12 members. Based on their differential substrate specificity, structural differences of their zymogens, preferred cellular localization, as well as known role in cellular processes, they can be divided into subfamilies with distinct role in cell (patho)physiology. Caspases are the key effector molecules in apoptosis. Their ability to proteolytically cleave selected cellular proteins, assures the progress and irreversibility of apoptosis.8,9 The semi-hierarchical and partially redundant organization of caspases (Fig. 1) guarantee strong amplification and rapid progression of the apoptotic process even if some family members are missing.7,10 Some caspase family members including caspase-1, 4 and 5, are primarily involved in proteolytic activation of various important cytokines. Maturation by proteolysis of some key activatory cytokines like IL-1β, IL-16 and IL-18 allows their immediate secretion without the time-consuming process of de novo synthesis. This way cells spare time, immediately mobilizing adequate immune response. Moreover, during viral attack, proteolytic signaling allows to mount a proper reaction under circumstances when shutting-off the cellular transcription and translation machinery is a powerful defense mechanism by itself. In addition to their well-established role in cell death and cytokine maturation, likely involvement in other crucial cellular processes, including activation, differentiation, and even in cell cycle progression emerges (for a review see refs: 4 and 11). Although these areas of caspase action still largely await exact definition, they may be responsible for an unexpected results of caspase-based pharmacological approaches. Efforts are on the way to negatively or positively modulate caspase activity for clinical purposes (Table 1). Discovery of drugs that selectively inhibit inflammatory caspases (caspase-1, -4 and 5) may help to control some autoimmunoaggressive diseases, like rheumatoid arthritis, as well as acute life threatening conditions such as sepsis. Inhibition of apoptotic caspases may be an approach of choice to slowdown, or even stop the progress of degenerative diseases like e.g., Alzheimer's disease or spinal lateral sclerosis. In contrast, selective activation of caspases, or at least lowering their activation threshold, may be a powerful approach to combat cancer and eradicate some chronic viral infections. As indicated in the Table 1, caspases are by far the most popular targets for the development of drugs that should modulate the apoptotic process. A very interesting and potentially promising approach is followed by Merck (Merck Frosst, Canada). The discovery that caspase-3, the key effector caspase in apoptosis, is inhibited by an intramollecular electrostatic interaction, so called “safety-catch”, a stretch of three aspartic acid molecules,12 raises hope for rush development of small pharmacologically-active molecules capable of interfering with the electrostatic interaction, thus lowering the threshold of activation, or even activating the caspase. Comparable approach towards activation of caspases by “small molecules” is followed by Maxim Pharmaceuticals Inc (Table 1). Most of other attempts that are designed to directly activate caspases are in rather preliminary experimental stage. Significant advancement has been made by companies searching for specific caspase inhibitors. “Prove of principle” experimental data in animal models indicate that caspase inhibitors may have therapeutic potential in the treatment of heart disease (Table 1), or stroke-related ischemia/reperfusion injury of the brain, liver and other organs.13,16 The protective effect of caspase inhibitors in these circumstances can be at least in part related to the limitation of the inflammatory response by caspase inhibition.17,18 Attempts to design inhibitors controlling the subfamily of inflammatory caspases have been made long before the role of caspases in apoptosis became obvious. Conducted later targeted disruption experiments in murine system fully confirmed the role of murine caspase-1 and 11 (the later is the murine homologue of human caspase-4 and 5) as crucial for propagation of acute inflammatory response that relies on IL-1β and other cytokines.19,20 Caspase-1(-/-) mice had a major defect in the production of mature IL-1β and impaired IL-1α synthesis (Fig. 2). Secretion of TNF and IL-6 in response to LPS stimulation was also diminished in these targeted animals. In addition, macrophages from caspase-1(-/-) mice were defective in LPS-induced IFN-γ production,21 and they were highly resistant to the lethal effects of endotoxin.22 Almost identical phenotype was observed by caspase-11(-/-) mice.23 The proinflammatory role of caspase-1 was strengthened by the finding that pharmacological blockade or genetic deletion of caspase-1 decreased necrosis, edema formation, and serum levels of amylase and lipase (both enzymes are indicators of pancreatic damage), during experimentally induced pancreatitis,23 which was associated with dramatic survival benefits. Caspase inhibitors were also very protective in murine and rat experimental sepsis model (cecal ligation and puncture).24 Application of either broad spectrum caspase inhibitor M-920, or the caspase-3 specific M-791 (both molecules synthesized by Merck) were equally protective. Both inhibitors protected 80–90% of animals whereas only 10–20% of control (solvent or inactive moleculetreated) animals survived the experiment. The protective effect was likely due to the prevention of sepsis-related apoptosis of T and B cells that undergo massive apoptosis during sepsis.25,26 Based on the striking phenotype similarity between caspase-1(-/-) and caspase-11(-/-), it has been proposed that caspase-1 is activated by a direct physical interaction with murine caspase-11.27 However, as caspase-11 does not directly cleave either procaspase-1 or pro-IL-1β in vitro, likely yet to be discovered adapter/chaperon molecules may be required to assist this process in the cell. This hypothetical multiprotein complex may appear to be an another promising target for selective modulation of the activity of inflammatory caspases without interference with the apoptotic cascade.
