

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Introduction
Activation of caspases is the key event of apoptosis as its initiates irreversible steps of the cell demise.1–9 Several methods, therefore, have been developed to monitor this event. Most frequently the caspases involvement is probed indirectly, by testing whether their specific inhibitors, when administered together with the inducer of apoptosis, can prevent particular apoptotic episodes. Also indirectly, caspases activation can be revealed by the presence of the specific cleavage products that can be identified electrophoretically by a characteristic change in molecular weight upon the cleavage, confirmed immunochemically on Western blots. Antibodies also are available that detect the specific cleavage products such as a 89 kD fragment of poly(ADP-ribose) polymerase-1 (PARP-1).10 The latter approach was adapted to cytometry and has been utilized to correlate caspases activation with the cell cycle position9 or collapse of the mitochondrial transmembrane electrochemical potential.11
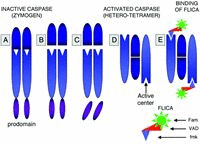
The approaches to directly probe activation of caspases also have been developed. Analysis of caspases molecular weight is one of them. Namely, because the activation involves cleavage of the zymogen procaspases (Fig. 1) the cleavage products by virtue of their lower molecular weight compared with the zymogen can be separated electrophoretically and identified on Western blots. As in the case of the cleavage products of caspases, antibodies recently become available that recognize the epitope that is characteristic of the activated form of these proteases. Their activation, thus, can also be detected directly in situ, immunocytochemically.12
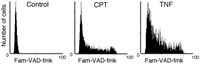
Still another approach utilizes peptide substrates that upon the caspase-induced cleavage generate colored or fluorescing products.13–16 Their use was primarily restricted to cell extracts and therefore provided no information on individual cells, heterogeneity of cell populations or correlation with other cell attributes, on a cell by cell basis. Recently, however, the use of these substrates was adapted to individual cells whose fluorescence was measured by flow cytometry.17
We have recently described the use of fluorochrome-labeled inhibitors of caspases (FLICA) to monitor activation of these enzymes in live cells.18–20 The use of enzyme active center specific inhibitors as affinity-labeling probes was introduced by us before to detect in situ activation of esterases,21 mast cell serine proteases22 and folate reductase.23 Instead of radioisotopelabeling that in these earlier studies was detected by autoradiography, we are now tagging the inhibitors with fluorochromes, that are detected by fluorescence microscopy and can be measured by flow- or laser scanning-cytometry. The FLICA ligands are carboxyfluorescein (Fam)- or sulforhodamine B (Sr)-labeled peptide fluoromethyl ketones (fmk) that with 1:1 stoichiometry covalently bind to active centers of caspases (Fig. 1). These labeled inhibitors, similar as the unlabeled ones (e.g., zVAD-fmk) are permeant, and at least during short-term incubation, appear to be relatively nontoxic to the cell. Actually, the unlabeled analogs have been reported to promote cell survival, protecting them from apoptosis.24–27 Exposure of live cells to FLICA results in uptake of these reagents followed by their binding to activated caspases within the cells that undergo apoptosis. Unbound FLICA are removed from the nonapoptotic cells that lack activated caspases by rinsing the cells with wash buffer. Cells labeled with FLICA can be examined by fluorescence microscopy, or subjected to quantitative analysis by flow- or laser scanning-cytometry (LSC).28,29 The present review describes principles of this FLICA approach and discusses some applications of the methodology.
Affinity Labeling of Enzyme Centers with FLICA
Procaspases contain N-terminal prodomain followed by a large (˜20 kD) then a small (˜10 kD) catalytic subunit (Fig. 1A). The size of prodomain varies between caspases. A large prodomain size have initiator caspases while a small size have effector caspases. Two related motifs are present in prodomains: the death effector domain (DED) and caspase recruitment domain (CARD).4–6 The sequential steps of activation, formation of the enzymatically active heterotetramer and binding of FLICA are described in the legend to Figure 1.
Several FLICA are commercially available (e.g., from Immunochemistry Technologies, Mn, USA; or Serologicals Corp. Norstar, Ga, USA), including fluorescein or sulforhodamine B-labeled VAD-fmk, which contains the valyl-alanyl-aspartic acid residue sequence. This three-amino-acid target sequence allows this inhibitor to irreversibly bind to activated caspases-1, -3, -4, -5, -7, -8 and -9 making it multi-caspases marker. Other inhibitors such as these that contain VDVAD, DEVD, VEID, YVAD, LETD, LEHD, and AEVD peptide residues preferentially bind to activated caspases-2, -3, -6, -1, -8, -9 and -10, respectively.
