

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

 and
and Summary
Nerve growth factor was the first identified protein with anti-apoptotic activity on neurons. This prototypic neurotrophic factor, together with the three structurally and functionally related growth factors, brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT3), and neurotrophin-4/5 (NT4/5), forms the neurotrophin protein family. Target T cells for neurotrophins include many neurons affected by neurodegenerative diseases such as Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis and peripheral polyneuropathies. In addition, the neurotrophins act on neurons affected by other neurological and psychiatric pathologies including ischemia, epilepsy, depression and eating disorders. Work with cell cultures and animal models provided solid support for the hypothesis that neurotrophins prevent neuronal death. While no evidence exists that a lack of neurotrophins underlies the etiology of any neurodegenerative disease, these studies have spurred on hopes that neurotrophins might be useful symptomatic therapeutic agents. However first clinical trials led to variable results and severe side effects were observed. For future therapeutic use of the neurotrophins it is therefore crucial to expand our knowledge about their physiological functions as well as their pharmacokinetic properties. A major challenge is to develop methods for their application in effective doses and in a precisely timed and localized fashion.
From Neurotrophin Physiology to Therapy
The human brain undergoes continuous structural remodeling in response to signals originating from inside and outside of the body. At a molecular level these changes can be very subtle and involve minor modifications of synaptic proteins. At a cellular level, dendritic spines or nerve cell arborizations are reshaped. Finally, entire nerve cells are newly generated or removed, even in the mature brain (reviewed in ref. 1). In the developing nervous system these changes are particularly drastic and hence are more easily observed and investigated. However, similar or identical molecular mechanisms are still at work in the adult mature brain.
An intricate molecular machinery comprised of many signaling proteins including protein growth factors and their receptors implements these changes in the nervous system. The neurotrophins, a family of protein growth factors, are prime candidates for molecular mediators of such neuronal plasticity. During embryonic and postnatal development these proteins regulate survival, differentiation and specification of neurons (reviewed by ref. 2). In the adult nervous system they modulate or trigger fast synaptic responses and cause changes in the function of synapses and in the morphology of neurites. Consequently they influence higher systemic functions such as behavior, learning, memory formation and cognition (reviewed in ref. 3).
In view of the important physiological function that the neurotrophins exert in the brain and in the peripheral nervous system, they are candidate agents for the treatment of neurological and psychiatric diseases. Hopes in these proteins were furthered by a large number of pre-clinical studies showing that most of the neuronal populations affected by neurodegenerative diseases respond to neurotrophins and express neurotrophin receptors, although there is no direct proof that a lack of neurotrophins is causally involved in the manifestation of any neurodegenerative pathology. Neurotrophins could inhibit or delay degenerative processes in neurons under numerous experimental conditions. Furthermore, the expression levels of neurotrophins and their receptors are strongly regulated in pathophysiological situations, arguing for their involvement in the cellular responses to pathological processes.
Unfortunately, the high therapeutic expectations in neurotrophins resulted in insufficient pre-clinical preparations of clinical trials (reviewed in ref. 4). In particular the need for precisely controlled, timed and localized delivery as well as the potential side effects have apparently been underestimated. In the following, we first describe the neurotrophin molecules, their receptors and the signaling pathways triggered by them. We then briefly summarize the evidence for neuroprotective effects of the neurotrophins with an emphasis on in vivo studies. In this context we give reference to the initial clinical trials performed with neurotrophins as well as to the problems encountered therein. Finally we attempt to point to possible solutions for these problems as an outlook.
Structure and Physiological Functions of Neurotrophins and Their Receptors
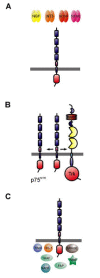
During the last decade, the knowledge of how cell survival and death is controlled has expanded at an ever increasing rate. In each cell, an intrinsic cell suicide program exists that is under constant control by cell-intrinsic and -extrinsic stimuli.5 Also, the survival and death of neurons depends strongly on extrinsic signaling by neighboring or interacting cell types. Many of these signals are soluble growth factors such as the neurotrophins nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), and neurotrophin-4/5 (NT-4/5) (Fig. 1). These neurotrophic factors signal by binding to two different types of transmembrane receptor proteins on responsive neurons. One class of neurotrophin receptors are the tropomyosin receptor kinase (Trk) proteins that form a small subfamily of the large family of receptor tyrosine kinases (Fig. 2). The second molecular entity that mediates neurotrophin signaling is the p75 neurotrophin receptor, a protein that binds all neurotrophins and is a member of the tumor necrosis factor receptor (TNF-R) family (Fig. 3).

Our view of the precise interplay between the neurotrophins and their receptors as well as of the receptor-mediated cellular signals in neurons is still fragmentary. In order to gain a better understanding of how neurotrophins protect neurons under pathophysiological conditions it is therefore of central importance to study the neurotrophin receptors and their intracellular signaling first under normal conditions. One important and largely enigmatic observation is that the response of neurons to neurotrophins can be very diverse depending on input from other signaling pathways and changes during development. Neurotrophin signaling is therefore often referred to as dependent on the “cellular context”. Consequently, results obtained with one type of neuron cannot reliably (and should not) be extrapolated onto other cells. As it will be discussed below, such an extrapolation of the effects of NGF on sensory neurons and of BDNF on spinal motoneurons had a negative impact on the first clinical studies with BDNF in amyotrophic lateral sclerosis (ALS).
The Neurotrophins
The neurotrophins are four distinct small basic proteins (Fig. 1a). In each case the active growth factor protein is a homodimer that is formed by identical peptide chains of about 120 amino acids each. The two monomers cohere by non-covalent chemical bonds. Highly conserved pairs of cysteine residues form a so-called cystein knot motif within each monomer that stabilizes the conformation of the proteins.6 Whereas the overall structure of the four neurotrophin molecules is very similar, differences between them become apparent upon closer inspection (Fig. 1b). Each molecule is uniquely characterized by a specific pattern of charged basic or acidic residues exposed on its surface. Differences between the individual neurotrophins are particularly evident within peptide loops protruding from the core of the molecules that is itself formed by beta sheets (reviewed in ref. 7). These loops mediate the main contacts between the neurotrophins and their specific receptors on the surface of responsive cells. This blend of structural similarities and differences in the structure of the molecules is reflected by the biological properties of the neurotrophins (reviewed in ref. 8). The specific molecular differences between the neurotrophins not only determine their receptor interactions, but also profoundly influence their biophysical and pharmacokinetic behavior. The neurotrophins have a tendency to stick unspecifically to surfaces, for example to cellular membranes. Their stickiness depends on a mixture of hydrophobic and hydrophilic interactions and is an important parameter for their use as therapeutics, since it is a limiting factor for their diffusion through organs and tissues such as the brain parenchyma. The diffusion behavior is different for each of the neurotrophins, for example NGF9 shows a higher diffusion rate compared to BDNF.10,11
Heterodimers of two different neurotrophin monomers can be formed in test tubes but were not detected under physiological conditions.12,13The generation and application of synthetic heterodimers is an interesting pharmacological approach that might broaden the spectrum of responding neurons. For example, for the treatment of sensory neurons in the peripheral nervous system heterodimers between NGF and NT3 might act simultaneously on NGF and NT3 responsive subpopulations (see below). Another interesting aspect of neurotrophin molecules is that their biosynthesis and secretion in the mature nervous system is controlled by neuronal activity. Frequently the neurotrophins seem to be made available to the surrounding tissue “on demand”. This mechanism enables the neurotrophins to adjust size and function of nerve cell populations to the precise requirement of the structures they innervate (reviewed by ref. 14). As discussed in the following sections, it will be important for the future therapeutic use of neurotrophins to devise strategies for a similarly controlled exposure of diseased neurons to these factors in order to minimize adverse side effects, as they have been observed following systemic treatment.
The Neurotrophin Receptors
The membrane receptors for neurotrophins comprise three receptor tyrosine kinases belonging to the tropomyosin receptor kinases (Trk) family ref. (reviewed by 15) and p75, a molecule that groups within the large family of TNF-receptor homologs (reviewed by ref. 16). The molecular interplay between these receptors is complex and incompletely understood,17 but as a net result it triggers cellular effects, which are highly ligand and cell type specific. The interactions of neurotrophins with their receptors are frequently analyzed on cell lines overexpressing neurotrophin receptors in a recombinant form. In these highly defined in vitro approaches strong binding interactions and strong activation of intracellular signaling pathways were observed which reflect in principle the mechanism occuring in primary nerve cells (reviewed in refs. 15,18). However, it should be kept in mind that the response of primary neurons to neurotrophins is under the control of fine-tuned cell specific parameters. In particular the antagonistic view that Trk receptors stimulate survival, whereas p75 signaling triggers apoptosis is certainly an over-simplified perspective.17 Another context-dependent and variable parameter is the affinity of neuronal receptors for their neurotrophin ligands, which can vary by several magnitudes between 10−12 M and 10−8M. The frequently expressed statement that Trk receptors form binding sites of high affinity for their neurotrophin ligands, whereas p75-binding sites are of low affinity is not correct. It derives from biochemical in vitro studies and is not thoroughly backed up by studies with primary neurons. As a matter of fact, data on the binding properties of defined receptor molecules on neurons in situ are sparse especially in the CNS. Therefore a precise dose-response curve has to be established for each cell type as a prerequisite for the establishment of treatment paradigms.
The spectra of responsive neurons are distinct and characteristic for each neurotrophin ligand, but they are also partly overlapping. For example, cholinergic forebrain neurons respond strongly to NGF and weakly to BDNF, whereas spinal motoneurons respond strongly to BDNF but are negatively affected by NGF (see below). Cellular responses to activation of different neurotrophin receptors by the same ligand can be very diverse: for example NGF can stimulate the survival of some cell types while it kills others (reviewed by ref. 19). Compared to other growth factor families, which typically have a much broader range of target T cells including numerous non-neuronal cell types, neurotrophins act predominantly on a restricted number of neuronal populations. This should be an advantage for their therapeutic use.
The interactions between the neurotrophins and their receptors are best analyzed on the sensory neurons on the dorsal root ganglia (DRG). A large number of biochemical, cell culture and genetic studies were performed with these cells. The expression pattern of the Trk-receptors was analyzed in the various sub-populations of sensory DRG neurons and was compared to the biological effects of their ligand and the results revealed an excellent correlation between the Trk receptor expression and responsiveness of subpopulations of DRG neurons. For example, the large sized, proprioceptive neurons express TrkC and respond to NT3 while small sized nociceptitive neurons express TrkA and depend on NGF. These findings were most convincingly corroborated in vivo by the analysis of mice carrying targeted mutations in receptor or ligand genes (reviewed by ref. 20).
In the peripheral nervous system (PNS), for almost all neuronal subpopulations expressing Trk receptors, biological functions could be defined for the corresponding ligands. By contrast, the correlation between Trk receptor expression and responsiveness is less obvious in the brain. One clearly defined case is that of the cholinergic forebrain neurons, which, like a small number of non-cholinergic nuclei express the NGF receptor TrkA.21 Consistently, cholinergic forebrain neurons respond strongly to NGF.22 By contrast, the BDNF and NT4/5 receptor TrkB is widely expressed in the mature CNS and is highly developmentally regulated.23 The finding that the TrkB gene is expressed in various protein isoforms further complicates a direct correlation between ligand function and TrkB receptor expression. The individual TrkB variants, such as TrkB.T1, with a truncatation in the intracellular domain, are generated by alternative splicing of the primary gene transcript and are regulated independently of each other.24,25 The situation for TrkC is complicated too. It is expressed in a variety of CNS structures preferentially during early ontogenetic development and down-regulated in the healthy mature brain.26
The second type of neurotrophin receptor is the p75 neurotrophin receptor, a 75 kDa glycoprotein. Despite having been the subject of a large number of recent studies, its function has only begun to be understood (reviewed by refs. 27,28). Studying this receptor is rendered difficult by the lack of a simple and reliable experimental read-out system for its activity. This is in contrast to the enzymatically active receptor of the Trk family, whose activity can easily be assessed by analysis of their tyrosine phosphorylation. The p75 neurotrophin receptor triggers signaling by non-covalent binding to intracellular molecules (Fig. 3C). The cellular effects of its signaling cascade are diverse. In certain biological structures, including neuronal precursor populations in the retina and spinal cord, p75 can cause the activation of apoptotic cell death (reviewed in ref.29). Furthermore, it exerts local regressive effects on neuritic growth.30 Besides its role as an independently signaling receptor, p75 also binds to all three Trk receptors and this physical interaction can modify the tyrosine kinase activity as well as the ligand specificity of the Trk receptors.31 In a reciprocal fashion the Trk receptors have a blocking effect on the downstream signaling of p75.32 The diverse biochemical and functional interactions between the Trk and p75 neurotrophin receptors render them a demanding object for future studies. Yet evidence is accumulating that the fine-tuning of these ligand-receptor interactions critically determines the distinct cellular responses. Hence the neurotrophin receptors should not merely be considered as simple mechanical triggers of intracellular signaling pathways. Consequently we have to realize, that based on our current fragmentary knowledge, the response of a cell to neurotrophin treatment cannot always be predicted, even when the expressed set of receptors has been determined.