Modulation of the Mitochondrial Death Pathway: Pro- and Antiapoptotic Bcl-2 Family Members and Their Perspectives in the Clinic
Bcl-2 family proteins are important regulators of apoptosis.28–31 The family comprises both antiapoptotic (e.g., Bcl-2, Bcl-XL) and proapoptotic proteins (e.g., Bax, Bid) with opposing biological functions, either inhibiting or promoting cell death (Fig. 1). Both subfamilies stay in equilibrium to each other in healthy cells. Antiapoptotic Bcl-2 family members inhibit apoptosis by blocking cytochrome c release from mitochondria,32,33 thus preventing activation of the apoptosome pathway. In contrast, Bax, and truncated form of Bid induce cytochrome c release and caspase activation in vitro34 and in vivo.35,36 Overexpression of Bcl2 could provide a survival advantage for cancer cells and it have been associated with increased frequency of lymphoma development in a murine model.37 Loss of the proapoptotic protein Bax function seems to be important in the pathogenesis of colorectal cancers.38
Chemotherapy, radiation and most of other death stimuli induce cell death by triggering cytochrome c release from mitochondria and activation of caspases through the apoptosome pathway (Fig. 1). Bcl-2 prevents cytochrome c release, thereby it blocks cell death, it is a suitable target for the development of the anticancer therapy. Cells that upregulate expression of the bcl-2 gene are significantly more resistant to a variety of noxious stimuli. Bcl-2 is frequently overexpressed in various malignancies, most commonly in a group of B-cell non-Hodgkin's lymphomas bearing t(14;18) chromosomal translocation. Thus, the antisense (AS) oligonucleotide, or phosphorothioate inhibition of Bcl-2 expression would shift the equilibrium in the cell towards proapoptotic family members. The AS approach shows high specificity of action for the selected target mRNA that is much higher than conventional small molecule drugs.
The development of therapy targeting Bcl-2 expression is in the most advanced stage among all apoptosis-based newly developed approaches. Genta Inc. has developed a series of AS sequences directed against different parts of the bcl-2 gene that inhibit Bcl-2 expression to different degree. Genasense, (Table 1) the most promising AS phosphorothioate is a very specific towards Bcl-2 mRNA. Preclinical studies have shown that in human xenografts in a SCID mouse lymphoma model, Genasense compares favorably with Cytoxan, a drug used for lymphoma treatment. Combination of Genasense with Cytoxan markedly potentiates the efficacy of treatment. Similar results were obtained in another studies where Genasense was combined with Taxetir (currently the most effective drug for breast cancer treatment), to treat nude mice with xenografts of human breast cancer. Both Taxetir and Genasense were equally effective in extending the mean lifetime of mice, but the combination of both drugs led to full reversal of the tumor in all treated mice. The treated animals were cured and remained tumor free for at least 180 days, while controls died at around day 10. In a model of human melanoma xenografted into nude mice, administration of dacarbazine (DTIC) markedly decreased the size of tumors. Genasense in combination with DTIC was found to abolish tumors. Phase I and II clinical trials has demonstrated a biological response towards Genasense treatment. Most promising data were obtained on lymphoma therapy, where a sustained and complete reversal of the disease was demonstrated. A patient with an advanced stage of lymphoma exhibited complete remission after 18 months of treatment. Ex vivo examination of the patient material confirmed the elimination of the Bcl-2 protein 5 days after the begin of Genasense administration. Genasense also effectively decreased Bcl-2 protein expression in melanoma tumors. In combination with chemotherapy it was capable to induce partial remission of latestage melanoma. In a study involving 25 patients with advanced-stage melanoma, with life expectancy <6 months, all patients responded to the combined therapy; treatment with Genasense increased life expectancy to ˜17 months. In patients with acute myeloid leukemia treatment with Genasense was also found to be promising. Treatment with Genasense for 5 to 7 days showed virtual elimination of Bcl-2, after which conventional chemotherapy was applied. This procedure resulted in complete remission in treated patients. The application of Genasense to treat bladder cancer resulted in a reduction in the size of the tumor after a single treatment of Genasense in a patient whose cancer was particularly resistant to chemotherapy. Phase I/II studies of Genasense have demonstrated an excellent safety profile with toxicity restricted to fever than 20% of patients, fatigue in 10% and rash in 5% (for a review see refs. 39 and 40). These are minor toxicities and are easily manageable and reverse quickly with the termination of treatment.
A significant limitation of Genasense and in general AS-based theropeutics is their inability to cross the blood-brain barrier. Thus, brain metastases are inaccessible for this sort of therapy. In addition, the clinical studies on the predictive value of Bcl-2 family proteins in hematological malignancies or solid tumors (for a review see ref. 41) when taking into consideration the influence of Bcl-2 family members on cell proliferation. It has been observed that phosphorylated at the G2/M transition Bcl-2 delays the reentry of resting NIH 3T3 cells into the cell cycle.42 Moreover, Bcl-2 transgenic mice have impaired T cell proliferation, whereas transgenic overexpression of Bax accelerates cell cycle progression and apoptosis.42,43 Cells overexpressing Bcl-2 also contain decreased levels of phosphorylated retinoblastoma protein, a key regulator of cell cycle progression.44 Moreover, downregulation of Bcl-2 by AS approaches enhances proliferation of acute myeloid leukemia cells.45 Finally, mutations that suppress the antiapoptotic activity of Bcl-2 also abolish the inhibitory effect on cell cycle transition. Thus the antiapoptotic activities of Bcl-2 may be linked and cell cycle suppressive activities of Bcl-2.11,42,46 The careful evaluation of a large-scale phase III clinical trials on Genasense and related AS-based approaches certainly will bring more light into the role of Bcl-2 family members in cell physiology and the efficacy of the respective AS therapy.