It is difficult to assess, however, how indeed specific is in situ binding of individual FLICA designed to be markers for the respective caspases. As mentioned, Fam-VAD-fmk lacks specificity and binds to all caspases, perhaps with an exception of caspase-2 to which it has a low binding affinity.25,26,30 The inhibitor with DEVD sequence designed to be caspase-3 specific is expected also to interact with several other caspases. Namely, the inhibitory constant (Ki ) of Ac-DEVD-CHO is 0.2–2.2 nM for caspase-3, 0.9 nM for caspase-8, and 1.6 nM for caspase-7.25 We observed that MCF-7 cells that are known to be caspase-3 null were quite strongly labeled with Fam-DEVD-fmk.19 This suggests that perhaps caspases-7 and -8, if not also other caspases, were labeled with Fam-DEVD-fmk in these cells. Other inhibitors also have strong affinity to more than a single caspase.26,30 Moreover, since little is known about effective concentration of the used FLICA within the cell and also about their binding constants to the respective caspases in situ, one has to be careful in drawing the conclusions about their specificity based on binding to live cells. We observed, however, that when the cells were pre-treated with a high concentration of the unlabeled inhibitor z-VAD-fmk the subsequent binding of Fam-VAD-fmk was reduced by over 90%.19 Likewise, in the presence of an excess of the caspase-3 substrate (Ac-DEVD-pNA) binding of Fam-DEVD-fmk was also diminished by over 90%.19 The FLICA binding sites within the cell, thus, can be competitively protected either by the unlabeled inhibitor or substrate. The specificity (or lack of it) of the FLICA with respect to the target caspases, thus, is comparable to that of their respective unlabeled analogs or substrates.
Subcellular Localization of FLICA-Binding Sites
Induction of apoptosis followed by cell exposure to FLICA makes them fluorochrome labeled (Fig. 2). It is apparent under fluorescence microscopy that preferentially labeled are the cells that have altered morphology, characteristic of apoptosis (Fig. 3). It is difficult, however, to assess in live cells, particularly in the cells that become spherical and detaching, as most apoptotic cells are, the intracellular localization of the fluorochrome.
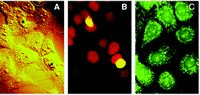
Because binding of FLICA to active centers of caspases is covalent, it is possible, after the binding occurred, to fix the cells (preferentially with formaldehyde), permeabilize them and counterstain their DNA with a fluorochrome of another color than FLICA. Following such a treatment cell morphology and nuclear chromatin can be assessed with better clarity while the FLICA (Fam-VAD-fmk) fluorescence remains. It is quite apparent that most FLICA-labeled cells are detaching from slide, are distinctly smaller and have condensed chromatin (Fig. 3A, the asterisk labeled cells). Their green FLICA fluorescence has both the cytoplasmic localization and overlaps with red nuclear fluorescence resulting that nuclei fluorescence in yellow. However, because of the spherical geometry of apoptotic cells, without a help of confocal microscopy, it is difficult to discern whether the yellow fluorescence seen over the nucleus is due to the presence of FLICA in the nucleus, colocalizing with DNA, or to the layer of the cytoplasm underneath and above of the nucleus.
Interestingly, when the cells are first fixed and subsequently subjected to labeling with FLICA, both apoptotic as well the nonapoptotic cells become labeled (Fig. 3C). The cell fixation makes the zymogen caspases in the nonapoptotic cells reactive with FLICA possibly by altering their conformation in such a way that the active centers become accessible to the inhibitors. The labeling has a very characteristic pattern: the most strongly and distinctly labeled are mitochondria and nucleoli. Nucleoplasm shows a diffuse and rather weak fluorescence while fluorescence of the cytoplasm outside of mitochondria is even less intense. This pattern is consistent with the reported localization of caspases in mitochondria.9,31,32 Although nuclear localization of several caspases also has been observed33–35 their presence in nucleoli has not been yet demonstrated. However, the indirect evidence suggests that caspases may be localized, at least following their activation, in nucleoli. Thus, at early stage of apoptosis the accumulation of the 89 kD product of caspase-3 mediated cleavage of PARP-1 was most preeminent in nucleoli and perinucleolar areas.10 Nucleolar segregation is also an early apoptotic event, and is associated with activation of caspases.36–39 Nucleolar segregation was shown to initiate separation of RNA from DNA that culminated in packaging these nucleic acids into separate apoptotic bodies.40 There is also evidence that the “upstream binding factor” (UBF), the protein regulating transcription of rDNA in the nucleolus, is the very early target of caspases during apoptosis.41
Detection of Caspase Activation Combined with Other Probes of Apoptosis
Simultaneous Analysis of Caspase Activation and Plasma Membrane Integrity
Multiparameter cytometry utilizing a combination of fluorochrome probes differing in emission or excitation wavelength allows one to correlate, within the same cells, the activation of caspases detected by FLICA with other apoptotic events. The evidence of a correlation, or lack of it, can reveal whether activation of caspases is, or is not, a prerequisite for the other event to take place. Such an analysis, if carried out sequentially at different time points after induction of apoptosis, also can reveal the time gap between the caspase activation and the observed event.
