

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Abstract
Neuronal viability is maintained through a complex interacting network of signaling pathways that can be perturbed in response to a multitude of cellular stresses. A shift in the balance of signaling pathways after stress or in response to pathology can have drastic consequences for the function or the fate of a neuron. There is significant evidence that acutely injured and degenerating neurons may die by an active mechanism of cell death. This process involves the activation of discrete signaling pathways that ultimately compromise mitochondrial structure, energy metabolism and nuclear integrity. In this review we examine recent evidence pertaining to the presence and activation of anti- and pro-cell death regulatory pathways in nervous system injury and degeneration.
Introduction
Neuronal viability is maintained through a complex interacting network of signaling pathways that can be perturbed in response to a multitude of cellular stresses. A shift in one or more of these signaling pathways can alter the fate of a neuron resulting in cell death or continued survival. The nature of the stresses affecting neurons, the duration of the stresses, the developmental stage of the neuron and a variety of other factors influence the signaling pathways that are ultimately affected. These diverse parameters may also regulate the temporal response as well as the final disposition of the affected neurons.
Apoptosis is a mechanism of cell death that plays a fundamental role during the development of many tissues including the central nervous system. Apoptosis has traditionally been distinguished in developing tissues on the basis of specific morphological and biochemical criteria, including perinuclear chromatin condensation, cell shrinkage and endonuclease-mediated internucleosomal DNA fragmentation into a “ladder” pattern. More recently, the term “apoptosis” has been used to describe the programmed biochemical pathways of cell death that accompany development, tissue injury and degeneration. In the context of this review, apoptosis will refer to the biochemical pathways of cell death. There is accumulating evidence that acutely injured and degenerating neurons may die by a process of apoptosis, contributing to the loss of neurons observed under these conditions. The possibility that neurons may succumb under some circumstances by an active mechanism of cell death has raised interest in the regulatory pathways governing these processes.
In this review we examine recent evidence pertaining to the presence, activation and contribution of these regulatory pathways to nervous system development, injury and degeneration. The review begins with a description of cell death signals initiating through plasma membrane receptors followed by a description of relevant pro- and anti-apoptotic signal transduction pathways activated by various cellular stresses and trophic factor receptors. The review then considers signal transduction cascades activated in the nucleus and finally the critical mediators of viability that are related to mitochondrial function and disassembly of essential cellular components.
Death Receptor-Mediated Neuronal Apoptosis
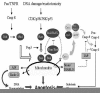
Members of the tumor necrosis factor receptor (TNFR) family are involved in a number of physiological processes, including neuronal cell death during development and after injury. This family of receptors includes Fas/CD95/Apo1, TNFR1, TNFR2, DR3/TRAMP/Wsl-1, TRAIL-R1/DR4, TRAIL-R2/DR5/Killer/TRICK2, TRAILR3/DcR1/TRID, TRAIL-R4/DcR2/TRUNDD, DcR3, osteoprotegerin, DR6, p75NTR and others.1 The prototypic members of the TNF receptor family are transmembrane proteins containing an extracellular cysteine-rich domain and an intracellular cytoplasmic protein-binding sequence called the death domain (DD). Activation of Fas/CD95, TNFR1, TRAIL-R1, TRAIL-R2 and DR6 receptors by their respective ligands leads to the recruitment of an intracellular protein complex known as the DISC (death-inducing signaling complex), followed by downstream caspase activation.