How to Control Already-Activated Caspases? -IAPs, Smac/DIABLO and Omi/HtrA2
The presence of activated caspases in a cell is not equivalent to the activation of the apoptotic process. Secretion of cytokines, or regulatory and effector functions of caspases during erythropoesis infers the presence of active caspases in cells under physiological conditions (for a review see refs. 4 and 11). It is the quantity and cellular localization of caspases that determines whether the cell will die.
IAPs (inhibitor of apoptosis proteins) are a family of proteins that contain BIR (baculoviral repeat) domains and in some cases, a zinc RING-finger domain.47 The family members, X-linked IAP (XIAP), Livin/ML-IAP, cIAP-1 and cIAP-2, are believed to inhibit apoptosis through direct inhibition of caspases, although some of these proteins are also involved in additional signaling pathways.48,49 XIAP, the most potent of these caspase inhibitors, selectively inhibits one of the active forms of caspase-9 (p35/p12 heterotetramer) through an interaction involving its BIR3 domain and the small subunit (p12) of caspase-9. In contrast, the BIR2 domain of XIAP, along with a few critical adjacent residues, is required to inhibit active caspases-3 and -7.5,50,51 Consequently, XIAP is thought to interfere with death receptor-induced apoptosis by inhibiting effector caspases and mitochondrial-induced apoptosis by inhibiting both initiator and effector caspases.
The antiapoptotic activity of IAPs is subjected to regulation by a structural homologue of the Drosophila proteins, Reaper, Hid and Grim, has been identified, termed Smac/DIABLO.52,53 This protein is normally localized to mitochondria, but like cytochrome c, is released into the cytosol during the early stages of apoptosis, where it promotes caspase activity by inhibiting IAPs, particularly XIAP. Smac does not resemble any protein with known function and represents a novel apoptosis regulator in mammalian cells. Although discovered quite recently Smac is among the most promising targets for tumor therapy. When overexpressed it sensitizes cells towards death stimuli,54 potentially offering low side effects combined with reversal of resistance towards cancer chemotherapeutica. Although the search for Smac inhibitors is in an early phase (Table 1), due to the relatively low molecular mass of 24 kD and relatively small interaction surface with IAPs the screen for a “small molecule” inhibitors has a great success chances.55
In contrast to Smac, the development of IAPs-based anticancer strategy may be more difficult. IAPs are the broadest caspase inhibitors in the cell, but due to their heterogeneity, and multiple caspase-inhibitory domains (eight human IAPs, containing in total 16 inhibitory domains, termed BIRs), it will be nearly impossible to target all of them with a single “small molecule” inhibitor. On the other hand, differential, tissue-specific expression pattern of IAPs may offer selective, tissue-specific modulation of caspase activity. For instance IAP-inhibiting molecules that do not target NAIP would spare central nervous system from adverse effects of cancer therapy. On the other hand, careful engineering of antisense molecules that target BIRs may reward in a “global” inhibitors of IAP expression.
Interestingly, another mitochondrial protein known as Omi/HtrA2 has just been identified which can bind XIAP.56–59 HtrA2 is a serine protease whose mitochondrial targeting signal is proteolytically removed upon import into the mitochondrion to reveal an N-terminus conserved IAP (AVPS) binding site. During apoptosis HtrA2 is released from the mitochondrion and inhibits the function of XIAP in analogous manner to Smac. Binding of Smac/DIABLO, HtrA2 and perhaps other as yet unidentified proteins can antagonise the binding of XIAP to caspase-9 and thereby modulate the caspase cleavage activity of the apoptosome. The magnitude of the apoptotic stimulus as well as cellular levels of Smac, HtrA2, XIAP and other as yet unidentified proteins may contribute to the sensitivity of a particular cell type to apoptosis. Thus, analogically to Smac, HtrA2 appears to be a potential target for pharmacological modulation of apoptosis.