Figure 4 illustrates an example of the analysis of caspases activation combined with the quest of integrity of plasma membrane. The green fluorescing pan-caspase inhibitor Fam-VAD-fmk was combined with the red fluorescing cationic fluorochrome propidium iodide (PI). The latter is excluded by live- and early-apoptotic but not by necrotic and late-apoptotic cells.42

Based on the differences in binding Fam-VAD-fmk (FLICA) and PI one can distinguish four cell subpopulations (compartments) on the bi-variate scattergrams representing cellular green versus red fluorescence. They are: (A) the cells that are both FLICA- and PI-negative (FLICA-/PI-); (B) the FLICA positive and PI negative cells (FLICA+/PI-); (C) the cells that are both FLICA and PI positive (FLICA+/PI+); and (D) the FLICA negative and PI positive cells (FLICA-/PI+).
These compartments represent the sequential changes that occur during apoptosis, involving activation of caspases followed by the loss of the plasma membrane integrity.43 Thus, the compartment A represents live, nonapoptotic cells with inactive caspases and intact plasma membrane. Activation of caspases with no changes in plasma membrane integrity characterizes early apoptotic cells (compartment B). The cells that are more advanced in apoptosis have their caspases still active, able to bind FLICA. Their capability to exclude PI, however, is lost (compartment C). Finally, the very late apoptotic cells are characterized by loss of the abilities to bind FLICA and to exclude PI.43
The loss of plasma membrane integrity that manifests in cell inability to exclude cationic dyes such as PI or trypan blue characterizes the so-called “necrotic stage” of apoptosis.44–46 The observed differential cell reactivity with FLICA allows one to subdivide the “necrotic stage” onto two sub-stages, the earlier one representing cells that still bind FLICA, and the late one, when the ability to bind these inhibitors is lost.43 It is possible that at this late stage the caspases become either inhibited or degraded to the point that their active centers do not react with FLICA anymore. Such cells, thus, are indistinguishable from the genuine necrotic cells, that die by mode of necrosis (“oncosis” or “accidental cell death”).44,46
Simultaneous Analysis of Caspase Activation and Annexin V Binding
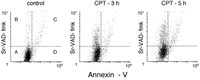
Early during apoptosis the asymmetry of plasma membrane phospholipids is broken and phosphatidylserine becomes exposed on the outer leaflet of the membrane.47,48 This event is considered to be one of the hallmarks of apoptosis. Because the anticoagulant protein annexin V binds with high affinity to phosphatidylserine, the fluorochrome-tagged annexin V is frequently used as a marker of apoptosis.48 While in most instances the annexin V binding is caspase activation-dependent, there are situations when it appears to be independent.49,50 It is desirable, therefore, to have an assay that simultaneously detects caspases activation and externalization of phosphatidylserine on plasma membrane of the same cells. Such an assay may also reveal the sequence and time relationship between caspases activation and loss of the asymmetry of the plasma membrane phospholipids, as reflected by the externalization of phospatidylserine.
The bivariate scatterplots (Fig. 5) illustrate changes in cell ability to bind annexin V and Sr-VAD-fmk following induction of apoptosis by CPT. Similar as in the case of cell labeling with Fam-VAD-fmk versus PI (Fig. 4), based on the difference in Sr-VAD-fmk versus annexin V binding one also can identify on these scatterplots four cell populations. In quadrant A are the nonapoptotic Sr-VAD-fmk and annexin V negative cells. In quadrant B are the cells that have activated caspases but do not bind annexin V. The cells that have both activated caspases and capability to bind annexin V are represented in quadrant C. Interestingly, the cells that show increased annexin V binding but no evidence of caspases activation are also apparent (quadrant D). Their presence may suggest that externalization of phosphatidylserine is caspase activation independent. However, we observed that the presence of active caspases cannot be detected at the late stage of apoptosis (Fig. 4). It is possible, therefore, that the cells that bind annexin V and are Sr-VAD-fmk negative (Fig. 5, quadrant D) are the same, very late apoptotic cells, that do not bind Fam-VAD-fmk and cannot exclude PI (Fig. 4, quadrant D).
The Cell Cycle Phase-Specific Activation of Caspases
FLICA binds covalently to the active centers of the respective caspases forming a thiomethyl ketone II bond. The bond is stable and withstands cell fixation.19 It is possible, therefore, to incubate live unfixed cells with FLICA in order to label the cells that activated their caspases, then rinse and fix them. The cells can be then probed with other fluorochromes, that require prior cell fixation and permeabilization. Cellular DNA, for example, can be stained with PI or 7-amino-actinomycin D (7-AAD) to correlate activation of caspases with the cell cycle position (Fig. 6). Likewise, DNA fragmentation (the presence of DNA strand breaks) can be detected by in situ labeling the 3'OH break termini with fluorochrome-tagged deoxynucleotides utilizing exogenous terminal deoxynucleotidyl transferase,51 to find out whether caspases activation correlates with activation of the apoptotic DNase.