Evidently, the biochemical properties of the cellular neurotrophin receptors are important for neurotrophin therapy. In vivo work has revealed that the effects of neurotrophins are very dose sensitive and strongly depend on the appropriate concentration.33 Treatment regimes should be adjusted to these findings, for example by developing spaced versus chronic infusion protocols.33,34 The underlying molecular mechanisms remain to be determined. One potential explanation could be down-regulation of the receptors. In particular the biological effects of BDNF at supraoptimal doses might be hampered by a specific molecular property of its receptor TrkB. Exposure to excess BDNF causes rapid internalization of TrkB followed by subsequent proteolytic degradation of the receptors.35 Consequently the neuron is desensitized for BDNF after prolonged periods of treatment.36 Desensitization is observed at different levels for the neurotrophin receptors and depends on so far unresolved molecular properties.37,38 In contrast to TrkB, TrkA is not downregulated. Consequently, TrkA phosphorylation is detected for a much longer time period after NGF injection into the brain and the treatment leads to a prolonged stimulation of receptor expression.21,39 The difference in cell membrane cycling between the TrkA and TrkB receptors was attributed to a short peptide stretch in their intracellular domains.35 Within the brain the truncated T1 isoform of the TrkB receptor appears to regulate the liquor concentrations of BDNF. It is strongly expressed in the ependymal cells and mediates internalization of BDNF.40 TrkB.T1 might form a ligand-specific removal or sequestering system for BDNF and NT4/5, but not for NGF potentially explaining the differences in diffusion of neurotrophins in the brain.
All neurotrophins interact with more than one functional receptor with different kinetics and affinities (Figs. 2a and 3a). At low, presumably physiological, concentrations neurotrophins bind to their preferred receptor and exert very specific effects. At elevated doses, their receptor spectrum is changed and consequently the biological response becomes less defined. Excessively applied neurotrophins can lead to adverse toxic effects through various receptor dependent mechanisms such as receptor competition or free radical formation.41–43 The degree of receptor promiscuity is different for each of the neurotrophins and involves both Trk and p75 neurotrophin receptors. In fact NT3, the most promiscuous ligand, binds to all known neurotrophin receptors. Yet it would be wrong to call these interactions unspecific since evidence is accumulating that such non-preferred interactions are indeed physiological.44,45 Obviously the variety of cellular responses at elevated neurotrophin concentration emphasizes the need for a precise control of an exposure regime.
Neurotrophin Signaling Mechanisms
Of all trk receptors the signaling cascades originating at the NGF receptor TrkA are best understood ref.(reviewed by 46), whereas our knowledge about the signaling of TrkB and TrkC is much less detailed.47 In many aspects Trk receptors are typical tyrosine kinases. Receptor dimerization upon ligand binding leads to autophosphorylation of the dimeric receptor complex at several defined tyrosine kinase residues.48 Three major intracellular signaling pathways originating at TrkA have been identified in biochemical and more recently in genetic experiments (Fig. 2b). The first pathway leads to the activation of the ERK MAP-kinase signaling module through a cascade that includes Ras. This pathway is primarily triggered by the association of Grb2/SOS complexes to phosphorylated adaptor proteins, most prominently Shc or FRS2. The activation of this pathway initiates predominantly differentiation and neuritogenesis, but also neuronal survival (reviewed by ref. 49). The second pathway stimulates predominantly neuronal survival by activation of protein kinase B (PKB)/AKT, which subsequently leads to inactivation of the pro-apoptotic protein Bad and to phosphorylation of the forkhead transcription factor.50 This anti-apoptotic pathway is activated by TrkA through its association with the large multi-adaptor proteins insulin receptor substrates 1 (IRS1) and IRS2, but also Gab-1.51,52 The IRS proteins then mediate PI3-kinase activation. The third pathway involves binding of PLC-γ to the carboxyl-terminal tyrosine residue 785 of TrkA. Activated PLC-γ stimulates production of the lipid second messenger IP3. The function of this neurotrophin-induced pathway is less defined compared to the other two. It appears to be important for synaptic plasticity and neurotrophin mediated neurotrophin release.53 Furthermore it also provides a potential link between the signaling of neurotrophins and electrical activity in the CNS through the control of intracellular Ca2+ levels. Physiological neuronal activity stimulates survival responses, while lack of activity promotes neuronal cell death. For example, blockade of preganglionic transmission onto sympathetic neurons increases the number of dying neurons during development.54 In the CNS, reduced neuronal survival has been observed in the developing and adult brain following blockade of electric activity.55 Neurotrophin mediated changes in cortical dendritic morphology also require neuronal activity.56
It is important to note that the cellular responses to signaling of the three trk receptors can be surprisingly diverse although these proteins share extensive primary sequence identities in their intracellular enzymatic domains.57 Hence, the same concept of diversity and similarity that applies to the structure and function of the ligand also is applicable to the receptors. The molecular explanation for the observed differences of Trk receptor signaling might reside in the differential use of intracellular adaptor proteins homologous to IRS, Shc or Gab-1. Distinct association kinetics of these molecules and binding to different consensus sites in the receptors may explain the observed specificity of Trk receptor signaling.
Our knowledge about the intracellular signaling mechanisms of the neurotrophin receptor p75 remain largely elusive. Recent data indicate that p75 mediates local effects on neurite outgrowth via interference with the activity of the small GTPase RhoA.30 Another p75-triggered pathway leads, via ceramide production by sphingomyelin hydrolysis,58,59 to the activation of the transcription factor NFκb.60 This pathway might serve as an anti-apoptotic signal balancing the activation of cell death cascades.61,62 That p75 activation can also actively kill cells is an important consideration for potential therapeutic use of neurotrophins as neuroprotective agent.63,64 The precise composition of the cell death pathway triggered by p75 remains to be established. Biochemical and genetic evidence involves several of the recently identified protein interactors (reviewed by ref.3). Downstream effects may include caspase 3 activation,65 activation of the JNK MAPK- module66 as well as activation of p53 and p73.67,68
Cellular Neuroprotective Mechanisms
It may be expected that neurotrophins would have two beneficial effects in neurodegenerative diseases. First, these proteins might enhance long term survival of damaged neurons and second, they might maintain or re-induce the physiological functional status of affected neurons.
A core problem for the treatment of neurological diseases is the incomplete knowledge about the underlying pathophysiological processes. It appears that neurons are killed by both necrotic and apoptotic mechanisms (see reviews in refs. 69,70). Interestingly, the same neurotrophic extracellular stimuli that regulate ontogenetic cell death and survival appear to be re-activated under pathophysiological situations in the mature nervous system. Therefore neurotrophins are valuable experimental tools for the study of neuroprotective mechanisms. They not only exert their activity on specific neuronal populations, they also prevent apoptotic or excitotoxic neuronal death by activating multiple signaling pathways. In various models of disease neurotrophins activate the tyrosine kinase receptors TrkA, TrkB and TrkC in order to prevent degeneration of injured mature CNS neurons in models of chronic degenerative diseases as well as in lesion paradigms. However, in many experimental conditions it is currently not known whether neurotrophin treatment interferes with apoptosis, necrosis or even directly with specific neuropathological mechanisms such as the generation of reactive oxygen species or excitatory cell death.71,72 For neurodegenerative diseases it remains a matter of debate to which degree the bcl2/caspase3 mechanism of apoptosis that regulates ontogenetic cell death is also causing pathophysiological neuronal death.73,74 The neuroprotective action of neurotrophins on neurons damaged by excitotoxins, toxic chemical agents or injury indicates that these factors also interfere with necrotic mechanisms (reviewed by ref. 75). Hence it appears that neurotrophins activate different cellular mechanisms to protect neurons in a cell specific manner.
Neurotrophins have broader biological effects on neurons than just regulation of cell death. They also regulate subcellular processes underlying neuronal plasticity and maintenance of structural neuronal integrity. Since neurodegenerative diseases are accompanied by substantial structural damage and reactive plasticity, neurotrophic therapy might be advantageous also in this regard. As outlined above, the effects of neurotrophins on survival and neuronal morphology are mediated by distinct intracellular signaling pathways and consequently these effects can be separated in animal models of neuronal cell death in vivo.76,77 In the cerebral cortex neurotrophins stimulate both growth of and retraction of dendritic branches indicating that the effect of neurotrophins on neuronal morphology is not invariably growth promoting.78 Again this observation emphasizes the necessity to further study the role of neurotrophins in maintenance and refinement of neuronal structures and at the same time it argues against systemic neurotrophin treatment, since the potential of interference of exogenously applied neurotrophins with functional cortical circuitries has to be a major concern (see below). In this context it is of interest to note that neurotrophins are themselves expressed in many neuronal subtypes and their production and secretion is itself regulated by exogenous neurotrophin supply.79–81 Transneuronal induction of neurotrophin gene expression is well documented in the autonomous nervous system and fulfills physiological functions there.82 Similar phenomena are likely to exist also inside the CNS, for example in the corticospinal tract.83 The possibility of transneuronal signaling also argues against systemic neurotrophin treatment.
Neurotrophins in Animal Models of Pathological Situations and Clinical Trials
Much of the hope that neurotrophins might be useful therapeutics is based on the dynamically regulated expression of these factors and their receptors in animal models of neurological diseases. Neurotrophins strongly inhibit or delay degenerative processes in a variety of in vivo and in vitro models of human neurodegenerative disease such as Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis and peripheral sensory neuropathy. Consequently neurotrophins have been used for clinical trials with the rationale that these proteins would inhibit degeneration of nerve cells and lead to re-establishment of lost synaptic connections. However, despite the convincing success in animal models of disease, clinical trials with neurotrophins were initially disappointing.
Cortical and Hippocampal Neurons
Strong neuroprotective effects have been observed with neurotrophins in models of acute neuronal injury. BDNF and TrkB are up-regulated as a part of the endogenous response to cerebral ischemia adjacent to the lesion84,85and the infarct size is reduced by application of BDNF or NT4/5.86–88 Neurotrophins and their receptors are strongly and differentially regulated after induction of seizures89–91 and soluble TrkB fusion proteins inhibit kindling development.92 In lesion models, BDNF protects neurons of the corticospinal tract93 and NT3 potentiates the axonal growth-promoting effect of the IN1 antibody after corticospinal tract lesion.94
Cholinergic Forebrain Neurons
All major neuronal subpopulations affected by Alzheimer's disease, in particular cholinergic cells in the basal forebrain, but also neurons of the hippocampus, cerebral cortex and the locus coeruleus, respond to at least one neurotrophin. The cholinergic deficit in the CNS might represent an ideal target for neurotrophin therapy, since most of the cholinergic neurons of the Nucleus Meynert complex respond very selective to NGF.95 The NGF receptor TrkA is expressed in most cholinergic neurons in the forebrain and a limited number of neurons in other non-cholinergic nuclei.96,97 In experiments with rodents and subsequently with primates, a protective effect of NGF against atrophy was demonstrated on cholinergic neurons after fimbria-fornix lesion.98 NGF prevents cholinergic atrophy not only if experimentally induced by a lesion, but also in aged animals. These cellular effects are paralleled by behavioral improvements of aged animals, whereas in normal younger animals NGF surprisingly had an adverse effect.99–101 A comparison with other growth factors shows that NGF is the most efficacious neurotrophin acting on cholinergic neurons.102–104 However, BDNF and NT4/5 also exerts significant neuroprotective effects on cholinergic forebrain neurons.105,106 Interestingly, BDNF gene expression levels are reduced in Alzheimer patients, concomitant with a loss of cholinergic basal forebrain neurons, although the causal relation to neurodegeneration remains open.107 NGF levels are increased in Alzheimer patients. Most likely it accumulates in the cortex as a consequence of the reduction in retrograde transport and removal by cholinergic fibers. There has been an initial report that mice with a partial p75 gene deletion scientific have a higher number of cholinergic neurons in the medial septum. This report caused a scientific dispute and later on had to be retracted.108,109 Recent results obtained with a novel mutant p75 mouse line generated in our laboratory indicate that p75 signaling can cause the death or atrophy of developing cholinergic neurons in the medial septum. A careful anatomical and histochemical analysis of ChAT-immunoreactive neurons revealed that mice in which the p75 gene is completely inactivated have a highly significant increase in cell number at postnatal day 15. Many of the surplus neurons are maintained for at least 3 months (T. Naumann, E. Casademunt, personal communication). It will be interesting to see, whether this increase in ChAT-positive neurons leads to detectable behavioral changes in these animals.