Much attention is currently being paid to survivin (Table 1), another IAP-family member that has been found to inhibit cell death by binding to caspases and the proapoptotic Smac.52,60 Survivin is specifically induced in the G2/M phase and it appears to function both as a cell cycle regulator and apoptosis suppressor.61 At the beginning of mitosis, survivin associates with microtubules of the mitotic spindle apparatus. Interestingly, caspase-3 and the CDK (cyclin-dependent kinase) inhibitor p21Waf1 also colocalizes with survivin at the centrosomes. Interference with survivin function induces caspase-3 activity, apoptosis and produces a defect characterized by hyperploidy, multinucleation and supernumerary centrosomes.61 However, the role of survivin as an apoptosis inhibitor has recently been challenged by some authors,62 claiming that its role is restricted to mitosis. Indeed, survivin-like proteins that play a role exclusively in caryokinesis, have been identified in yeasts and C. elegans. Several of these genes show similar intronexon structure, particularly around the BIR-encoding sequences.63,64 Mouse embryos lacking survivin closely resemble the C. elegans BIR1(-/-) phenotype. BIR1 is the homologue of survivin, in C. elegans. In both species, the chromatin replicates, but cytokinesis is abnormal because cleavage furrows that begin to form are not completed. The phenotypes in both the mouse and C. elegans resemble those of embryos lacking INCENP (INner CENtromere Protein) homologues.65 Furthermore, the localization of survivin homologues and INCENP homologues in the worm and in vertebrates is similar. These proteins localize to the centromeres until the metaphase-anaphase transition but then remain in the equatorial zone as the chromosomes separate, eventually localizing to the midbody at telophase, after which they are degraded.63,65 The function of the survivin and the INCENP-like proteins is conserved from yeast to vertebrates. These proteins are required to coordinate chromosome segregation with cytokinesis.65
Nevertheless, regardless if the survivin in addition to control of caryokinesis-related events, functions also as a caspase inhibitor, its downregulation may prove to be a new powerful anticancer tool. Initial experiments targeting survivin by specific ribozymes or with the antisense nucleotides, induced apoptosis in various cell lines or abolished cisplatin resistance.66,67 Based on these promising results Isis Pharmaceuticals and Abbott Laboratories (Table 1) have launched the development of AS-based therapies targeting survivin.67
Receptor-Mediated PCD-Prospects and Limitations
Due to the experience of severe systemic toxicity of TNF and hepatotoxicity of CD95L/FasL in mice, these molecules are unlikely to be used in the clinic for cancer treatment. Therefore we focus on the third member of the TNF-related apoptosis inducing ligand family, TRAIL. This death ligand, known also as the Apo-2L, was cloned and preliminary characterized in mid-90s.68 Unlike the other death receptor/ligand systems, the TRAIL appears to be more complex. Since the discovery of TRAIL, five receptors have been found to bind to TRAIL, and several intracellular checkpoints have been identified that regulate TRAIL sensitivity.69,70 Numerous promising reports describe that TRAIL potently induces apoptosis in tumor or virally infected cells, but has little or no detectable cytotoxic effects on normal and nontransformed cells. Moreover, no overall toxicity was observed during different in vivo studies in mice and monkeys.71,72 Preclinical safety studies in primates (cynomolgus monkeys) did not show adverse reactions even when substantial doses of recombinant TRAIL (10 mg/kg/day) were used.72 The extreme liver toxicity (massive haemorrhagic liver necrosis) that has barred the in vivo testing of CD95L and TNF is not observed upon TRAIL treatment. Therefore, TRAIL is believed to be safe for use as anticancer agent without causing damage to nontransformed tissues.73 From a therapeutic point of view, even more exciting is the finding that TRAIL induces apoptosis in a highly synergistic manner when combined with anticancer drugs or irradiation. The potentiation of cytotoxicity is especially observed in those tumor cells that are refractory to the treatment with either agent alone. The mechanisms which account for this potentiating effect may include the transcriptional induction of DR4 and DR5 TRAIL-receptors, the reduced expression of antiapoptotic molecules such as Bcl-2, Bcl-XL or c-FLIP, and the upregulation of proteins with proapoptotic effects such as caspase-8 and FADD. A variety of malignancies, including common ones, like acute leukemia, breast cancer, colon cancer, lung cancer, melanoma and other malignant proliferative disorders refractory to standard treatment, regained sensitivity when cotreated with TRAIL.74–79 What really determines the susceptibility of tumor cells and the resistance of nontransformed cells to TRAIL, is still largely unknown.
Another novel mechanism of action underlies the synergic effect of all-trans-retinoic acid and its derivatives (Table 1). It has been shown that 6-[3-(1-adamantyl)-4-hydroxyphenyl]-2-naphthalene carboxylic acid (code name: CD-437) not only synergizes with TRAIL, but furthermore it induces the expression of TRAIL, thus killing target cells in a TRAIL-autocrine or paracrine fashion.80 The synergic effects of TRAIL and retinoids have been shown by a number of cancers including acute leukemia, lung and prostate cancer.78,80–82 Concerns have been raised after some primary cells, in particular hepatocytes, were found to be sensitive to TRAIL, however this is now considered to be dependent on the way the recombinant protein was engineered and also on the manner the primary cells were prepared. Although the data on TRAIL-clinical trials are not available yet, the bench and animal experiments place this molecule among the most promising cancertherapy approaches to be tested in the decade.
Perspectives
The development of “smart” cancer therapeutics, that selectively target only malignant cells will be one of the mayor challenges in cancer research during the 21st century. The exploitation of oncogene-directed treatment approaches targeting the mechanisms of uncontrolled proliferation will require prior better understanding of the numerous mechanisms underlying malignant diseases. Combinations of agents targeting different functions of a given oncoprotein complex, or different physiological processes, such as differentiation and apoptosis (CD-437, TRAIL, chemotherapeutica), are clearly bound to be more effective than single-agent protocols. Execution of clinical trials using multiagent protocols without the necessity of testing them individually should help to bring effective therapies into the clinical setting at a faster rate.