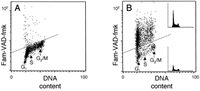
Figure 6 presents the measurement of fluorescence of HL-60 cells that, to induce apoptosis, were treated with tumor necrosis factor alpha (TNF) then labeled with Fam-VAD-fmk, fixed and subsequently stained with PI. Their green and red fluorescence was measured by flow cytometry. The bi-variate distribution (scatterplot) representing cell labeling with these fluorochromes allows one to identify the population of cells with activated caspases (increased Fam-VAD-fmk fluorescence) and through the gating analysis, to reveal the cell cycle distribution (based on intensity of PI fluorescence) separately, in subpopulations of cells with activated and non-activated caspases. (Fig. 6). The DNA content frequency histograms of these subpopulations, as shown in this figure, can be deconvoluted to reveal whether activation of caspases is cell cycle-phase specific. As it is evident, in the case induction of apoptosis by TNF, caspases are activated in all phases of the cell cycle (Fig. 6).
Stathmo-Apoptosis: The Use of FLICA to Arrest the Process of Apoptosis
The extent (incidence) of apoptosis in a cell population is commonly estimated based on the frequency of apoptotic cells (apoptotic index, AI). AI, thus, is a “snapshot” estimate of the percentage or fraction of apoptotic cells in that population at a particular time-point. However, because apoptosis is a transient, kinetic event, AI is an inaccurate measure of the incidence of apoptosis (Fig. 7). Namely, the entire apoptotic process, from its onset to the final cell disintegration, when the cell is no longer detectable, is often short and may be of variable duration.46,52,53 For example, upon induction, apoptosis of cells of hematopoietic tumor lines (e.g., Jurkat, HL-60, U-937) progresses rapidly and the cells disintegrate within 3–5 h. The duration of apoptosis in vivo appears to be even shorter. This is reflected by the fact that in tissues, under conditions of homeostasis, AI often approximates the mitotic index.46,52,53 Because mitosis lasts ˜1 h, apoptosis must be of comparable length of time. On the other hand apoptosis of epithelial or fibroblast cell lineage occurs with a 24-h delay upon induction, and the apoptotic process is much longer.54 In addition, some inducers of apoptosis or factors in cellular environment may alter the duration of the apoptotic process e.g., by modulating formation and/or shedding of apoptotic bodies, rate of proteolysis or DNA degradation. Serine protease inhibitors, for example, significantly prolong apoptosis by preventing internucleosomal DNA degradation51 and nuclear fragmentation.55 The length of time-window during which the apoptotic cell can be identified also varies, as it depends on the marker (assay) that is being used, and expression of different markers is variable in time. In all these situations, therefore, when either duration of apoptosis or of the time-window of its detection, varies, AI cannot accurately represent incidence of apoptosis (Fig. 7).
To obtain a more accurate assessment of the incidence of apoptosis, we have developed a stathmoapoptosis assay56 in which Fam-VAD-fmk is used to arrest cells in apoptosis thereby preventing their disintegration and loss from analysis (Fig. 8). Because the arrested (apoptotic) cells become fluorochrome labeled, they could easily be identified by fluorescence microscopy, as well as flow- or laser scanning-cytometry.56 Fam-VAD-fmk, thus, has dual function in this assay, namely arresting the process of apoptosis and also serving as marker apoptotic cells. This approach is analogous to stathmokinesis, the assay that is used to estimate cell birth rate from a slope of the plot representing mitotic cell accumulation during their arrest by a mitotic poison.57
Because the process of apoptosis is halted at the stage of caspase activation the caspase-mediated events either do not occur in the arrested cell, or occur but at a much slower rate. Indeed, we observed that at 20 μM and higher concentration of Fam-VAD-fmk, the HL-60 or MCF-7 cells did not disintegrate for up to 48 h.56 The arrested cells have still relatively high level of interference contrast when examined by microscopy and exhibit higher intensity light scatter signal than the apoptotic cells growing in the absence of Fam-VAD-fmk, when analyzed by flow cytometry.56 These arrested cells, however, are unable to exclude PI or trypan blue and cannot be revived when rinsed free of the inhibitor and grown in fresh medium.56 It should be noted that the rate of cell entry to apoptosis is not affected by Fam-VAD-fmk.56
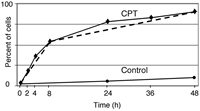
Arresting cells in apoptosis by Fam-VAD-fmk enables one to plot cumulative apoptotic index (CAI) as a function of time after administration of the inducer of apoptosis (Fig. 8). The plot reveals the rate of cell entry (kinetics) into apoptosis during the treatment. As it is evident in Figure 8, two distinct rates characterize the cells treated with camptothecin (CPT). During the initial 6 h approximately 50% cells from the culture undergoes apoptosis at a rate of about 8% of cells per hour. The remaining cells are entering apoptosis for up to 48 h at a rate of ˜1% of cells per hour. Control cell cultures exposed to Fam-VAD-fmk in the absence of CPT show a minor increase in the percentage of labeled cells after a 48 h culture period (˜9%), consistent with the rate of spontaneous apoptosis in untreated cell cultures approximating of 0.2% cells per hour.