Fas/CD95/Apo1
Fas receptors and Fas ligand (FasL) are expressed on both astrocytes and neurons in normal rat and human brain.2 Exogenous application of FasL can cause neuronal apoptosis in vivo and in vitro, and this death can be blocked by a selective caspase-8 inhibitor.3 Embryonic motoneurons co-express Fas and FasL at the time of programmed cell death, and their death may be reduced by blocking antibodies to the Fas receptor or by the use of a specific caspase-8 inhibitor.4 Other studies have revealed a role for Fas in neuronal death after injury. In spinal cord ischemia, studies suggest the rapid formation of a complex containing the Fas receptor and pro-caspase-8, followed by activation of caspases -8 and -3 and neuronal apoptosis.5 FasL is also up-regulated in cortical neurons after cerebral ischemia.6,7 In lpr mice expressing a dysfunctional Fas receptor, infarct volume after ischemia was reduced when compared to wild-type control mice.7 Nervous system infections may also be a stimulus for activation of the Fas/FasL pathway. Both Fas and FasL are up-regulated in the brains of patients with HIV.8 In vitro studies indicate that neuronal death in HIV dementia is not due to direct viral infection of neurons but may be due to the release from activated microglia of other cytokines such as TNF and/or FasL.9
Most of the studies elucidating the mechanisms underlying Fas/FasL receptor signaling have been performed in non-neuronal cells (for review, see Refs. 10,11). The binding of FasL to its receptor leads to trimerization of the receptor and results in the recruitment of an intracellular adapter protein, FADD (Fas-associated death domain). FADD binds to the DD of the Fas/CD95/Apo1 receptor via homophilic DD-DD interactions. FADD also contains a separate death effector domain (DED) at its N-terminal, which interacts directly with a homologous region in the prodomain of pro-caspase-8. The association of the trimerized Fas/CD95/Apo1 receptor, FADD and pro-caspase-8 forms the DISC. Pro-caspase-8 subsequently undergoes autocatalytic cleavage to yield its active form. Caspase-8 may then cleave and activate caspases -3, -6 and -7 directly, thereby leading to cell death.12 Alternatively, caspase-8 may cleave Bid to form truncated Bid (t-Bid; see Ref. 13). Some investigators have argued that there are at least two Fas/CD95/Apo1L signaling pathways that occur after DISC formation.14 One pathway involves mitochondrial amplification of caspase activation, while the other results in mitochondrial dysfunction that occurs only after activation of caspases -8 and -3. Whereas the former apoptotic pathway is inhibitable by bcl-2 overexpression, the latter is not. The reasons for these cell type-specific differences in the order of caspase activation and mitochondrial dysfunction after Fas activation are not completely known. Nevertheless, it is apparent that caspase-8 is the primary apical caspase involved in signaling by Fas/CD95/Apo1L and other members of the TNFR superfamily. Caspase-10 also contains several DEDs and can be recruited to the DISC after activation of TNF and TRAIL (TNF-related apoptosis-inducing ligand) receptors.15 Thus, caspase-10 may also serve as an apical caspase in some cells, as has been demonstrated in lymphocytes.16 However, the fact that caspase-8 gene deletion completely abrogates TNFR1 and Fas receptor-induced apoptosis indicates that caspase-8 is primarily responsible for death receptor signaling by the TNFR superfamily.12
TNF/TNFRs
Recent studies suggest that TNF may play an important role in the control of neuronal survival. For example, TNFa induced apoptosis in cultured primary rat cortical neurons and in differentiated PC12 cells.17 Function-blocking antibodies to TNFa or TNFR1 partially protected embryonic mouse sympathetic and sensory neurons from apoptosis induced by NGF withdrawal.18 Moreover, fewer sensory and sympathetic neurons died during the phase of naturally occurring cell death in TNFa-deficient mice than in wild-type mice, and sympathetic neurons derived from such mice survived better than their wild-type counterparts. Other investigators have found that TNFR1-/- and TNFR2-/- mice have an increased number of apoptotic cells in the injured mouse spinal cord.19 The proposed explanation for this result was that activation of TNFRs in the wild-type animals resulted in NF-κB activation and increased expression of c-IAP2, thereby inhibiting caspase-mediated apoptosis.