The future of cancer therapy lays in treatment individualization and target selection. Doctors need to pick these patients who would benefit from a particular therapeutic approach. DNA chip-based genetic diagnostic will allow proper identification of molecular targets that differentiate even among histologically-judged the same cancer. Malignancies with a common histologic origin and characteristics, need to receive an individualized therapy depending on the genetic trait of the particular group of cancer cells. Drugs that target molecular or genetic derangements in tumors should be the primary goal for exploiting the knowledge of cancer biology that will continue to be elaborated in the coming decades. Prominent among the drug classes that will address this goal will be these targeting apoptotic pathways as well as other signal transduction cascades, which offer the potential for great activity with low toxicity. The final common pathway of tumorigenesis contains shared derangements of cell cycle control, apoptosis, invasion and metastasis that will be relevant to all tumor types. More discrete, probably secondary changes are responsible for the individual clinical manifestation and they underlay the individual characteristic of a particular cancer case. Thus simultaneous targeting of both common and individual characteristics of a given tumor will be the foundation of a successful clinical treatment.
Acknowledgments
This work was supported by research grants from the German Research Council (DFG), IZKF-Univ. Münster, IMF and “Deutsche Krebshilfe”, issued to M.L.
References
- 1.
- Raff M C. Social controls on cell survival and cell death. Nature. 1992;356:397–400. [PubMed: 1557121]
- 2.
- Sadowski-Debbing K, Coy J F, Mier W. et al. Caspases—their role in apoptosis and other physiological processes as revealed by knock-out studies. Arch Immunol Ther Exp (Warsz). 2002;50:19–34. [PubMed: 11916306]
- 3.
- Yuan J, Shaham S, Ledoux S. et al. The C. elegans cell death gene ced3 encodes a protein similar to mammalian interleukin-1 β-converting enzyme. Cell. 1993;75:641–52. [PubMed: 8242740]
- 4.
- Michalke M, Stroh C, Chlichlia C. et al. The emerging role of caspases in signal transduction as revealed by knockout studiesnot only apoptosis. Signal Transd. 2001;2:51–65.
- 5.
- Huang Y, Park Y C, Rich R L. et al. Structural basis of caspase inhibition by XIAP: differential roles of the linker versus the BIR domain. Cell. 2001;104:781–90. [PubMed: 11257231]
- 6.
- Kischkel F C, Hellbardt S, Behrmann I. et al. Cytotoxicity-dependent APO1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 1995;14:5579–88. [PMC free article: PMC394672] [PubMed: 8521815]
- 7.
- Los M, Wesselborg S, Schulze Osthoff K. The role of caspases in development, immunity, and apoptotic signal transduction: lessons from knockout mice. Immunity. 1999;10:629–39. [PubMed: 10403638]
- 8.
- Slee E A, Adrain C, Martin S J. Serial killers: ordering caspase activation events in apoptosis. Cell Death Differ. 1999;6:1067–74. [PubMed: 10578175]
- 9.
- Slee E A, Harte M T, Kluck R M. et al. Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner. J Cell Biol. 1999;144:281–292. [PMC free article: PMC2132895] [PubMed: 9922454]
- 10.
- Zheng T S, Hunot S, Kuida K. et al. Deficiency in caspase-9 or caspase-3 induces compensatory caspase activation. Nat Med. 2000;6:1241–7. [PubMed: 11062535]
- 11.
- Los M, Stroh C, Janicke R U. et al. Caspases: more than just killers? Trends Immunol. 2001;22:31–4. [PubMed: 11286689]
- 12.
- Roy S, Bayly C I, Gareau Y. et al. Maintenance of caspase-3 proenzyme dormancy by an intrinsic “safety catch” regulatory tripeptide. Proc Natl Acad Sci U S A. 2001;98:6132–7. [PMC free article: PMC33434] [PubMed: 11353841]
- 13.
- Cursio R, Gugenheim J, Ricci J E. et al. Caspase inhibition protects from liver injury following ischemia and reperfusion in rats. Transplant Int. 2000;13:S568–72. [PubMed: 11112076]
- 14.
- Mocanu M M, Baxter G F, Yellon D M. Caspase inhibition and limitation of myocardial infarct size: protection against lethal reperfusion injury. Br J Pharmacol. 2000;130:197–200. [PMC free article: PMC1572087] [PubMed: 10807653]
- 15.
- Farber A, Connors J P, Friedlander R M. et al. A specific inhibitor of apoptosis decreases tissue injury after intestinal ischemia-reperfusion in mice. J Vasc Surg. 1999;30:752–60. [PubMed: 10514215]
- 16.
- Yaoita H, Ogawa K, Maehara K. et al. Attenuation of ischemia/reperfusion injury in rats by a caspase inhibitor. Circulation. 1998;97:276–81. [PubMed: 9462530]
- 17.
- Daemen M A, van 't Veer C, Denecker G. et al. Inhibition of apoptosis induced by ischemia-reperfusion prevents inflammation. J Clin Invest. 1999;104:541–9. [PMC free article: PMC408540] [PubMed: 10487768]
- 18.
- Pomerantz B J, Reznikov L L, Harken A H. et al. Inhibition of caspase 1 reduces human myocardial ischemic dysfunction via inhibition of IL-18 and IL-1beta. Proc Natl Acad Sci USA. 2001;98:2871–6. [PMC free article: PMC30232] [PubMed: 11226333]
- 19.