To reveal whether the observed differences in rate of cell death may be related to the cell cycle phase the cell cycle distribution of the cells undergoing apoptosis (Fam-VAD-fmk labeled) at a faster rate i.e. early during the treatment (05 h), can be compared with that of the nonapoptotic cells in the same culture, as shown in Figure 6. When such a comparison was done we observed that in the CPT-treated cultures predominantly the S phase cells were undergoing apoptosis at the faster (˜8 % cells per hour) rate.56 The observation that the cells progressing through S phase are particularly sensitive to CPT is consistent with the wealth of the published data,58–60 and with the mechanism of cell death that involves a collision of the DNA replication forks with the CPT-generated DNA lesions (“cleavable complexes”) transforming these lesions into double-stranded breaks that trigger apoptosis.61 The slow rate of entrance to apoptosis observed between 8 and 48 h (˜1 % cells per hour) represents predominantly G1 cells that most likely were entering S phase during treatment with CPT. The stochastic nature of both slopes of the stathmoapoptotic plot is consistent with the kinetics of cell progression through the cycle.57,62
The ability by FLICA to arrest in apoptosis and through an estimate of CAI to measure frequency of cells that at are committed to die may be of special value as a prognostic marker in analysis of sensitivity of tumor cells (e.g., leukemias, lymphomas, myelomas) to the treatment. Namely, during chemotherapy tumor cells are dying predominantly by mode of apoptosis.63 Because they undergo apoptosis asynchronously, the single “snapshot” estimate of AI as it is conventionally done,63 cannot reveal an incidence of apoptosis with adequate accuracy. In contrast, the ex vivo studies of the CAI of the patient's blood or bone marrow blasts taken after drug administration (at the time when the drug blood or bone marrow level already starts to fall) are expected to reveal the fraction of cells committed in vivo to die in response to the treatment, and thus be of predictive value.
It should be noted, however, that the stathmoapoptosis approach based on the use of FLICA is applicable in analysis of the caspase-mediated apoptosis only. Other means of prevention of apoptotic cell disintegration and other means of their detection have to be used to apply this principle in studies of apoptosis that is not caspase mediated.49,50,64 Since serine proteases, in addition to caspases, appear to be also essential for completion of apoptosis51,65,66 it is likely that inhibitors of serine proteases also may be used to ensure stathmo-apoptosis.
Acknowledgments
Supported by NCI Grant CA 28 704, Chemotherapy Foundation and “This Close” Foundation for Cancer Research.
References
- 1.
- Alnemri E S, Livingston D I, Nicholson D W. et al. Human ICE/CED-3 protease nomenclature. Cell. 1996;87:171. [PubMed: 8861900]
- 2.
- Kaufmann S H, Desnoyers S, Ottaviano Y. et al. Specific proteolytic cleavage of poly(ADPribose) polymerase: an early marker of chemotherapy-induced apoptosis. Cancer Res. 1993;53:3976–85. [PubMed: 8358726]
- 3.
- Lazebnik Y A, Kaufmann S H, Desnoyers S. et al. Cleavage of poly(ADPribose) polymerase by proteinase with properties like ICE. Nature. 1994;371:346–7. [PubMed: 8090205]
- 4.
- Budihardjo I, Olsiver H, Lutter M. et al. Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol. 1999;15:269–90. [PubMed: 10611963]
- 5.
- Earnshaw W C, Martins L M, Kaufmann S H. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu Rev Biochem. 1999;68:383–424. [PubMed: 10872455]
- 6.
- Nicholson D W. Caspase structure, proteolytic substrates and function during apoptotic cell death. Cell Death Differ. 1999;6:1028–42. [PubMed: 10578171]
- 7.
- Zhang T S, Hunort S, Kuida K. et al. Caspase knockouts: matters of life and death. Cell Death Differ. 1999;6:1043–53. [PubMed: 10578172]
- 8.
- Stennicke H R, Salvesen G S. Catalytic properties of caspases. Cell Death Differ. 1999;6:1060–6. [PubMed: 10578173]
- 9.
- Zhivotovsky B, Samali A, Gahm A. et al. Caspases: their intracellular localization and translocation during apoptosis. Cell Death Differ. 1999;6:644–51. [PubMed: 10453075]
- 10.
- Li X, Darzynkiewicz Z. Cleavage of poly(ADPribose) polymerase measured in situ in individual cells: relationship to DNA fragmentation and cell cycle position during apoptosis. Exp Cell Res. 2000;255:125–32. [PubMed: 10666341]
- 11.
- Li X, Du L, Darzynkiewicz Z. During apoptosis of HL-60 andU937 cells caspases are activated independently of dissipation of mitochondrial electrochemical potential. Exp Cell Res. 2000;257:290–7. [PubMed: 10837143]
- 12.