Two studies report on the effect of NGF applied by intraventricular injection to patients suffering from Alzheimer's disease. In both reports some beneficial effects were observed, which were opposed, however, by adverse side effects such as weight loss and induction of painful sensations.110,111 Hence it appears that this form of NGF application is not well suited for treatment of Alzheimer's disease.
Mesencephalic Dopaminergic neurons
BDNF and NT4/5 support the survival of dopaminergic neurons isolated from the substantia nigra in cell cultures and protect them against 6-OHDA and MPP toxicity.112,113 However, in lesion experiments in adult animals no protection by neurotrophins against the loss of dopamine was observed following MPTP application.114 On adult axotomized cells BDNF and NT3 promote survival and differentiation of dopaminergic neurons.115 In unlesioned animals chronic injection of BDNF into the substantia nigra lead to conflicting results with either hypo- or hyperfunction of the dopaminergic system indicating complex interactions and a potential for significant side-effects.116,117 Furthermore, the expression of the BDNF/NT4/5 receptor TrkB expression is low under physiological conditions in the cells of the substantia nigra.118,119
Spinal and Facial Motoneurons
Spinal motoneurons express the BDNF receptors TrkB120 and mice with a targeted deletion of the TrkB gene have less motoneurons.121 These results are in conflict with the analysis of mice carrying a targeted deletion in the TrkB ligands. Mice carrying a targeted mutation in the genes encoding BDNF and NT4/5 show no reduction in motoneuron number.122,123 Motoneuron axotomy increases BDNF and TrkB gene expression in motoneurons.124 In neuronal cell cultures, motoneuron survival is mediated by both NT4/5 and BDNF.125 In vivo pharmacological administration of BDNF rescues embryonic motoneurons from naturally occurring death and axotomized neonatal motoneurons are also rescued by BDNF in vivo.126–128 Most notably, adult rat motoneurons in the facial nucleus can be rescued with BDNF after axotomy, indicating that mature neurons maintain their responsiveness to this neurotrophin.129 The common neurotrophin receptor p75 is expressed on embryonic motoneurons and on mature cells after injury.130,131 During early development p75 triggers cell death of some immature neurons in the spinal cord.132 Therefore, it should be considered that neurotrophins promote not only cell survival but also could trigger regressive phenomena on motoneurons as described above for cholinergic neurons in the basal forebrain. In newborn rats increased cell death is observed after application of NGF.126 This NGF mediated cell death is dependent on p75 and likely to be caused by ligand competition for the binding to functional Trk receptors.42 The effect of the absence of p75 receptor on the regeneration of adult axotomized facial motoneurons has recently been tested in our complete knock out animals. No differences were observed in neuronal survival and speed of axonal regeneration (A. Gschwendtner, G. Raivich, personal communication), which is in contrast to a previous report that describes improved motoneuron regeneration in the absence of full-length p75.133
No indication exists that the degeneration of motoneurons in the anterior horn and in the brainstem in amyotrophic lateral sclerosis is linked to a lack of trophic support. Nevertheless the strong response during development and in lesion paradigms of spinal motoneurons to growth factors such as insulin-like growth factor 1 (IGF1), glial cell line derived neurotrophic factor (GDNF), ciliary neuronotrophic factor (CNTF) and in particular BDNF renders ALS a good candidate disease for neurotrophic therapy.134 However, a clinical phase III trial of systemically applied BDNF in ALS was recently abandoned. It is questionable whether therapeutically effective concentrations of BDNF have reached the soma of spinal motoneurons in this clinical trial. Motoneurons of all central neurons were considered attractive target T cells since their axons in the periphery were thought to have free access to growth factors applied systemically. Subsequently those factors could be retrogradly transported and exert their activity on the cell soma in the spinal cord. The hypothesis of such a retrograde activity of BDNF is mainly based on the findings that sensory and sympathetic neurons effectively take up and transport the neurotrophin NGF.135 However, the retrograde transport of NGF might be strongly facilitated by some of the specific properties of peripheral NGF-responsive neurons, which are characterized by unmyelinated fibers and free nerve endings. By contrast the BDNF-sensitive motoneurons have thickly myelinated fibers and synapses deeply buried in the muscle. While BDNF is effectively retrogradely transported by motoneuron axons upon injection into the sciatic nerve,136 motoneuron endplates are very poor entry points for systemically applied BDNF. Intraventricular injection of BDNF provides an alternative application route and work with rodents has demonstrated that adult facial motoneurons were equally rescued by BDNF administered intraventricularly and systemically.129 However the pre-clinical studies performed in rodents are difficult to compare with the situation in humans, where the distances between peripherally applied factor and the cell soma are considerably longer. Therefore, large animals, such as sheep, which have diffusion distances comparable to human beings, have been employed in pre-clinical studies.11 Unfortunately the planned clinical trials based on these experiments are now in jeopardy after the disappointing result with systemically applied BDNF.137
Peripheral Neurons
Peripheral sensory neurons represent the best-investigated target T cells for neurotrophins. Basically all embryonic and most adult sensory neurons express Trk receptors often in combination with p75. Of special interest are cells in the dorsal root ganglia.138,139 Surprisingly, adult, but not embryonic, sensory neurons survive in culture in the absence of neurotrophins, while they maintain their neurotrophin responsiveness.140,141 These findings indicate a role of neurotrophins in the regulation of specific functional properties of adult sensory neurons. This is corroborated by in vivo results with application of neurotrophins or blocking antibodies directed against them.142 In axotomized cells the expression of genes encoding neurotrophins and their receptor are downregulated and the expression is restored by neurotrophin treatment.143 In patients with diabetes neurotrophin levels are increased in skin under neuropathological conditions.144 NGF primarily acts on adult nociceptors characterized by small fiber diameter and substance P expression (reviewed by ref. 145). NT3 acts preferentially on large-fiber neurons, prevents demyelination of Fast proprioceptive fibers under pathological conditions and reverses the characteristic electrophysiological slowing down of conduction velocity.146 Furthermore it protects rat sensory neurons from cisplatin-induced toxicity.147 Another potential peripheral target for neurotrophin treatment are the structures of the auditory system. Interestingly not only the auditory and vestibular neurons, but also the hair cells depend on neurotrophin support during development.148 Both BDNF and NT3 have strong protective effects against ototoxic treatment.149,150 Finally NT3 acts potently on the neurons of developing enteric nervous system151 and TrkC is expressed on mature enteric neurons152 suggesting this factor as a potential treatment for gut diseases.
Recombinant human nerve growth factor has been tested in phase II and phase III clinical trials for the treatment of patients with small fiber neuropathies. In a phase II trial with AIDS patients NGF-treatment caused significant relief of deafferentiation pain.153 The trials with patients suffering from diabetic polyneuropathy gave rise to variable results. A first phase II trial initially indicated beneficial effects on sensory neurological scores,154 a result that could not be corroborated in a subsequent phase III trial.155 In all studies NGF caused mild myalgia and injection pain as an adverse effect.156 The underlying cellular mechanism for these painful sensations is not fully elucidated. But it is well documented that NGF treatment up-regulates Substance P expression in sensory neurons and induces mast cell degranulation.157 Recently it was shown that NGF activates the capsaicin receptor through TrkA mediated activation of phospholipase C.158
Topical application of NGF has substantial beneficial effects on ulcer healing, potentially in an indirect manner as a consequence of its activity on sensory innervation. In a study with patients suffering from corneal keratitis local NGF application restored corneal integrity in all cases159 and wound healing was induced in rheumatoid arthritis patients with foot ulcers.160
Non-Neuroprotective and Side Effects of Neurotrophins
Besides their cellular neuroprotective function, the neurotrophins influence also higher brain function and modulate the functions of non-neuronal organs, in particular the immune and the vascular system. While these effects on non-neuronal cells might well be exploitable for therapies such as autoimmune diseases, they could give rise to adverse side effects during treatment of neurodegenerative diseases.
Changes in Monoaminergic Brain Functions and Behavior
In addition to their neuroprotective role, neurotrophins are also potent regulators of neuronal gene transcription. In the serotonergic system BDNF regulates the expression of serotonergic markers including tryptophan hydroxylase and the serotonine transporter SERT, but not neuronal survival.161,162 Similarly, neurotrophin-induced changes of neurotransmitter function might occur on many nerve cells, including noradrenergic neurons,163 dopaminergic and cholinergic neurons. BDNF infusion alters serotonin levels and serotonergic axonal sprouting in the brain164,165 and modifies firing patterns of serotonergic and dopaminergic neurons.166,167 Therefore neurotrophin treatment might result in psychogenic or behavioral effects. BDNF intraventricular infusion indeed induces behavioral changes in mice.168 Heterozygote BDNF mutant mice are hyperaggressive and hyperphagic.169,170 Conversely, infusion of BDNF into normal animals causes abrupt weight loss.164 Taken together these findings indicate that satiety and locomotor nuclei in the hypothalamus are controlled by BDNF.
Effects on Memory Formation and Mental State
Neurotrophins change the shape and functional properties of central neurons in numerous ways. For example, already milliseconds after applications of BDNF to slice cultures, ion channel properties are modified and action potentials are triggered.171 After seconds and minutes the release of neurotransmitter is facilitated and biochemical modifications of synaptic components are observed, such as phosphorylation of synapsins.172,173 After days to weeks axons and dendrites change their shape.78,174,175 Experiments with knockout mice have demonstrated that in hippocampal neurons the activity of TrkB and BDNF are required for long term potentiation (LTP) of synapses, a molecular mechanism associated with memory formation.176,177 Furthermore, hippocampal mechanisms of pathological depression might be controlled by BDNF.178,179 In animal models of depression BDNF infusion had antidepressant effects.180 For cortical neurons it is predicted that their exposure to excess neurotrophins leads to more and stronger synapses, for example in GABA-ergic inhibitory neurons responding to BDNF (reviewed by ref. 181). These changes may result in unbalanced local neuronal circuitries in several brain areas including in particular the neocortex. Indeed, neurotrophins effectively change the induction phase of the kindling model of epilepsy.182–184
Stimulation of Non-Neuronal Cells
While neurotrophins exert their activity pre-dominantly on neurons, there are also effects described on non-neuronal cells. Schwann cells express large amounts of the p75 receptor and are known targets for neurotrophins. In particular, NGF activates the transcription factor NFκb in Schwann cells60 and leads to changes in Schwann cell migration.185 Following NGF treatment in vivo Schwann cell hyperplasia has been observed.186 Another glial target for neurotrophin actions are oligodendrocytes. NT3 is a mitogen for oligodendrocyte precursor187 and both BDNF and NT3 stimulate oligodendrocyte proliferation during axonal regeneration.188 In contrast, NGF kills cultured mature oligodendrocytes via a p75-mediated mechanism.63 A similar pro-apoptotic effect of NGF was also recently described for Schwann cells.189
In the immune system NGF is an autocrine survival factor for memory B cells190 and stimulates IgG4 production of human B cells.191 NGF also mediates profound anti-inflammatory effects on T cells and antigen presenting cells such as macrophages and microglia. Recently, we demonstrated that NGF suppresses the inducibility of MHC class II on microglial cells.192 Furthermore, NGF directly acts on monocytes and inhibits their trans-endothelium migration into the cerebrum.193 The role and the effect of NGF in experimental autoimmune encephalomyelitis were analyzed in several studies. Intraventricular application of NGF protected marmosets against EAE.194 T cells overexpressing NGF inhibited the clinical symptoms and reduced the infiltration of monocytes in EAE193 (Fig. 4).