- Kuida K, Lippke J A, Ku G. et al. Altered cytokine export and apoptosis in mice deficient in interleukin1ß converting enzyme. Science. 1995;267:2000–2003. [PubMed: 7535475]
- 20.
- Li P, Allen H, Banerjee S. et al. Mice deficient in IL-1b-converting enzyme are defective in production of mature IL-1b and resistant to endotoxic shock. Cell. 1995;80:401–411. [PubMed: 7859282]
- 21.
- Fantuzzi G, Puren A J, Harding M W. et al. Interleukin 18 regulation of interferon gamma production and cell proliferation as shown in interleukin-1beta converting enzyme (caspase 1)deficient mice. Blood. 1998;91:2118–25. [PubMed: 9490698]
- 22.
- Li P, Allen H, Banerjee S. et al. Characterization of mice deficient in interleukin-1b converting enzyme. J Cell Biochem. 1997b;64:27–32. [PubMed: 9015751]
- 23.
- Norman J, Yang J, Fink G. et al. Severity and mortality of experimental pancreatitis are dependent on interleukin-1b converting enzyme (ICE). J Interferon Cytokine Res. 1997;17:113–8. [PubMed: 9058318]
- 24.
- Hotchkiss R S, Chang K C, Swanson P E. et al. Caspase inhibitors improve survival in sepsis: a critical role of the lymphocyte. Nat Immunol. 2000;1:496–501. [PubMed: 11101871]
- 25.
- Ayala A, Herdon C D, Lehman D L. et al. Differential induction of apoptosis in lymphoid tissues during sepsis: variation in onset, frequency, and the nature of the mediators. Blood. 1996;87:4261–75. [PubMed: 8639785]
- 26.
- Wang S D, Huang K J, Lin Y S. et al. Sepsis-induced apoptosis of the thymocytes in mice. J Immunol. 1994;152:5014–21. [PubMed: 8176219]
- 27.
- Wang S, Miura M, Jung Y K. et al. Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell. 1998;92:501–9. [PubMed: 9491891]
- 28.
- Adams J M, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998;281:1322–6. [PubMed: 9735050]
- 29.
- Antonsson B, Martinou J C. The Bcl-2 protein family. Exp Cell Res. 2000;256:50–7. [PubMed: 10739651]
- 30.
- Bouillet P, Huang D C, O'Reilly L A. et al. The role of the proapoptotic Bcl-2 family member bim in physiological cell death. Ann N Y Acad Sci. 2000;926:83–9. [PubMed: 11193044]
- 31.
- Reed J C. Double identity for proteins of the Bcl-2 family. Nature. 1997;387:773–6. [PubMed: 9194558]
- 32.
- Kluck R M, Bossy-Wetzel E, Green D R. et al. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–6. [PubMed: 9027315]
- 33.
- Yang J, Liu X, Bhalla K. et al. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–32. [PubMed: 9027314]
- 34.
- Jurgensmeier J M, Xie Z, Deveraux Q. et al. Bax directly induces release of cytochrome c from isolated mitochondria. Proc Natl Acad Sci USA. 1998;95:4997–5002. [PMC free article: PMC20202] [PubMed: 9560217]
- 35.
- Rosse T, Olivier R, Monney L. et al. Bcl-2 prolongs cell survival after Bax-induced release of cytochrome c. Nature. 1998;391:496–9. [PubMed: 9461218]
- 36.
- Stepczynska A, Lauber K, Engels I H. et al. Staurosporine and conventional anticancer drugs induce overlapping, yet distinct pathways of apoptosis and caspase activation. Oncogene. 2001;20:1193–202. [PubMed: 11313863]
- 37.
- McDonnell T J, Deane N, Platt F M. et al. Bcl-2 immunoglobulin transgenic mice demonstrate extended B cell survival and follicular lymphoproliferation. Cell. 1989;57:79–88. [PubMed: 2649247]
- 38.
- Rampino N, Yamamoto H, Ionov Y. et al. Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science. 1997;275:967–9. [PubMed: 9020077]
- 39.
- Banerjee D. Genasense (Genta Inc). Curr Opin Investig Drugs. 2001;2:574–80. [PubMed: 11566020]
- 40.
- Nicholson D W. From bench to clinic with apoptosis-based therapeutic agents. Nature. 2000;407:810–6. [PubMed: 11048733]
- 41.
- Hamilton A, Piccart M. The contribution of molecular markers to the prediction of response in the treatment of breast cancer: a review of the literature on HER2, p53 and Bcl-2. Ann Oncol. 2000;11:647–63. [PubMed: 10942052]
- 42.
- O'Reilly L A, Huang D C, Strasser A. The cell death inhibitor Bcl-2 and its homologues influence control of cell cycle entry. EMBO J. 1996;15:6979–90. [PMC free article: PMC452524] [PubMed: 9003774]
- 43.
- Brady H J, Gil Gomez G, Kirberg J. et al. Bax alpha perturbs T cell development and affects cell cycle entry of T cells. EMBO J. 1996;15:6991–7001. [PMC free article: PMC452525] [PubMed: 9003775]
- 44.
- Mazel S, Burtrum D, Petrie H T. Regulation of cell division cycle progression by Bcl-2 expression: a potential mechanism for inhibition of programmed cell death. J Exp Med. 1996;183:2219–26. [PMC free article: PMC2192544] [PubMed: 8642331]
- 45.