- Tanaka M, Momoi T, Marunouchi T. In situ detection of activated caspase-3 in apoptotic granule neurons in the developing cerebellum in slice cultures and in vivo. Brain Res Dev Brain Res. 2000;121:223–8. [PubMed: 10876036]
- 13.
- Gorman A M, Hirt U A, Zhivotovsky B. et al. Application of a fluorometric assay to detect caspase activity in thymus tissue undergoing apoptosis in vivo. J Immunol Methods. 1999;226:43–8. [PubMed: 10410970]
- 14.
- Liu J, Bhalgat M, Zhang C. et al. Fluorescent molecular probes V: a sensitive caspase-3 substrate for fluorometric assays. Bioorg Med Chem Lett. 1999:3231–6. [PubMed: 10576694]
- 15.
- Hug H, Los M, Hirt W. et al. Rhodamine 110-linked amino acids and peptides as substrates to measure caspase activity upon apoptosis induction in intact cells. Biochemistry. 1999;38:13906–11. [PubMed: 10529236]
- 16.
- Komoriya A, Packard B Z, Brown M J. et al. Assessment of caspase activities in intact apoptotic thymocytes using cell-permeable fluorogenic caspase substrates. J Exp Med. 2000;191:1819–28. [PMC free article: PMC2213522] [PubMed: 10839799]
- 17.
- Belloc F, Belaund-Rotureau M A, Lavignolle V. et al. Flow cytometry of caspase-3 activation in preapoptotic leukemic cells. Cytometry. 2000;40:151–60. [PubMed: 10805935]
- 18.
- Bedner E, Smolewski P, Amstad P. et al. Activation of caspases measured in situ by binding of fluorochrome-labeled inhibitors of caspases (FLICA): correlation with DNA fragmentation. Exp Cell Res. 2000;259:308–13. [PubMed: 10942603]
- 19.
- Smolewski P, Bedner E, Du L. et al. Detection of caspases activation by fluorochrome-labeled inhibitors: Multiparameter analysis by laser scanning cytometry. Cytometry. 2001;44:73–82. [PubMed: 11309811]
- 20.
- Amstad P A, Yu G, Johnson G L. et al. Detection of caspase activation in situ by fluorochrome-labeled caspase inhibitors. Biotechniques. 2001;31:608–16. [PubMed: 11570504]
- 21.
- Ostrowski K, Barnard E, Stocka Z. et al. Localization of acetylcholinesterase activity using a 3H-labeled irreversible inhibitor. Exp Cell Res. 1963;31:89–99. [PubMed: 14043853]
- 22.
- Darzynkiewicz Z, Barnard E A. Specific proteases of mast cells. Nature. 1966;212:1198–203.
- 23.
- Darzynkiewicz Z, Rogers A W, Barnard E A. Autoradiography with tritiated methotrexate and the cellular distribution of folate reductase. Science. 1966;151:1528–30. [PubMed: 5909582]
- 24.
- Kunstle G, Leist M, Uhlig S. et al. ICE-protease inhibitors block murine liver injury and apoptosis caused by CD95 or by TNF-alpha. Immunol Lett. 1997;55:5–10. [PubMed: 9093874]
- 25.
- Ekert P G, Silke J, Vaux D L. Caspase inhibitors. Cell Death Differ. 1999;6:1081–6. [PubMed: 10578177]
- 26.
- Thornberry N A, Peterson E P, Zhao J J. et al. Inactivation of interleukin-1 beta converting enzyme by peptide (acyloxy)methyl ketones. Biochemistry. 1994;33:3934–40. [PubMed: 8142397]
- 27.
- Garcia-Calvo M, Peterson E P, Leiting B. et al. Inhibition of human caspases by peptide-based and macromolecular inhibitors. J Biol Chem. 1998;273:32608–13. [PubMed: 9829999]
- 28.
- Kamentsky L A, Burger D E, Gershman R J. et al. Slide-based laser scanning cytometry. Acta Cytol. 1997;41:123–43. [PubMed: 9022736]
- 29.
- Darzynkiewicz Z, Bedner E, Li X. et al. Laser scanning cytometry. A new instrumentation with many applications. Exp Cell Res. 1999;249:1–12. [PubMed: 10328948]
- 30.
- Thornberry N A, Rano T A, Peterson E P. et al. A combinatorial approach defines specificities of members of the caspase family and granzyme B. J Biol Chem. 1997;272:17907–11. [PubMed: 9218414]
- 31.
- Mancini M, Nicholson D W, Roy S. et al. The caspase-3 precursor has a cytosolic and mitochondrial distribution: implications for apoptotic signaling . J Cell Biol. 1998;140:1485–95. [PMC free article: PMC2132665] [PubMed: 9508780]
- 32.
- Susin S A, Lorenzo H K, Zamzani N. et al. Mitochondrial release of caspase-2 and 9 during apoptotic process. J Exp Med. 1999;189:381–93. [PMC free article: PMC2192979] [PubMed: 9892620]
- 33.