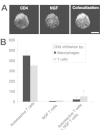
Another tissue responsive to neurotrophins is the cardiovascular system. Recently, BDNF was identified as a potent survival regulator of endothelial cells in the heart.195 Therefore its systemic application might change the properties of mature large vessels. We have recently analyzed the vascular system of p75 null mutant mice and found that the walls of the large elastic vessels in embryos lacking p75 are thinner compared to wild-type animals with a corresponding strong tendency to form hemorrhages (von Schack et al, unpublished observation). NT3 mutant mouse embryos display severe heart malformations.196 Neurotrophins and their receptors are expressed under pathophysiological conditions in adult blood vessels.197 Taken together these results strongly indicate a role for the neurotrophins in the development and maintenance of large blood vessels. Hence cardiovascular problems after systemic application of neurotrophins need to be considered as potential side effects.
Potential Improvements of Neurotrophin Therapy
The physiology of neurotrophins and their receptors is complex and, as outlined above, the list of observed and potential side effects is steadily growing. It appears that this complexity has been underestimated during the first clinical trials. At the same time it has become apparent that systemic neurotrophin treatment is not very effective. New methods of local application and means to control adverse side effects should be developed.
Modification of Neurotrophins and Their Pharmacology
The interactions between neurotrophins and their receptors can be modulated by the generation of mutant growth factors by recombinant DNA technology. In one approach mutated neurotrophin proteins have been developed that bind to multiple Trk receptors and to p75.198–200 Mutant growth factors with enhanced receptor specificity compared to the naturally occurring proteins have also been generated.201 In order to solve the problem of the short half-lives of neurotrophins in vivo, new agents are required which are more resistant to proteolytic degradation.202 These could, for example, consist of synthetic peptides or antibody agonists for neurotrophin receptors.203,204 The potential use of heteromeric neurotrophins combined from two different monomers has already been mentioned above.205 In addition, conjugation of neurotrophins with other proteins such as the transferrin receptor might enable their passage through the blood-brain barrier.206 However, the existing conjugates cross the blood-brain barrier only very inefficiently.
Stimulation of local endogenous neurotrophin synthesis or release with small molecular weight compounds has to be considered as an alternative to the more invasive application of exogenous growth factors (reviewed by ref. 207). Endogenous neurotrophin production and release is part of the response of the nervous system to injuries. In the brain, the mechanism that links neuronal activity to the expression of the BDNF gene has begun to be understood.208,209 However the neurotrophin genes have multiple promoters and are expressed in tissue specific transcripts indicating a complex network of gene regulatory mechanisms.210 Correspondingly multiplex regulatory mechanisms appear to determine the expression of the Trk receptor genes.211 One advantage of activating the endogenous neurotrophin receptor expression or activity might be the stimulation of autocrine loops. Improved responsiveness to neurotrophic factors can be expected to strengthen existing connections or to generate additional ones. This might lead to increased overall electric activity, which in turn stimulates neurons to produce and secrete endogenous neurotrophins by increasing intracellular Ca2+ levels.53 Consequently such an autocrine or paracrine cellular system, within limits, might stabilize itself.
Alternative Application Methods and Growth Factor Cocktails
Neurotrophins, like other large polypeptides, can only poorly cross the blood-brain barrier unless it breaks down in diseases such as ischemia.212 Alternative methods of neurotrophin application in the CNS must therefore be developed. Current approaches to solve this problem include gene therapy using different viral vectors such as adenoviruses, adeno-associated virus, and lentiviruses.213–215 Activated and genetically engineered T-cell lines, which secrete recombinant neurotrophins after transmigration into the brain parenchyma, have been experimentally used as therapeutic vehicles.216 For example, T cells retrovirally transduced with the NGF gene migrate into the brain of a rat with experimental autoimmune encephalomyelitis improving the disease and inhibiting immune cells infiltration into the brain tissue (Fig. 4). Other strategies include implantation of gelfoam pledgets from which neurotrophins are slowly released217,218 or insertion of small capsules containing neurotrophin-secreting cells.219,220
An important therapeutic concept for future studies will be to exploit synergistic mechanisms between the signaling cascades of neurotrophins and other growth factors. This appears to be particularly important for CNS neurons, which typically depend on more than one growth factor for their survival. In long term it seems likely that combinations of neurotrophins and other growth factor families with neurotrophic activity, such as IGFs, GDNF-like molecules, FGFs or neuropoietic cytokines, in particular CNTF, will be more effective than a single growth factor. Cocktails of growth factors can be designed in which each growth factor is present at suboptimal concentrations, which would limit receptor availability and consequently side effects. Such cocktails have already been shown to act synergistically on motoneurons.221-223 It might also turn out to be desirable to block adverse effects of endogenous cytokines, which act antagonistically to neurotrophins.224 Thus, blocking action of TNF or CD95 with neutralizing antibodies might be a useful therapeutic approach to support neurotrophin effects.
Neurotrophins and Cell Replacement Therapy
Apart from the immediate use of the neurotrophins as therapeutic agents, it is also likely that one or more of the neurotrophins will be an integral part of treatment protocols for stem cells with the aim to differentiate them into specific neuronal cell types for cell replacement therapy.225–227
From the rather disappointing results of the clinical trials performed so far the conclusion can be drawn, that systemic and probably also intraventricular infusion of neurotrophins are rather ineffective and prone to give rise to severe side effects. One alternative approach could be to combine cell replacement therapy with neurotrophin gene transfer into stem cells prior to transplantation.228,229 This approach appears particularly attractive since neurotrophins have the potential to not only prevent or slow down neurodegeneration, but also to foster the establishment of synaptic connections of stem cell derived neurons. Stem cells can be transplanted to defined brain structures and even more importantly they can be engineered to produce neurotrophin transgenes under exogenous control dependent on the absence or presence of small molecular weight substances such as doxycycline, ecdysone or tamoxifen.230 Such an approach would warrant localized expression and at the same time allow to interrupt the therapy as soon as side effects are observed. However, in light of the current excitement about the great potential of cell replacement therapies for neurodegenerative diseases, it is recommended to take a look back on the clinical trials with neurotrophins. They also have been prematurely advertised as miracle drugs. As desirable as it would be, it cannot reasonably be expected that cell replacement therapies will escape the phases of experimental problems and frustration that the neurotrophins went through.
Acknowledgments
The authors are grateful to Dr. Alexander Flügel, Dr. Hans Lassmann and Ingeborg Haarmann for help in the experiments with GFP transduced T cells. We thank Dr. Hartmut Wekerle and Dr. Yves-Alain Barde for continuous support and Dr. Hans Thoenen and Dr. Michael Sendtner for their helpful comments on the manuscript.
References
- 1.
- Gage FH. Mammalian neural stem cells. Science. 2000;287:1433–1438. [PubMed: 10688783]
- 2.
- Barde Y -A. The Nerve Growth Factor Family. Prog Growth Factor Res. 1990;2:237–348. [PubMed: 2133291]
- 3.
- Bibel M, Barde Y -A. Neurotrophins: Key regulators of cell fate and cell shape in the vertebrate nervous system. Genes Dev. 2000;14:2919–2937. [PubMed: 11114882]
- 4.
- Thoenen H. Treatment of degenerative disorders of the nervous system: From helpless descriptive categorization to rational therapeutic approaches In: Ingoglia NA, Murray N, eds. Axonal Regeneration in the Central Nervous System New York: Dekker M,2001675–697.
- 5.
- Goyal L. Cell death inhibition: keeping caspases in check. Cell. 2001;104:805–808. [PubMed: 11290317]
- 6.
- McDonald NQ, Lapatto R, Murray-Rust J. et al. New protein fold revealed by a 2.3-Å resolution crystal structure of nerve growth factor. Nature. 1991;354:411–414. [PubMed: 1956407]
- 7.
- McDonald NQ, Chao MV. Structural determinants of neurotrophin action. J Biol Chem. 1995;270:19669–19672. [PubMed: 7649974]
- 8.
- Lewin GR, Barde YA. Physiology of the Neurotrophins. Annu Rev Neurosci. 1996;19:289–317. [PubMed: 8833445]
- 9.
- Tria MA, Fusco M, Vantini G. et al. Pharmacokinetics of nerve growth factor (NGF) following different routes of administration to adult rats. Exp Neurol. 1994;127:178–183. [PubMed: 8033961]
- 10.
- Yan Q, Matheson C, Sun J. et al. Distribution of intracerebral ventricularly administered neurotrophins in rat brain and its correlation with trk receptor expression. Exp Neurol. 1994;127:23–36. [PubMed: 8200435]
- 11.
- Dittrich F, Ochs G, Grosse-Wilde A. et al. Pharmacokinetics of intrathecally applied BDNF and effects on spinal motoneurons. Exp Neurol. 1996;141:225–239. [PubMed: 8812156]
- 12.
- Radziejewski C, Robinson RC. Heterodimers of the neurotrophic factors: Formation, isolation, and differential stability. Biochemistry. 1993;32:13350–13356. [PubMed: 8241191]
- 13.
- Arakawa T, Haniu M, Narhi LO. et al. Formation of heterodimers from three neurotrophins, nerve growth factor, neurotrophin-3, and brain-derived neurotrophic factor. J Biol Chem. 1994;269:27833–27839. [PubMed: 7961712]
- 14.
- Thoenen H. Neurotrophins and activity-dependent plasticity. Prog Brain Res. 2000;128:183–191. [PubMed: 11105678]
- 15.
- Barbacid M. The Trk family of neurotrophin receptors. J Neurobiol. 1994;25:1386–1403. [PubMed: 7852993]
- 16.
- Grell M, Clauss M. TNF and TNF receptor superfamily In: Bona CA, Revillard J-P, eds. Cytokines and Cytokine Receptors New York: Harwood Academic Press, 2000118–148.
- 17.
- Dechant G. Molecular interactions between neurotrophin receptors Cell Tissue Res 2001. (online version http://dx.doi.org/10.1007/s004410100378) [PubMed: 11545260]
- 18.
- Dechant G, Rodríguez-Tébar A, Barde Y -A. Neurotrophin Receptors. Prog Neurobiol. 1994;42:347–352. [PubMed: 8008834]
- 19.
- Davies AM. Neurotrophins: The yin and yang of nerve growth factor. Curr Biol. 1997;7:R38–R40. [PubMed: 9072168]
- 20.
- Snider WD. Functions of the neurotrophins during nervous system development: What the knockouts are teaching us. Cell. 1994;77:627–638. [PubMed: 8205613]
- 21.
- Holtzman DM, Li Y, Parada LF. et al. p140trk mRNA marks NGF-responsive forebrain neurons: evidence that trk gene expression is induced by NGF. Neuron. 1992;9:465–478. [PubMed: 1524827]
- 22.
- Hefti F. Nerve growth factor promotes survival of septal cholinergic neurons after fimbrial transections. J Neurosci. 1986;6:2155–2162. [PMC free article: PMC6568758] [PubMed: 3746405]
- 23.
- Klein R, Martin-Zanca D, Barbacid M. et al. Expression of the tyrosine kinase receptor gene TrkB is confined to the murine embryonic and adult nervous system. Development. 1990;109:845–850. [PubMed: 2171894]
- 24.
- Klein R, Conway D, Parada LF. et al. The TrkB tyrosine protein kinase gene codes for a second neurogenic receptor that lacks the catalytic kinase domain. Cell. 1990;61:647–656. [PubMed: 2160854]
- 25.
- Middlemas DS, Lindberg RA, Hunter T. trk B, a neural receptor protein-tyrosine kinase: evidence for a full-length and two truncated receptors. Mol Cell Biol. 1991;11:143–153. [PMC free article: PMC359604] [PubMed: 1846020]
- 26.
- Tessarollo L, TsoulFas P, Martin-Zanca D. et al. TrkC, a receptor for neurotrophin-3, is widely expressed in the developing nervous system and in non-neuronal tissues. Development. 1993;118:463–475. [PubMed: 8223273]
- 27.
- Barker PA. p75NTR: A study in contrasts [see comments] Cell Death Differ. 1998;5:346–356. [PubMed: 10200483]
- 28.
- Dobrowsky RT, Carter BD. p75 neurotrophin receptor signaling: Mechanisms for neurotrophic modulation of cell stress? J Neurosci Res. 2000;61:237–243. [PubMed: 10900070]
- 29.
- Frade JM, Barde YA. Nerve growth factor: Two receptors, multiple functions. BioEssays. 1998;20:137–145. [PubMed: 9631659]
- 30.