- Konopleva M, Tari A M, Estrov Z. et al. Liposomal Bcl-2 antisense oligonucleotides enhance proliferation, sensitize acute myeloid leukemia to cytosine arabinoside, and induce apoptosis independent of other antiapoptotic proteins. Blood. 2000;95:3929–38. [PubMed: 10845930]
- 46.
- O'Connor L, Huang D C, O'Reilly L A. et al. Apoptosis and cell division. Curr Opin Cell Biol. 2000;12:257–63. [PubMed: 10819542]
- 47.
- Deveraux Q L, Reed J C. IAP family proteins-suppressors of apoptosis. Genes Dev. 1999;13:239–52. [PubMed: 9990849]
- 48.
- Deveraux Q L, Roy N, Stennicke H R. et al. IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J. 1998;17:2215–23. [PMC free article: PMC1170566] [PubMed: 9545235]
- 49.
- Kasof G M, Gomes B C. Livin, a novel inhibitor of apoptosis protein family member. J Biol Chem. 2001;276:3238–46. [PubMed: 11024045]
- 50.
- Deveraux Q L, Leo E, Stennicke H R. et al. Cleavage of human inhibitor of apoptosis protein XIAP results in fragments with distinct specificities for caspases. EMBO J. 1999;18:5242–51. [PMC free article: PMC1171595] [PubMed: 10508158]
- 51.
- Sun C, Cai M, Meadows R P. et al. NMR structure and mutagenesis of the third Bir domain of the inhibitor of apoptosis protein XIAP. J Biol Chem. 2000;275:33777–81. [PubMed: 10934209]
- 52.
- Du C, Fang M, Li Y. et al. Smac, a mitochondrial protein that promotes cytochrome cdependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. [PubMed: 10929711]
- 53.
- Ekert P G, Silke J, Hawkins C J. et al. DIABLO promotes apoptosis by removing MIHA/XIAP from processed caspase 9. J Cell Biol. 2001;152:483–90. [PMC free article: PMC2195997] [PubMed: 11157976]
- 54.
- Srinivasula S M, Datta P, Fan X J. et al. Molecular determinants of the caspase-promoting activity of Smac/DIABLO and its role in the death receptor pathway. J Biol Chem. 2000;275:36152–7. [PubMed: 10950947]
- 55.
- Liu Z, Sun C, Olejniczak E T. et al. Structural basis for binding of Smac/DIABLO to the XIAP BIR3 domain. Nature. 2000;408:1004–8. [PubMed: 11140637]
- 56.
- Suzuki Y, Imai Y, Nakayama H. et al. A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol Cell. 2001;8:613–21. [PubMed: 11583623]
- 57.
- Martins L M, Iaccarino I, Tenev T. et al. The serine protease Omi/HtrA2 regulates apoptosis by binding XIAP through a reaper-like motif. J Biol Chem. 2002;277:439–44. [PubMed: 11602612]
- 58.
- Verhagen A M, Silke J, Ekert P G. et al. HtrA2 promotes cell death through its serine protease activity and its ability to antagonize inhibitor of apoptosis proteins. J Biol Chem. 2002;277:445–54. [PubMed: 11604410]
- 59.
- Hegde R, Srinivasula S M, Zhang Z. et al. Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein-caspase interaction. J Biol Chem. 2002;277:432–8. [PubMed: 11606597]
- 60.
- Tamm I, Wang Y, Sausville E. et al. IAP-family protein survivin inhibits caspase activity and apoptosis induced by Fas (CD95), Bax, caspases, and anticancer drugs. Cancer Res. 1998;58:5315–20. [PubMed: 9850056]
- 61.
- Li F, Ackermann E J, Bennett C F. et al. Pleiotropic celldivision defects and apoptosis induced by interference with survivin function. Nat Cell Biol. 1999;1:461–6. [PubMed: 10587640]
- 62.
- Silke J, Vaux D L. Two kinds of BIR-containing protein inhibitors of apoptosis, or required for mitosis. J Cell Sci. 2001;114:1821–7. [PubMed: 11329368]
- 63.
- Speliotes E K, Uren A, Vaux D. et al. The survivin-like C. elegans BIR1 protein acts with the Aurora-like kinase AIR2 to affect chromosomes and the spindle midzone. Mol Cell. 2000;6:211–23. [PubMed: 10983970]
- 64.
- Uren A G, Coulson E J, Vaux D L. Conservation of baculovirus inhibitor of apoptosis repeat proteins (BIRPs) in viruses, nematodes, vertebrates and yeasts. Trends Biochem Sci. 1998;23:159–62. [PubMed: 9612077]
- 65.
- Uren A G, Wong L, Pakusch M. et al. Survivin and the inner centromere protein INCENP show similar cell cycle localization and gene knockout phenotype. Curr Biol. 2000;10:1319–28. [PubMed: 11084331]
- 66.
- Pennati M, Colella G, Folini M. et al. Ribozyme-mediated attenuation of survivin expression sensitizes human melanoma cells to cisplatin-induced apoptosis. J Clin Invest. 2002;109:285–6. [PMC free article: PMC150847] [PubMed: 11805141]
- 67.