- Collusi P A, Harvey N L, Kumar S. Prodomain-dependent nuclear localization of the caspase-2 (Nedd2) precursor. A novel function for a caspase prodomain. J Biol Chem. 1998;273: 24535–42. [PubMed: 9733748]
- 34.
- Mao P L, Jiang Y, Wee B Y. et al. Activation of caspase-1 in the nucleus requires nuclear translocation of procaspase-1 mediated by its prodomain. J Biol Chem. 1998;273:23621–4. [PubMed: 9726961]
- 35.
- Ritter P M, Marti A, Blanc C. et al. Nuclear localization of procaspase-9 and processing by caspase-3-like activity to mammary epithelial cells. Eur J Cell Biol. 2000;79:358–64. [PubMed: 10887967]
- 36.
- Imazawa T, Nishikawa A, Tada M. et al. Nucleolar segregation as an early marker for DNA damage; an experimental study in rats treated with 4-hydroxyaminoquinolone-1 oxide. Virchows Arch. 1995;426:295–300. [PubMed: 7773509]
- 37.
- Raipert S, Bennion G, Hickman J A. et al. Nucleolar segregation during apoptosis of hematopoietic stem cell line FDCPMix. Cell Death Differ. 1999;6: 334–41. [PubMed: 10381627]
- 38.
- Miller M L, Andriga A, Dixon K. et al. Insights into UV-induced apoptosis: ultrastructure, trichrome staining and spectral imaging. Micron. 2002;33:157–66. [PubMed: 11567885]
- 39.
- Horky M, Wurzer G, Kotala V. et al. Segregation of nucleolar components coincides with caspase-3 activation in cisplatin-treated HeLa cells. J Cell Sci. 2001;114:663–70. [PubMed: 11171371]
- 40.
- Halicka H D, Bedner E, Darzynkiewicz Z. Segregation of RNA and separate packaging of DNA and RNA in apoptotic bodies during apoptosis. Exp Cell Res. 2000;260:248–56. [PubMed: 11035919]
- 41.
- Torres-Montaner A, Bolivar J, Astole A. et al. Immunohistochemical detection of ribosomal transcription factor UBF and AgNOR staining identify apoptotic events in neoplastic cells of Hodgkin's disease and in other lymphoid cells. J Histochem Cytochem. 200;48:1521–30. [PubMed: 11036095]
- 42.
- Darzynkiewicz Z, Li X, Gong J. Assays of cell viability: discrimination of cells dying by apoptosis. Methods Cell Biol. 1994;41:15–38. [PubMed: 7861963]
- 43.
- Smolewski P, Grabarek J, Halicka H D. et al. Assay of caspase activation in situ combined with probing plasma membrane integrity to detect three distinct stages of apoptosis. J Immunol Methods. 2002;265:111–21. [PubMed: 12072182]
- 44.
- Darzynkiewicz Z, Juan G, Li X. et al. Cytometry in cell necrobiology: analysis of apoptosis and accidental cell death (necrosis). Cytometry. 1997;27:1–20. [PubMed: 9000580]
- 45.
- Darzynkiewicz Z, Bruno S, Del Bino G. et al. Features of apoptotic cells measured by flow cytometry. Cytometry. 1992;13:795–808. [PubMed: 1333943]
- 46.
- Majno G, Joris I. Apoptosis, oncosis and necrosis. An overview of cell death. Am J Pathol. 1995;146:3–15. [PMC free article: PMC1870771] [PubMed: 7856735]
- 47.
- Koopman G, Reutelingsperger C P M, Kuijten G A M. et al. Annexin V for flow cytometric detection of phosphatidylserine expression of B cells undergoing apoptosis. Blood. 1994;84:1415–20. [PubMed: 8068938]
- 48.
- Vermes I, Haanen C, Reutelingsperger C. Flow cytometry of apoptotic cell death. J Immunol Methods. 2000;243:167–90. [PubMed: 10986414]
- 49.
- Harter L, Keel M, Hentze H. et al. Spontaneous in contrast to CD95-induced neutrophil apoptosis is independent of caspase activity. J Trauma. 2001;50:982–8. [PubMed: 11426111]
- 50.
- Qi L, Sit K H. CpG-specific common commitment in caspase-dependent and independent cell death. Mol Cell Biol Res Commun. 2000;3:33–41. [PubMed: 10683315]
- 51.
- Gorczyca W, Bruno S, Darzynkiewicz R J. et al. DNA strand breaks occurring during apoptosis: their early in situ detection by the terminal deoxynucleotidyl transferase and nick translation assays and prevention by serine protease inhibitors. Int J Oncol. 1992;1:639–48. [PubMed: 21584593]
- 52.
- Kerr J F R, Wyllie A H, Curie A R. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–57. [PMC free article: PMC2008650] [PubMed: 4561027]
- 53.