- Yamashita T, Tucker KL, Barde YA. Neurotrophin binding to the p75 receptor modulates Rho activity and axonal outgrowth. Neuron. 1999;24:585–593. [PubMed: 10595511]
- 31.
- Bibel M, Hoppe E, Barde YA. Biochemical and functional interactions between the neurotrophin receptors trk and p75NTR. EMBO J. 1999;18:616–622. [PMC free article: PMC1171154] [PubMed: 9927421]
- 32.
- Majdan M, Miller FD. Neuronal life and death decisions functional antagonism between the Trk and p75 neurotrophin receptors. Int J Dev Neurosci. 1999;17:153–161. [PubMed: 10452359]
- 33.
- Vejsada R, Sagot Y, Kato AC. BDNF-mediated rescue of axotomized motor neurones decreases with increasing dose. NeuroReport. 1994;5:1889–1892. [PubMed: 7841370]
- 34.
- Vejsada R, Sagot Y, Kato AC. Quantitative comparison of the transient rescue effects of neurotrophic factors on axotomized motoneurons in vivo. Eur J Neurosci. 1995;7:108–115. [PubMed: 7711927]
- 35.
- Sommerfeld MT, Schweigreiter R, Barde YA. et al. Down-regulation of the neurotrophin receptor TrkB following ligand binding. Evidence for an involvement of the proteasome and differential regulation of TrkA and TrkB. J Biol Chem. 2000;275:8982–8990. [PubMed: 10722747]
- 36.
- Carter BD, Zirrgiebel U, Barde Y -A. Differential regulation of p21ras activation in neurons by nerve growth factor and brain-derived neurotrophic factor. J Biol Chem. 1995;270:21751–21757. [PubMed: 7665594]
- 37.
- Frank L, Ventimiglia R, Anderson K. et al. BDNF down-regulates neurotrophin responsiveness, TrkB protein and TrkB mRNA levels in cultured rat hippocampal neurons. Eur J Neurosci. 1996;8:1220–1230. [PubMed: 8752592]
- 38.
- Widmer HR, Ohsawa F, Knusel B. et al. Down-regulation of phosphatidylinositol response to BDNF and NT-3 in cultures of cortical neurons. Brain Res. 1993;614:325–334. [PubMed: 8348325]
- 39.
- Knusel B, Kaplan DR, Hefti F. Intraparenchymal NGF injections in adult and aged rats induce long-lasting Trk tyrosine phosphorylation. Exp Neurol. 1996;139:121–130. [PubMed: 8635559]
- 40.
- Biffo S, Offenhäuser N, Carter BD. et al. Selective binding and internalisation by truncated receptors restrict the availability of BDNF during development. Development. 1995;121:2461–2470. [PubMed: 7671810]
- 41.
- Von Bartheld CS, Kinoshita Y, Prevette D. et al. Positive and negative effects of neurotrophins on the isthmo-optic nucleus in chick embryos. Neuron. 1994;12:639–654. [PubMed: 8155324]
- 42.
- Wiese S, Metzger F, Holtmann B. et al. The role of p75NTR in modulating neurotrophin survival effects in developing motoneurons. Eur J Neurosci. 1999;11:1668–1676. [PubMed: 10215920]
- 43.
- Klocker N, Cellerino A, Bahr M. Free radical scavenging and inhibition of nitric oxide synthase potentiates the neurotrophic effects of brain-derived neurotrophic factor on axotomized retinal ganglion cells In vivo. J Neurosci. 1998;18:1038–1046. [PMC free article: PMC6792783] [PubMed: 9437024]
- 44.
- Davies AM, Minichiello L, Klein R. Developmental changes in NT3 signalling via TrkA and TrkB in embryonic neurons. EMBO J. 1995;14:4482–4489. [PMC free article: PMC394540] [PubMed: 7556091]
- 45.
- Wyatt S, Piñón L G P, Ernfors P. et al. Sympathetic neuron survival and TrkA expression in NT3-deficient mouse embryos. EMBO J. 1997;16:3115–3123. [PMC free article: PMC1169930] [PubMed: 9214629]
- 46.
- Friedman WJ, Greene LA. Neurotrophin signaling via Trks and p75. Exp Cell Res. 1999;253:131–142. [PubMed: 10579918]
- 47.
- Yuen EC, Mobley WC. Early BDNF, NT-3, and NT-4 signaling events. Exp Neurol. 1999;159:297–308. [PubMed: 10486198]
- 48.
- Cunningham ME, Greene LA. A function-structure model for NGF-activated TRK. EMBO J. 1998;17:7282–7293. [PMC free article: PMC1171074] [PubMed: 9857185]
- 49.
- Kaplan DR, Miller FD. Signal transduction by the neurotrophin receptors. Curr Opin Cell Biol. 1997;9:213–221. [PubMed: 9069267]
- 50.
- Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. [PubMed: 10579998]
- 51.
- Holgado-Madruga M, Moscatello DK, Emlet DR. et al. Grb2-associated binder-1 mediates phosphatidylinositol 3-kinase activation and the promotion of cell survival by nerve growth factor. Proc Natl Acad Sci USA. 1997;94:12419–12424. [PMC free article: PMC24976] [PubMed: 9356464]
- 52.
- Yamada M, Ohnishi H, Sano S. et al. Insulin receptor substrate (IRS)-1 and IRS-2 are tyrosine-phosphorylated and associated with phosphatidylinositol 3-kinase in response to brain-derived neurotrophic factor in cultured cerebral cortical neurons. J Biol Chem. 1997;272:30334–30339. [PubMed: 9374521]
- 53.
- Canossa M, Gartner A, Campana G. et al. Regulated secretion of neurotrophins by metabotropic glutamate group I (mGluRI) and Trk receptor activation is mediated via phospholipase C signalling pathways. EMBO J. 2001;20:1640–1650. [PMC free article: PMC145457] [PubMed: 11285228]
- 54.
- Maderdrut JL, Oppenheim RW, Prevette D. Enhancement of naturally occurring cell death in the sympathetic and parasympathetic ganglia of the chicken embryo following blockade of ganglionic transmission. Brain Res. 1988;444:189–194. [PubMed: 2834023]
- 55.
- Ghosh A, Carnahan J, Greenberg ME. Requirement for BDNF in activity-dependent survival of cortical neurons. Science. 1994;263:1618–1623. [PubMed: 7907431]
- 56.
- McAllister AK, Katz LC, Lo DC. Neurotrophin regulation of cortical dendritic growth requires activity. Neuron. 1996;17:1057–1064. [PubMed: 8982155]
- 57.
- Brodski C, Schnürch H, Dechant G. Neurotrophin-3 promotes the cholinergic differentiation of sympathetic neurons. Proc Natl Acad Sci USA. 2000;97:9683–9688. [PMC free article: PMC16925] [PubMed: 10931939]
- 58.
- Dobrowsky RT, Jenkins GM, Hannun YA. Neurotrophins induce sphingomyelin hydrolysisModulation by Co- expression of p75NTR with Trk receptors. J Biol Chem. 1995;270:22135–22142. [PubMed: 7673191]
- 59.
- Dobrowsky RT, Carter BD. Coupling of the p75 neurotrophin receptor to sphingolipid signaling. Ann NY Acad Sci. 1998;845:32–45. [PubMed: 9668341]
- 60.
- Carter BD, Kaltschmidt C, Kaltschmidt B. et al. Selective activation of NF-kappaB by nerve growth factor through the neurotrophin receptor p75. Science. 1996;272:542–545. [PubMed: 8614802]
- 61.
- Hamanoue M, Middleton G, Wyatt S. et al. p75-mediated NF-kappaB activation enhances the survival response of developing sensory neurons to nerve growth factor. Mol Cell Neurosci. 1999;14:28–40. [PubMed: 10433815]
- 62.
- Goldhawk DE, Meakin SO, Verdi JM. Subpopulations of rat B2 neuroblasts exhibit differential neurotrophin responsiveness during sympathetic development. Dev Biol. 2000;218:367–377. [PubMed: 10656776]
- 63.
- Casaccia-Bonnefil P, Carter BD, Dobrowsky RT. et al. Death of oligodendrocytes mediated by the interaction of nerve growth factor with its receptor p75. Nature. 1996;383:716–719. [PubMed: 8878481]
- 64.
- Frade JM, Rodríguez-Tébar A, Barde YA. Induction of cell death by endogenous nerve growth factor through its p75 receptor. Nature. 1996;383:166–168. [PubMed: 8774880]
- 65.
- Agerman K, Baudet C, Fundin B. et al. Attenuation of a caspase-3-dependent cell death in NT4- and p75-deficient embryonic sensory neurons. Mol Cell Neurosci. 2000;16:258–268. [PubMed: 10995552]
- 66.
- Yoon SO, Casaccia-Bonnefil P, Carter B. et al. Competitive signaling between TrkA and p75 nerve growth factor receptors determines cell survival. J Neurosci. 1998;18:3273–3281. [PMC free article: PMC6792655] [PubMed: 9547236]
- 67.
- Aloyz RS, Bamji SX, Pozniak CD. et al. P53 is essential for developmental neuron death as regulated by the TrkA and p75 neurotrophin receptors. J Cell Biol. 1998;143:1691–1703. [PMC free article: PMC2132983] [PubMed: 9852160]
- 68.
- Pozniak CD, Radinovic S, Yang A. et al. An anti-apoptotic role for the p53 family member, p73, during developmental neuron death. Science. 2000;289:304–306. [PubMed: 10894779]
- 69.
- Budd SL, Nicholls DG. Mitochondria in the life and death of neurons. Essays Biochem. 1998;33:43–52. [PubMed: 10488440]
- 70.
- Martin LJ. Neuronal cell death in nervous system development, disease, and injury. Int J Mol Med. 2001;7:455–478. [PubMed: 11295106]
- 71.
- Dugan LL, Creedon DJ, Johnson E M Jr. et al. Rapid suppression of free radical formation by nerve growth factor involves the mitogen-activated protein kinase pathway. Proc Natl Acad Sci USA. 1997;94:4086–4091. [PMC free article: PMC20572] [PubMed: 9108109]
- 72.
- Koh JY, Gwag BJ, Lobner D. et al. Potentiated necrosis of cultured cortical neurons by neurotrophins. Science. 1995;268:573–575. [PubMed: 7725105]
- 73.
- Davies AM. The Bcl-2 family of proteins, and the regulation of neuronal survival. Trends Neurosci. 1995;18:355–358. [PubMed: 7482798]
- 74.
- Martin LJ. Neuronal death in amyotrophic lateral sclerosis is apoptosis: Possible contribution of a programmed cell death mechanism. J Neuropathol Exp Neurol. 1999;58:459–471. [PubMed: 10331434]
- 75.
- Lindholm D. Role of neurotrophins in preventing glutamate induced neuronal cell death. J Neurol. 1994;242:S16–S18. [PubMed: 7699402]
- 76.
- Sagot Y, Dubois-Dauphin M, Tan SA. et al. Bcl-2 overexpression prevents motoneuron cell body loss but not axonal degeneration in a mouse model of a neurodegenerative disease. J Neurosci. 1995;15:7727–7733. [PMC free article: PMC6578059] [PubMed: 7472523]
- 77.
- Patel TD, Jackman A, Rice FL. et al. Development of sensory neurons in the absence of NGF/TrkA signaling in vivo. Neuron. 2000;25:345–357. [PubMed: 10719890]
- 78.
- McAllister AK, Katz LC, Lo DC. Opposing roles for endogenous BDNF and NT-3 in regulating cortical dendritic growth. Neuron. 1997;18:767–778. [PubMed: 9182801]
- 79.
- Leingärtner A, Heisenberg C -P, Kolbeck R. et al. Brain-derived neurotrophic factor increases neurotrophin-3 expression in cerebellar granule neurons. J Biol Chem. 1994;269:828–830. [PubMed: 8288635]
- 80.
- Canossa M, Griesbeck O, Berninger B. et al. Neurotrophin release by neurotrophins: Implications for activity-dependent neuronal plasticity. Proc Natl Acad Sci USA. 1997;94:13279–13286. [PMC free article: PMC24300] [PubMed: 9371837]
- 81.
- Krüttgen A, Möller JC, Heymach J V Jr. et al. Neurotrophins induce release of neurotrophins by the regulated secretory pathway. Proc Natl Acad Sci USA. 1998;95:9614–9619. [PMC free article: PMC21387] [PubMed: 9689129]
- 82.