- Chen J, Wu W, Tahir S K. et al. Downregulation of survivin by antisense oligonucleotides increases apoptosis, inhibits cytokinesis and anchorage-independent growth. Neoplasia. 2000;2:235–41. [PMC free article: PMC1507573] [PubMed: 10935509]
- 68.
- Wiley S R, Schooley K, Smolak P J. et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. 1995;3:673–82. [PubMed: 8777713]
- 69.
- Sheridan J P, Marsters S A, Pitti R M. et al. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science. 1997;277:818–21. [PubMed: 9242611]
- 70.
- Thome M, Schneider P, Hofmann K. et al. Viral FLICE inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature. 1997;386:517–21. [PubMed: 9087414]
- 71.
- Walczak H, Miller R E, Ariail K. et al. Tumoricidal activity of tumor necrosis factor-related apoptosis inducing ligand in vivo. Nat Med. 1999;5:157–63. [PubMed: 9930862]
- 72.
- Ashkenazi A, Pai R C, Fong S. et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest. 1999;104:155–62. [PMC free article: PMC408479] [PubMed: 10411544]
- 73.
- El-Deiry W S. The TRAIL to an anticancer agent. Drug Resist Updat. 1999;2:79–80. [PubMed: 11504473]
- 74.
- Wen J, Ramadevi N, Nguyen D. et al. Antileukemic drugs increase death receptor 5 levels and enhance Apo2L-induced apoptosis of human acute leukemia cells. Blood. 2000;96:3900–6. [PubMed: 11090076]
- 75.
- Mori S, MurakamiMori K, Nakamura S. et al. Sensitization of AIDS-Kaposi's sarcoma cells to Apo2 ligand-induced apoptosis by actinomycin D. J Immunol. 1999;162:5616–23. [PubMed: 10228045]
- 76.
- Hernandez A, Wang Q D, Schwartz S A. et al. Sensitization of human colon cancer cells to TRAIL-mediated apoptosis. J Gastrointest Surg. 2001;5:56–65. [PubMed: 11309649]
- 77.
- Keane M M, Ettenberg S A, Nau M M. et al. Chemotherapy augments TRAIL-induced apoptosis in breast cell lines. Cancer Res. 1999;59:734–41. [PubMed: 9973225]
- 78.
- Sun S Y, Yue P, Hong W K. et al. Augmentation of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis by the synthetic retinoid 6[3-(1adamantyl)-4-hydroxyphenyl]-2-naphthalene carboxylic acid (CD-437) through upregulation of TRAIL receptors in human lung cancer cells. Cancer Res. 2000;60:7149–55. [PubMed: 11156424]
- 79.
- Griffith T S, Chin W A, Jackson G C. et al. Intracellular regulation of TRAIL-induced apoptosis in human melanoma cells. J Immunol. 1998;161:2833–40. [PubMed: 9743343]
- 80.
- Altucci L, Rossin A, Raffelsberger W. et al. Retinoic acid-induced apoptosis in leukemia cells is mediated by paracrine action of tumor-selective death ligand TRAIL. Nat Med. 2001;7:680–6. [PubMed: 11385504]
- 81.
- Bradbury J. TRAIL leads to apoptosis in acute promyelocytic leukaemia. Lancet. 2001;357:1770. [PubMed: 11403822]
- 82.
- Sun S Y, Yue P, Lotan R. Implication of multiple mechanisms in apoptosis induced by the synthetic retinoid CD-437 in human prostate carcinoma cells. Oncogene. 2000;19:4513–22. [PubMed: 11002424]
- Introduction
- Modulation of Caspase Activity, Implications for Apoptosis and Cytokine Maturation
- Modulation of the Mitochondrial Death Pathway: Pro- and Antiapoptotic Bcl-2 Family Members and Their Perspectives in the Clinic
- How to Control Already-Activated Caspases? -IAPs, Smac/DIABLO and Omi/HtrA2
- Receptor-Mediated PCD-Prospects and Limitations
- Perspectives
- Acknowledgments
- References
- Caspases as Targets for Drug Development - Madame Curie Bioscience DatabaseCaspases as Targets for Drug Development - Madame Curie Bioscience Database
- Insights into the Modulation of Ceramide Metabolism by Naturally Occurring and S...Insights into the Modulation of Ceramide Metabolism by Naturally Occurring and Synthetic Sphingolipid Analogs as Monitored by Electrospray Tandem Mass Spectrometry - Madame Curie Bioscience Database
- Modeling Structure-Activity Relationships - Madame Curie Bioscience DatabaseModeling Structure-Activity Relationships - Madame Curie Bioscience Database
- Regional Differences and Variability in Left Ventricular Wall Motion - Madame Cu...Regional Differences and Variability in Left Ventricular Wall Motion - Madame Curie Bioscience Database
- UI-H-BW1-anf-g-03-0-UI.s1 NCI_CGAP_Sub7 Homo sapiens cDNA clone IMAGE:3082180 3'...UI-H-BW1-anf-g-03-0-UI.s1 NCI_CGAP_Sub7 Homo sapiens cDNA clone IMAGE:3082180 3', mRNA sequencegi|11599688|gnl|dbEST|7041874|gb|BF 9.1|Nucleotide
Your browsing activity is empty.
Activity recording is turned off.
See more...