- Kerr J F R, Winterford C M, Harmon V. Apoptosis. Its significance in cancer and cancer therapy. Cancer. 1994;73:2013–26. [PubMed: 8156506]
- 54.
- Del Bino G, Darzynkiewicz Z, Degraef C. et al. Comparison of methods based on annexin V binding, DNA content or TUNEL for evaluating cell death in HL-60 and adherent MCF-7 cells. Cell Prolif. 1999;32:25–37. [PMC free article: PMC6726319] [PubMed: 10371301]
- 55.
- Hara S, Halicka H D, Bruno S. et al. Effect of protease inhibitors on early events of apoptosis. Exp Cell Res. 1996;232:372–84. [PubMed: 8601414]
- 56.
- Smolewski P, Grabarek J, Phelps D J. et al. Stathmoapoptosis: arresting apoptosis by fluorochrome-labeled inhibitor of caspases. Int J Oncol. 2001;19:657–63. [PubMed: 11562738]
- 57.
- Darzynkiewicz Z, Traganos F, Kimmel M. Assay of cell cycle kinetics by multivariate flow cytometry using the principle of stathmokinesis In:Techniques in Cell cycle Analysis JW Gray and ZDarzynkiewicz Eds. Humana Press, Clifton, NJ, USA,1997291–336.
- 58.
- Del Bino G, Lassota P, Darzynkiewicz Z. The S-phase cytotoxicity of camptothecin. Exp Cell Res. 1991;193:27–35. [PubMed: 1995300]
- 59.
- Deptala A, Li X, Bedner E. et al. Differences in induction of p53, p21WAF1and apoptosis in relation to cell cycle phase of MCF-7 cells treated with camptothecin. Int J Oncol. 1999;15:861–71. [PubMed: 10536167]
- 60.
- Liu W, Zhang R. Upregulation of p21WAF1/CIP1 inhuman breast cancer cell lines MCF-7 and MDAMB468 undergoing apoptosis induced by natural product anticancer drug 1-hydroxycamptothecin through p53-dependent and -independent pathways. Int J Oncol. 1998;12:793–804. [PubMed: 9499438]
- 61.
- Liu L F, Duann P, Lin C P. et al. Mechanism of action of camptothecin. Ann NY Acad Sci. 1996;803:44–9. [PubMed: 8993499]
- 62.
- Smith J A, Martin L. Do cells cycle? Proc Natl Acad Sci U S A. 1973;70:1263–7. [PMC free article: PMC433472] [PubMed: 4515625]
- 63.
- Gorczyca W, Bigman K, Mittelman A. et al. Induction of DNA strand breaks associated with apoptosis during treatment of leukemias. Leukemia. 1993;7:659–70. [PubMed: 8483318]
- 64.
- Joza N, Susin S A, Daugas E. et al. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature. 2001;410:549–54. [PubMed: 11279485]
- 65.
- Dong Z, Saikumar P, Patel Y. et al. Serine protease inhibitors suppress cytochrome c-mediated caspase-9 activation and apoptosis during hypoxia-reoxygenation. Biochem J. 2000;347:669–77. [PMC free article: PMC1221002] [PubMed: 10769169]
- 66.
- Grabarek J, Du L, Johnson G L, Lee B W. et al. Sequential activation of caspases and serine proteases (serpases) during apoptosis. Cell Cycle. 2002;1:124–31. [PubMed: 12429921]
- 67.
- Grabarek J, Dragan M, Lee B W. et al. Activation of chymotrypsinlike serine protease(s) during apoptosis detected by affinity labeling of the enzymatic center with fluoresceinated inhibitor. Int J Oncol. 2002;20:225–33. [PubMed: 11788882]
- 68.
- Darzynkiewicz Z, Williamson B, Carswell E A. et al. Cell cycle-specific effects of tumor necrosis factor. Cancer Res. 1984;44:83–90. [PubMed: 6690064]
- In Situ Activation of Caspases Revealed by Affinity Labeling Their Enzymatic Sit...In Situ Activation of Caspases Revealed by Affinity Labeling Their Enzymatic Sites - Madame Curie Bioscience Database
- The Origins of Species - Madame Curie Bioscience DatabaseThe Origins of Species - Madame Curie Bioscience Database
- Telomerase Activity as a Marker of Tumor Cell Survival to Evaluate Sensitivity o...Telomerase Activity as a Marker of Tumor Cell Survival to Evaluate Sensitivity of Neoplastic Cells to Cancer Treatment - Madame Curie Bioscience Database
- Nuclear Envelope Dynamics in Drosophila Pronuclear Formation and in Embryos - Ma...Nuclear Envelope Dynamics in Drosophila Pronuclear Formation and in Embryos - Madame Curie Bioscience Database
- Cytokines in the Pathogenesis of Rheumatoid Arthritis and Collagen-Induced Arthr...Cytokines in the Pathogenesis of Rheumatoid Arthritis and Collagen-Induced Arthritis - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...