- Causing CG, Gloster A, Aloyz R. et al. Synaptic innervation density is regulated by neuron-derived BDNF. Neuron. 1997;18:257–267. [PubMed: 9052796]
- 83.
- Giehl KM. Trophic dependencies of rodent corticospinal neurons. Rev Neurosci. 2001;12:79–94. [PubMed: 11236067]
- 84.
- Merlio JP, Ernfors P, Kokaia Z. et al. Increased production of the TrkB protein tyrosine kinase receptor after brain insults. Neuron. 1993;10:151–164. [PubMed: 8439408]
- 85.
- Kokaia Z, Andsberg G, Yan Q. et al. Rapid alterations of BDNF protein levels in the rat brain after focal ischemia: Evidence for increased synthesis and anterograde axonal transport. Exp Neurol. 1998;154:289–301. [PubMed: 9878168]
- 86.
- Beck T, Lindholm D, Castren E. et al. Brain-derived neurotrophic factor protects against ischemic cell damage in rat hippocampus. J Cereb Blood Flow Metab. 1994;14:689–692. [PubMed: 8014217]
- 87.
- Tsukahara T, Yonekawa Y, Tanaka K. et al. The role of brain-derived neurotrophic factor in transient forebrain ischemia in the rat brain. Neurosurgery. 1994;34:323–331. [PubMed: 8177394]
- 88.
- Chan KM, Lam DT, Pong K. et al. Neurotrophin-4/5 treatment reduces infarct size in rats with middle cerebral artery occlusion. Neurochem Res. 1996;21:763–767. [PubMed: 8873080]
- 89.
- Gall C, Lauterborn J, Bundman M. et al. Seizures and the regulation of neurotrophic factor and neuropeptide gene expression in brain. Epilepsy Res Suppl. 1991;4:225–245. [PubMed: 1815605]
- 90.
- Ernfors P, Bengzon J, Kokaia Z. et al. Increased levels of messenger RNAs for neurotrophic factors in the brain during kindling epileptogenesis. Neuron. 1991;7:165–176. [PubMed: 1829904]
- 91.
- Mudo G, Jiang XH, Timmusk t. et al. Change in neurotrophins and their receptor mRNAs in the rat forebrain after status epilepticus induced by pilocarpine. Epilepsia. 1996;37:198–207. [PubMed: 8635431]
- 92.
- Binder DK, Routbort MJ, Ryan TE. et al. Selective inhibition of kindling development by intraventricular administration of TrkB receptor body. J Neurosci. 1999;19:1424–1436. [PMC free article: PMC6786043] [PubMed: 9952419]
- 93.
- Giehl KM, Tetzlaff W. BDNF and NT-3, but not NGF, prevent axotomy-induced death of rat corticospinal neurons in vivo. Eur J Neurosci. 1996;8:1167–1175. [PubMed: 8752586]
- 94.
- Schnell L, Schneider R, Kolbeck R. et al. Neurotrophin-3 enhances sprouting of corticospinal tract during development and after adult spinal cord lesion. Nature. 1994;367:170–173. [PubMed: 8114912]
- 95.
- Hefti F, Weiner WJ. Nerve growth factor and Alzheimer's disease. Ann Neurol. 1986;20:275–281. [PubMed: 3532929]
- 96.
- Vazquez ME, Ebendal T. Messenger RNAs for trk and the low-affinity NGF receptor in rat basal forebrain. NeuroReport. 1991;2:593–596. [PubMed: 1661622]
- 97.
- Venero JL, Hefti F. TrkA NGF receptor expression by non-cholinergic thalamic neurons. NeuroReport. 1993;4:959–962. [PubMed: 8369489]
- 98.
- Hefti F, Lapchak PA. Pharmacology of nerve growth factor in the brain. Adv Pharmacol. 1993;24:239–273. [PubMed: 8504065]
- 99.
- Will B, Hefti F. Behavioural and neurochemical effects of chronic intraventricular injections of nerve growth factor in adult rats with fimbria lesions. Behav Brain Res. 1985;17:17–24. [PubMed: 4041219]
- 100.
- Fischer W, Wictorin K, Bjoerklund A. et al. Amelioration of cholinergic neuron atrophy and spatial memory impairment in aged rats by nerve growth factor. Nature. 1987;329:65–68. [PubMed: 3627243]
- 101.
- Markowska AL, Koliatsos VE, Breckler SJ. et al. Human nerve growth factor improves spatial memory in aged but not in young rats. J Neurosci. 1994;14:4815–4824. [PMC free article: PMC6577162] [PubMed: 8046452]
- 102.
- Widmer HR, Knusel B, Hefti F. BDNF protection of basal forebrain cholinergic neurons after axotomy: complete protection of p75NGFR-positive cells. NeuroReport. 1993;4:363–366. [PubMed: 8499589]
- 103.
- Koliatsos VE, Price DL, Gouras GK. et al. Highly selective effects of nerve growth factor, brain-derived neurotrophic factor, and neurotrophin-3 on intact and injured basal forebrain magnocellular neurons. J Comp Neurol. 1994;343:247–262. [PubMed: 8027442]
- 104.
- Fischer W, Sirevaag A, Wiegand SJ. et al. Reversal of spatial memory impairments in aged rats by nerve growth factor and neurotrophins 3 and 4/5 but not by brain-derived neurotrophic factor. Proc Natl Acad Sci USA. 1994;91:8607–8611. [PMC free article: PMC44655] [PubMed: 8078930]
- 105.
- Knüsel B, Winslow JW, Rosenthal A. et al. Promotion of central cholinergic and dopaminergic neuron differentiation by brain-derived neurotrophic factor but not neurotrophin 3. Proc Natl Acad Sci USA. 1991;88:961–965. [PMC free article: PMC50934] [PubMed: 1992488]
- 106.
- Alderson RF, Wiegand SJ, Anderson KD. et al. Neurotrophin-4/5 maintains the cholinergic phenotype of axotomized septal neurons. Eur J Neurosci. 1996;8:282–290. [PubMed: 8714699]
- 107.
- Phillips HS, Hains JM, Armanini M. et al. BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer's disease. Neuron. 1991;7:695–702. [PubMed: 1742020]
- 108.
- Peterson DA, Leppert JT, Lee KF. et al. Basal forebrain neuronal loss in mice lacking neurotrophin receptor p75. Science. 1997;277:837–838. [PubMed: 9273702]
- 109.
- Peterson DA, Leppert JT, Lee KF, Gage FH. et al. Basal forebrain neuronal loss in mice lacking neurotrophin receptor p75. Science. 1997;277:837–839. [PubMed: 9273702]
- 110.
- Olson L, Nordberg A, von Holst H. et al. Nerve growth factor affects 11C-nicotine binding, blood flow, EEG, and verbal episodic memory in an Alzheimer patient (case report). J Neural Transm Park Dis Dement Sect. 1992;4:79–95. [PubMed: 1540306]
- 111.
- Eriksdotter JM, Nordberg A, Amberla K. et al. Intracerebroventricular infusion of nerve growth factor in three patients with Alzheimer's disease. Dement Geriatr Cogn Disord. 1998;9:246–257. [PubMed: 9701676]
- 112.
- Hyman C, Hofer M, Barde Y -A. et al. BDNF is a neurotrophic factor for dopaminergic neurons of the substantia nigra. Nature. 1991;350:230–232. [PubMed: 2005978]
- 113.
- Spina MB, Squinto SP, Miller J. et al. Brain-derived neurotrophic factor protects dopamine neurons against 6-hydroxydopamine and N-methyl-4-phenylpyridinium ion toxicity: Involvement of the glutathione system. J Neurochem. 1992;59:99–106. [PubMed: 1613515]
- 114.
- Knusel B, Beck KD, Winslow JW. et al. Brain-derived neurotrophic factor administration protects basal forebrain cholinergic but not nigral dopaminergic neurons from degenerative changes after axotomy in the adult rat brain. J Neurosci. 1992;12:4391–4402. [PMC free article: PMC6576000] [PubMed: 1432101]
- 115.
- Hagg T. Neurotrophins prevent death and differentially affect tyrosine hydroxylase of adult rat nigrostriatal neurons in vivo. Exp Neurol. 1998;149:183–192. [PubMed: 9454627]
- 116.
- Altar CA, Boylan CB, Jackson C. et al. Brain-derived neurotrophic factor augments rotational behavior and nigrostriatal dopamine turnover in vivo. Proc Natl Acad Sci USA. 1992;89:11347–11351. [PMC free article: PMC50547] [PubMed: 1454818]
- 117.
- Lapchak PA, Beck KD, Araujo DM. et al. Chronic intranigral administration of brain-derived neurotrophic factor produces striatal dopaminergic hypofunction in unlesioned adult rats and fails to attenuate the decline of striatal dopaminergic function following medial forebrain bundle transection. Neuroscience. 1993;53:639–650. [PubMed: 8098137]
- 118.
- Ringstedt T, Lagercrantz H, Persson H. Expression of members of the trk family in the developing postnatal rat brain. Brain Res Dev Brain Res. 1993;72:119–131. [PubMed: 8453762]
- 119.
- Numan S, Seroogy KB. Expression of TrkB and TrkC mRNAs by adult midbrain dopamine neurons: A double-label in situ hybridization study. J Comp Neurol. 1999;403:295–308. [PubMed: 9886032]
- 120.
- Koliatsos VE, Clatterbuck RE, Winslow JW. et al. Evidence that brain-derived neurotrophic factor is a trophic factor for motor neurons in vivo. Neuron. 1993;10:359–367. [PubMed: 8080464]
- 121.
- Klein R, Smeyne RJ, Wurst W. et al. Targeted disruption of the TrkB neurotrophin receptor gene results in nervous system lesions and neonatal death. Cell. 1993;75:113–122. [PubMed: 8402890]
- 122.
- Liu X, Ernfors P, Wu H. et al. Sensory but not motor neuron deficits in mice lacking NT4 and BDNF. Nature. 1995;375:238–241. [PubMed: 7746325]
- 123.
- Conover JC, Erickson JT, Katz DM. et al. Neuronal deficits, not involving motor neurons, in mice lacking BDNF and/or NT4. Nature. 1995;375:235–238. [PubMed: 7746324]
- 124.
- Kobayashi NR, Bedard AM, Hincke MT. et al. Increased expression of BDNF and TrkB mRNA in rat facial motoneurons after axotomy. Eur J Neurosci. 1996;8:1018–1029. [PubMed: 8743749]
- 125.
- Sendtner M, Holtmann B, Hughes RA. The response of motoneurons to neurotrophins. Neurochem Res. 1996;21:831–841. [PubMed: 8873088]
- 126.
- Sendtner M, Holtmann B, Kolbeck R. et al. Brain-derived neurotrophic factor prevents the death of motoneurons in newborn rats after nerve section. Nature. 1992;360:757–759. [PubMed: 1465147]
- 127.
- Yan Q, Elliott J, Snider WD. Brain-derived neurotrophic factor rescues spinal motor neurons from axotomy-induced cell death. Nature. 1992;360:753–755. [PubMed: 1281520]
- 128.
- Johnson JE, Qin-Wei Y, Prevette D. et al. Brain-derived proteins that rescue spinal motoneurons from cell death in the chick embryo: Comparisons with target-derived and recombinant factors. J Neurobiol. 1995;27:573–589. [PubMed: 7561835]
- 129.
- Yan Q, Matheson C, Lopez OT. et al. The biological responses of axotomized adult motoneurons to brain-derived neurotrophic factor. J Neurosci. 1994;14:5281–5291. [PMC free article: PMC6577076] [PubMed: 8083736]
- 130.
- Ernfors P, Henschen A, Olson L. et al. Expression of nerve growth factor receptor mRNA is developmentally regulated and increased after axotomy in rat spinal cord motoneurons. Neuron. 1989;2:1605–1613. [PubMed: 2560649]
- 131.
- Koliatsos VE, Crawford TO, Price DL. Axotomy induces nerve growth factor receptor immunoreactivity in spinal motor neurons. Brain Res. 1991;549:297–304. [PubMed: 1653083]
- 132.
- Frade JM, Barde YA. Genetic evidence for cell death mediated by nerve growth factor and the neurotrophin receptor p75 in the developing mouse retina and spinal cord. Development. 1999;126:683–690. [PubMed: 9895316]
- 133.
- Ferri CC, Moore FA, Bisby MA. Effects of facial nerve injury on mouse motoneurons lacking the p75 low-affinity neurotrophin receptor. J Neurobiol. 1998;34:1–9. [PubMed: 9469614]
- 134.
- Yan Q, Miller JA. The use of trophic factors in degenerative motoneuron diseases. Exp Neurol. 1993;124:60–63. [PubMed: 8282082]
- 135.
- Stoeckel K, Guroff G, Schwab M. et al. The significance of retrograde axonal transport for the accumulation of systemically administered nerve growth factor (NGF) in the rat superior cervical ganglion. Brain Res. 1976;109:271–284. [PubMed: 58700]
- 136.
- Curtis R, Tonra JR, Stark JL. et al. Neuronal injury increases retrograde axonal transport of the neurotrophins to spinal sensory neurons and motor neurons via multiple receptor mechanisms. Mol Cell Neurosci. 1998;12:105–118. [PubMed: 9790733]
- 137.
- Ochs G, Penn RD, Giess R. et al. A trial of recombinant methionyl human Brain-Derived Neurotrophic Factor (BDNF) given by intrathecal (IT) infusion to patients with Amyotrophic Lateral Sclerosis (ALS). Neurology. 1998:A246. [PubMed: 11464953]
- 138.
- Phillips HS, Armanini MP. Expression of the trk family of neurotrophin receptors in developing and adult dorsal root ganglion neurons. Philos Trans R Soc Lond B Biol Sci. 1996;351:413–416. [PubMed: 8730779]
- 139.
- Karchewski LA, Kim FA, Johnston J. et al. Anatomical evidence supporting the potential for modulation by multiple neurotrophins in the majority of adult lumbar sensory neurons. J Comp Neurol. 1999;413:327–341. [PubMed: 10524342]
- 140.
- Lindsay RM. Nerve growth factors (NGF, BDNF) enhance axonal regeneration but are not required for survival of adult sensory neurons. J Neurosci. 1988;8:2394–2405. [PMC free article: PMC6569525] [PubMed: 3249232]
- 141.
- Lindsay RM, Harmar AJ. Nerve growth factor regulates expression of neuropeptide genes in adult sensory neurons. Nature. 1989;337:362–364. [PubMed: 2911387]
- 142.
- Mendell LM. Neurotrophin action on sensory neurons in adults: an extension of the neurotrophic hypothesis. Pain. 1999;Suppl 6:S127–S132. [PubMed: 10491981]
- 143.
- Verge VM, Gratto KA, Karchewski LA. et al. Neurotrophins and nerve injury in the adult. Philos Trans R Soc Lond B Biol Sci. 1996;351:423–430. [PubMed: 8730781]
- 144.
- Anand P. Neurotrophins and peripheral neuropathy. Philos Trans R Soc Lond B Biol Sci. 1996;351:449–454. [PubMed: 8730784]
- 145.
- Lewin GR. Neurotrophins and the specification of neuronal phenotype. Philos Trans R Soc Lond B Biol Sci. 1996;351:405–411. [PubMed: 8730778]
- 146.
- Gao WQ, Dybdal N, Shinsky N. et al. Neurotrophin-3 reverses experimental cisplatin-induced peripheral sensory neuropathy. Ann Neurol. 1995;38:30–37. [PubMed: 7611721]
- 147.
- Apfel SC, Arezzo JC, Lipson L. et al. Nerve growth factor prevents experimental cisplatin neuropathy. Ann Neurol. 1992;31:76–80. [PubMed: 1543351]
- 148.
- Ernfors P, Van De WT, Loring J. et al. Complementary roles of BDNF and NT-3 in vestibular and auditory development. Neuron. 1995;14:1153–1164. [PubMed: 7605630]
- 149.
- Ernfors P, Duan ML, Elshamy WM. et al. Protection of auditory neurons from aminoglycoside toxicity by neurotrophin-3. Nat Med. 1996;2:463–467. [PubMed: 8597959]
- 150.
- Staecker H, Kopke R, Malgrange B. et al. NT-3 and/or BDNF therapy prevents loss of auditory neurons following loss of hair cells. NeuroReport. 1996;7:889–894. [PubMed: 8724667]
- 151.
- Chalazonitis A, Rothman TP, Chen J. et al. Neurotrophin-3 induces neural crest-derived cells from fetal rat gut to develop in vitro as neurons or glia. J Neurosci. 1994;14:6571–6584. [PMC free article: PMC6577287] [PubMed: 7965061]
- 152.
- Sternini C, Su D, Arakawa J. et al. Cellular localization of Pan-trk immunoreactivity and TrkC mRNA in the enteric nervous system. J Comp Neurol. 1996;368:597–607. [PubMed: 8744446]
- 153.
- McArthur JC, Yiannoutsos C, Simpson DM. et al. A phase II trial of nerve growth factor for sensory neuropathy associated with HIV infection. AIDS Clinical Trials Group Team 291. Neurology. 2000;54:1080–1088. [PubMed: 10720278]
- 154.
- Apfel SC, Kessler JA, Adornato BT. et al. Recombinant human nerve growth factor in the treatment of diabetic polyneuropathy. NGF Study Group. Neurology. 1998;51:695–702. [PubMed: 9748012]
- 155.
- Apfel SC, Schwartz S, Adornato BT. et al. Efficacy and safety of recombinant human nerve growth factor in patients with diabetic polyneuropathy: A randomized controlled trial. rhNGF Clinical Investigator Group. JAMA. 2000;284:2215–2221. [PubMed: 11056593]
- 156.
- Petty BG, Cornblath DR, Adornato BT. et al. The effect of systemically administered recombinant human nerve growth factor in healthy human subjects. Ann Neurol. 1994;36:244–246. [PubMed: 8053664]
- 157.
- Mazurek N, Weskamp G, Erne P. et al. Nerve growth factor induces mast cell degranulation without changing intracellular calcium levels. FEBS Lett. 1986;198:315–320. [PubMed: 2420641]
- 158.
- Chuang H, Prescott ED, Kong H. et al. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature. 2001;411:957–962. [PubMed: 11418861]
- 159.
- Lambiase A, Rama P, Bonini S. et al. Topical treatment with nerve growth factor for corneal neurotrophic ulcers. N Engl J Med. 1998;338:1174–1180. [PubMed: 9554857]
- 160.
- Tuveri M, Generini S, Matucci-Cerinic M. et al. NGF, a useful tool in the treatment of chronic vasculitic ulcers in rheumatoid arthritis. Lancet. 2000;356:1739–1740. [PubMed: 11095266]
- 161.
- Siuciak JA, Clark MS, Rind HB. et al. BDNF induction of tryptophan hydroxylase mRNA levels in the rat brain. J Neurosci Res. 1998;52:149–158. [PubMed: 9579405]
- 162.
- Galter D, Unsicker K. Sequential activation of the 5-HT1A serotonin receptor and TrkB induces the serotonergic neuronal phenotype. Mol Cell Neurosci. 2000;15:446–455. [PubMed: 10833301]
- 163.
- Arenas E, Persson H. Neurotrophin-3 prevents the death of adult central noradrenergic neurons in vivo. Nature. 1994;367:368–371. [PubMed: 8114936]
- 164.
- Pelleymounter MA, Cullen MJ, Wellman CL. Characteristics of BDNF-induced weight loss. Exp Neurol. 1995;131:229–238. [PubMed: 7534721]
- 165.
- Mamounas LA, Altar CA, Blue ME. et al. BDNF promotes the regenerative sprouting, but not survival, of injured serotonergic axons in the adult rat brain. J Neurosci. 2000;20:771–782. [PMC free article: PMC6772430] [PubMed: 10632606]
- 166.
- Shen RY, Altar CA, Chiodo LA. Brain-derived neurotrophic factor increases the electrical activity of pars compacta dopamine neurons in vivo. Proc Natl Acad Sci USA. 1994;91:8920–8924. [PMC free article: PMC44718] [PubMed: 8090745]
- 167.
- Celada P, Siuciak JA, Tran TM. et al. Local infusion of brain-derived neurotrophic factor modifies the firing pattern of dorsal raphe serotonergic neurons. Brain Res. 1996;712:293–298. [PubMed: 8814905]
- 168.
- Martin-Iverson MT, Todd KG, Altar CA. Brain-derived neurotrophic factor and neurotrophin-3 activate striatal dopamine and serotonin metabolism and related behaviors: Interactions with amphetamine. J Neurosci. 1994;14:1262–1270. [PMC free article: PMC6577524] [PubMed: 8074747]
- 169.
- Lyons WE, Mamounas LA, Ricaurte GA. et al. Brain-derived neurotrophic factor-deficient mice develop aggressiveness and hyperphagia in conjunction with brain serotonergic abnormalities. Proc Natl Acad Sci USA. 1999;96:15239–15244. [PMC free article: PMC24804] [PubMed: 10611369]
- 170.
- Kernie SG, Liebl DJ, Parada LF. BDNF regulates eating behavior and locomotor activity in mice. EMBO J. 2000;19:1290–1300. [PMC free article: PMC305670] [PubMed: 10716929]
- 171.
- Kafitz KW, Rose CR, Thoenen H. et al. Neurotrophin-evoked rapid excitation through TrkB receptors. Nature. 1999;401:918–921. [PubMed: 10553907]
- 172.
- Lohof AM, Ip NY, Poo M. Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature. 1993;363:350–353. [PubMed: 8497318]
- 173.
- Jovanovic JN, Czernik AJ, Fienberg AA. et al. Synapsins as mediators of BDNF-enhanced neurotransmitter release. Nat Neurosci. 2000;3:323–329. [PubMed: 10725920]
- 174.
- Ruit KG, Osborne PA, Schmidt RE. et al. Nerve growth factor regulates sympathetic ganglion cell morphology and survival in the adult mouse. J Neurosci. 1990;10:2412–2419. [PMC free article: PMC6570366] [PubMed: 2376779]
- 175.
- McAllister AK, Lo DC, Katz LC. Neurotrophins regulate dendritic growth in developing visual cortex. Neuron. 1995;15:791–803. [PubMed: 7576629]
- 176.
- Korte M, Carroll P, Wolf E. et al. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci USA. 1995;92:8856–8860. [PMC free article: PMC41066] [PubMed: 7568031]
- 177.
- Minichiello L, Korte M, Wolfer D. et al. Essential role for TrkB receptors in hippocampus-mediated learning. Neuron. 1999;24:401–414. [PubMed: 10571233]
- 178.
- Duman RS, Vaidya VA, Nibuya M. et al. Stress, antidepressant treatments, and neurotrophic factors: Molecular and cellular mechanisms. The Neuroscientist. 1995;6:351–360.
- 179.
- Duman RS, Heninger GR, Nestler EJ. A molecular and cellular theory of depression. Arch Gen Psychiatry. 1997;54:597–606. [PubMed: 9236543]
- 180.
- Siuciak JA, Lewis DR, Wiegand SJ. et al. Antidepressant-like effect of brain-derived neurotrophic factor (BDNF). Pharmacol Biochem Behav. 1997;56:131–137. [PubMed: 8981620]
- 181.
- Marty S, Berzaghi MD, berninger b. Neurotrophins and activity-dependent plasticity of cortical interneurons. Trends Neurosci. 1997;20:198–202. [PubMed: 9141194]
- 182.
- Adams B, Sazgar M, Osehobo P. et al. Nerve growth factor accelerates seizure development, enhances mossy fiber sprouting, and attenuates seizure-induced decreases in neuronal density in the kindling model of epilepsy. J Neurosci. 1997;17:5288–5296. [PMC free article: PMC6793837] [PubMed: 9204913]
- 183.
- Van der Ze CE, Rashid K, Le K. et al. Intraventricular administration of antibodies to nerve growth factor retards kindling and blocks mossy fiber sprouting in adult rats. J Neurosci. 1995;15: 5316–5323. [PMC free article: PMC6577871] [PubMed: 7623154]
- 184.
- Reibel S, Larmet Y, Le BT. et al. Brain-derived neurotrophic factor delays hippocampal kindling in the rat. Neuroscience. 2000;100:777–788. [PubMed: 11036211]
- 185.
- Anton ES, Weskamp G, Reichardt LF. et al. Nerve growth factor and its low-affinity receptor promote Schwann cell migration. Proc Natl Acad Sci USA. 1994;91:2795–2799. [PMC free article: PMC43457] [PubMed: 8146193]
- 186.
- Winkler J, Ramirez GA, Kuhn HG. et al. Reversible Schwann cell hyperplasia and sprouting of sensory and sympathetic neurites after intraventricular administration of nerve growth factor. Ann Neurol. 1997;41:82–93. [PubMed: 9005869]
- 187.
- Barres BA, Raff MC, Gaese F. et al. A crucial role for neurotrophin-3 in oligodendrocyte development. Nature. 1994;367:371–375. [PubMed: 8114937]
- 188.
- McTigue DM, Horner PJ, Stokes BT. et al. Neurotrophin-3 and brain-derived neurotrophic factor induce oligodendrocyte proliferation and myelination of regenerating axons in the contused adult rat spinal cord. J Neurosci. 1998;18:5354–5365. [PMC free article: PMC6793495] [PubMed: 9651218]
- 189.
- Syroid DE, Maycox PJ, Soilu-Hanninen M. et al. Induction of postnatal Schwann cell death by the low-affinity neurotrophin receptor in vitro and after axotomy. J Neurosci. 2000;20:5741–5747. [PMC free article: PMC6772552] [PubMed: 10908614]
- 190.
- Torcia M, Bracci-Laudiero L, Lucibello M. et al. Nerve growth factor is an autocrine survival factor for memory B lymphocytes. Cell. 1996;85:345–356. [PubMed: 8616890]
- 191.
- Kimata H, Yoshida A, Ishioka C. et al. Nerve growth factor specifically induces human IgG4 production. Eur J Immunol. 1991;21:137–141. [PubMed: 1991484]
- 192.
- Neumann H, Misgeld T, Matsumuro K. et al. Neurotrophins inhibit major histocompatibility class II inducibility of microglia: involvement of the p75 neurotrophin receptor. Proc Natl Acad Sci USA. 1998;95:5779–5784. [PMC free article: PMC20456] [PubMed: 9576961]
- 193.
- Flugel A, Matsumuro K, Neumann H. et al. Anti-inflammatory activity of nerve growth factor in experimental autoimmune encephalomyelitis: inhibition of monocyte transendothelial migration. Eur J Immunol. 2001;31:11–22. [PubMed: 11169433]
- 194.
- Villoslada P, Hauser SL, Bartke I. et al. Human nerve growth factor protects common marmosets against autoimmune encephalomyelitis by switching the balance of T helper cell type 1 and 2 cytokines within the central nervous system. J Exp Med. 2000;191:1799–1806. [PMC free article: PMC2193155] [PubMed: 10811872]
- 195.
- Donovan MJ, Lin MI, Wiegn P. et al. Brain-derived neurotrophic factor is an endothelial cell survival factor required for intramyocardial vessel stabilization. Development. 2000;127:4531–4540. [PubMed: 11023857]
- 196.
- Donovan MJ, Hahn R, Tessarollo L. et al. Identification of an essential nonneuronal function of neurotrophin 3 in mammalian cardiac development. Nat Genet. 1996;14:210–213. [PubMed: 8841198]
- 197.
- Donovan MJ, Miranda RC, Kraemer R. et al. Neurotrophin and neurotrophin receptors in vascular smooth muscle cells. Regulation of expression in response to injury. Am J Pathol. 1995;147:309–324. [PMC free article: PMC1869811] [PubMed: 7639328]
- 198.
- Ilag LL, Curtis R, Glass D. et al. Pan-neurotrophin 1: a genetically engineered neurotrophic factor displaying multiple specificities in peripheral neurons in vitro and in vivo. Proc Natl Acad Sci USA. 1995;92:607–611. [PMC free article: PMC42791] [PubMed: 7831338]
- 199.
- Urfer R, TsoulFas P, Soppet D. et al. The binding epitopes of neurotrophin-3 to its receptors TrkC and gp75 and the design of a multifunctional human neurotrophin. EMBO J. 1994;13:5896–5909. [PMC free article: PMC395565] [PubMed: 7529173]
- 200.
- Friedman WJ, Black IB, Persson H. et al. Synergistic trophic actions on rat basal forebrain neurons revealed by a synthetic NGF/BDNF chimaeric molecule. Eur J Neurosci. 1995;7:656–662. [PubMed: 7620616]
- 201.
- Ryden M, Murray-Rust J, Glass D. et al. Functional analysis of mutant neurotrophins deficient in low-affinity binding reveals a role for p75LNGFR in NT-4 signalling. EMBO J. 1995;14:1979–1990. [PMC free article: PMC398297] [PubMed: 7744005]
- 202.
- Saragovi HU, Gehring K. Development of pharmacological agents for targeting neurotrophins and their receptors. Trends Pharmacol Sci. 2000;21:93–98. [PubMed: 10689362]
- 203.
- LeSauteur L, Maliartchouk S, Le Jeune H. et al. Potent human p140-TrkA agonists derived from an anti-receptor monoclonal antibody. J Neurosci. 1996;16:1308–1316. [PMC free article: PMC6578545] [PubMed: 8778282]
- 204.
- Maliartchouk S, Feng Y, Ivanisevic L. et al. A designed peptidomimetic agonistic ligand of TrkA nerve growth factor receptors. Mol Pharmacol. 2000;57:385–391. [PubMed: 10648649]
- 205.
- Treanor J J S, Schmelzer C, Knusel B. et al. Heterodimeric neurotrophins induce phosphorylation of Trk receptors and promote neuronal differentiation in PC12 cells. J Biol Chem. 1995;270:23104–23110. [PubMed: 7559453]
- 206.
- Friden PM, Walus LR, Watson P. et al. Blood-brain barrier penetration and in vivo activity of an NGF conjugate. Science. 1993;259:373–377. [PubMed: 8420006]
- 207.
- Hefti F. Pharmacology of neurotrophic factors. Annu Rev Pharmacol Toxicol. 1997;37:239–267. [PubMed: 9131253]
- 208.
- Saudou F, Finkbeiner S, Devys D. et al. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. [PubMed: 9778247]
- 209.
- Shieh PB, Ghosh A. Molecular mechanisms underlying activity-dependent regulation of BDNF expression. J Neurobiol. 1999;41:127–134. [PubMed: 10504200]
- 210.
- timmusk t, Palm K, Metsis M. et al. Multiple promoters direct tissue-specific expression of the rat BDNF gene. Neuron. 1993;10:475–489. [PubMed: 8461137]
- 211.
- Ma L, Merenmies J, Parada LF. Molecular characterization of the TrkA/NGF receptor minimal enhancer reveals regulation by multiple cis elements to drive embryonic neuron expression. Development. 2000;127:3777–3788. [PubMed: 10934022]
- 212.
- Pan W, Banks WA, Kastin AJ. Permeability of the blood-brain barrier to neurotrophins. Brain Res. 1998;788:87–94. [PubMed: 9554964]
- 213.
- Gravel C, Gotz R, Lorrain A. et al. Adenoviral gene transfer of ciliary neurotrophic factor and brain- derived neurotrophic factor leads to long-term survival of axotomized motor neurons. Nat Med. 1997;3:765–770. [PubMed: 9212104]
- 214.
- Mandel RJ, Gage FH, Clevenger DG. et al. Nerve growth factor expressed in the medial septum following in vivo gene delivery using a recombinant adeno-associated viral vector protects cholinergic neurons from fimbria-fornix lesion-induced degeneration. Exp Neurol. 1999;155:59–64. [PubMed: 9918705]
- 215.
- Kordower JH, Emborg ME, Bloch J. et al. Neurodegeneration prevented by lentiviral vector delivery of GDNF in primate models of Parkinson's disease. Science. 2000;290:767–773. [PubMed: 11052933]
- 216.
- Flugel A, Willem M, Berkowicz T. et al. Gene transfer into CD4+ T lymphocytes: Green fluorescent protein-engineered, encephalitogenic T cells illuminate brain autoimmune responses. Nat Med. 1999;5:843–847. [PubMed: 10395334]
- 217.
- Hoffman D, Wahlberg L, Aebischer P. NGF released from a polymer matrix prevents loss of ChAT expression in basal forebrain neurons following a fimbria-fornix lesion. Exp Neurol. 1990;110:39–44. [PubMed: 2209780]
- 218.
- Shibayama M, Hattori S, Himes BT. et al. Neurotrophin-3 prevents death of axotomized Clarke's nucleus neurons in adult rat. J Comp Neurol. 1998;390:102–111. [PubMed: 9456179]
- 219.
- Hoffman D, Breakefield XO, Short MP. et al. Transplantation of a polymer-encapsulated cell line genetically engineered to release NGF. Exp Neurol. 1993;122:100–106. [PubMed: 8339781]
- 220.
- Winn SR, Hammang JP, Emerich DF. et al. Polymer-encapsulated cells genetically modified to secrete human nerve growth factor promote the survival of axotomized septal cholinergic neurons. Proc Natl Acad Sci USA. 1994;91:2324–2328. [PMC free article: PMC43363] [PubMed: 8134395]
- 221.
- Haase G, Kennel P, Pettmann B. et al. Gene therapy of murine motor neuron disease using adenoviral vectors for neurotrophic factors. Nat Med. 1997;3:429–436. [PubMed: 9095177]
- 222.
- Sendtner M. Gene therapy for motor neuron disease. Nat Med. 1997;3:380–381. [PubMed: 9095166]
- 223.
- Sagot Y, Rosse T, Vejsada R. et al. Differential effects of neurotrophic factors on motoneuron retrograde labeling in a murine model of motoneuron disease. J Neurosci. 1998;18:1132–1141. [PMC free article: PMC6792750] [PubMed: 9437033]
- 224.
- Nagatsu T, Mogi M, Ichinose H. et al. Changes in cytokines and neurotrophins in Parkinson's disease. J Neural Transm Suppl. 2000:277–290. [PubMed: 11205147]
- 225.
- Vicario-Abejon C, Johe KK, Hazel TG. et al. Functions of basic fibroblast growth factor and neurotrophins in the differentiation of hippocampal neurons. Neuron. 1995;15:105–114. [PubMed: 7619514]
- 226.
- Vicario-Abejon C, Collin C, TsoulFas P. et al. Hippocampal stem cells differentiate into excitatory and inhibitory neurons. Eur J Neurosci. 2000;12:677–688. [PubMed: 10712648]
- 227.
- Leventhal C, Rafii S, Rafii D. et al. Endothelial trophic support of neuronal production and recruitment from the adult mammalian subependyma. Mol Cell Neurosci. 1999;13:450–464. [PubMed: 10383830]
- 228.
- Rubio F, Kokaia Z, Arco A. et al. BDNF gene transfer to the mammalian brain using CNS-derived neural precursors. Gene Ther. 1999;6:1851–1866. [PubMed: 10602381]
- 229.
- Martinez-Serrano A, Lundberg C, Horellou P. et al. CNS-derived neural progenitor cells for gene transfer of nerve growth factor to the adult rat brain: Complete rescue of axotomized cholinergic neurons after transplantation into the septum. J Neurosci. 1995;15:5668–5680. [PMC free article: PMC6577617] [PubMed: 7643209]
- 230.
- Moutsatsos IK, Turgeman G, Zhou S. et al. Exogenously regulated stem cell-mediated gene therapy for bone regeneration. Mol Ther. 2001;3:449–461. [PubMed: 11319905]
- Summary
- From Neurotrophin Physiology to Therapy
- Structure and Physiological Functions of Neurotrophins and Their Receptors
- Neurotrophins in Animal Models of Pathological Situations and Clinical Trials
- Non-Neuroprotective and Side Effects of Neurotrophins
- Potential Improvements of Neurotrophin Therapy
- Acknowledgments
- References
- Neurotrophins - Madame Curie Bioscience DatabaseNeurotrophins - Madame Curie Bioscience Database
- In Vitro Endothelialization: Its Contribution Towards an Ideal Vascular Replacem...In Vitro Endothelialization: Its Contribution Towards an Ideal Vascular Replacement - Madame Curie Bioscience Database
- Cryptosporidium parvum isolate kurnool small subunit ribosomal RNA gene, partial...Cryptosporidium parvum isolate kurnool small subunit ribosomal RNA gene, partial sequencegi|2806264502|gb|PQ345813.1|Nucleotide
- PREDICTED: Megachile rotundata uncharacterized LOC100879616 (LOC100879616), tran...PREDICTED: Megachile rotundata uncharacterized LOC100879616 (LOC100879616), transcript variant X4, mRNAgi|805827549|ref|XM_012298214.1|Nucleotide
Your browsing activity is empty.
Activity recording is turned off.
See more...