Like the Fas/CD95/Apo1L receptor, TNF receptors (TNFRs) also signal cell death via the formation of an intracellular protein complex.15 After binding its ligand, TNFR1 trimerizes and recruits the intracellular DD-containing protein, TRADD (TNFR-associated death domain). TRADD, in turn, recruits FADD, pro-caspase-8 and the serine-threonine kinase RIP (receptor-interacting protein). TNFR1 also couples to a number of other intracellular signaling pathways. The serine-threonine kinase interacting protein (RIP) also interacts with the NF-κB pathway.20 Activation of NF-κB is thought to oppose TNF-induced apoptosis in many cell types. TNFR1 can also associate with another factor, TRAF2 (TNF receptor-associated factor 2), which appears to couple TNFR1 to both NF-κB and c-Jun N-terminal kinase (JNK) activation.21 Like TNFR1, DR6 has also been shown to activate NF-κB and JNK (Pan et al., 1998). Additional studies indicate that TNFRs may even couple to the nuclear transcription factor AP-1.22
TNFR2 lacks a cytoplasmic death domain. Thus, it was initially unclear how activation of the receptor leads to cell death. However, a recent study by Grell et al.23 has indicated that activation of TNFR2 leads to increased synthesis of TNF, which then acts in an autocrine fashion on TNFR1 receptors to initiate cell death.
TRAIL/TRAIL-Rs
TRAIL/Apo-2L is a type II transmembrane protein which, based on homology to TNF, was predicted (and subsequently shown) to be most effective in activating TRAIL receptors as a multimer.24 The tissue distribution of TRAIL receptors and their ligand varies widely, with both being commonly expressed in the same tissues.24,25 There are currently five known TRAIL receptors, including TRAIL-R1/DR4, TRAILR2/DR5/Killer, TRAIL-R3/DcR1/LIT, TRAIL-R4/DcR2/TRUNDD, DcR3 and osteoprotegerin, a soluble receptor that also binds osteoclast differentiation factor.15,24,26 TRAIL-R1 and TRAIL-R2 are 58% identical and are type I transmembrane proteins that contain extracellular cysteine-rich domains and intracellular cytoplasmic domains containing a DD similar to that of the TNFRs.24 TRAILR3/DcR1 and TRAIL-R4/DcR2 have a somewhat different structure than that seen in TRAIL-R1 or TRAIL-R2. TRAIL-R3/DcR1 contains only a partial cytoplasmic DD, while TRAIL-R4/DcR2 lacks any transmembrane component and is instead attached to the cell surface by a glycosylphosphatidylinositol linker.1,24,27
Interestingly, TRAIL preferentially induces apoptosis in tumorigenic cells and not in most normal tissues, including the brain.28,29 However, this finding has recently been challenged by reports of TRAIL-induced apoptosis in normal human hepatocytes and in epileptic human brain.30 Messenger RNA for TRAIL and its receptors have been localized in many tissues, including normal human brain tissue.31 Exposure of epileptic human brain slices to TRAIL induces apoptosis,32 and TRAIL induces cell death in primary mouse cortical neurons and neuroblastoma cells in vitro.6,33
The mechanisms underlying TRAIL/Apo-2L receptor signaling are not as well understood as those for CD95/Fas/Apo1 and TNFRs. As mentioned previously, TRAIL receptor activation leads to activation of caspase-8,1,34 and caspase inhibition blocks TRAIL-induced apoptosis.35 Studies have demonstrated inhibition of TRAIL signaling in FADD-deficient cells, as well as direct binding of FADD and TRADD to TRAIL-R1 and TRAIL-R2.34,36 Thus, it seems likely that TRAIL receptor activation results in DISC formation, caspase-8 activation and cell death in a manner similar to that observed with TNFRs and the Fas receptor. There may be other signaling pathways coupled to TRAIL receptors as well, although their functional role remains to be determined. For example, TRAIL-R4 has been shown to activate NF-κB, despite having an incomplete DD.27
The factors determining sensitivity to TRAIL are only now being elucidated. TRAIL itself is up-regulated after cerebral ischemia,6 and both Trail-R2/DR5 and TRAIL-R3/DcR1 can be induced in a variety of tissues in a p53-dependent or p53-independent manner.37-39 These findings suggest that sensitivity to TRAIL-mediated apoptosis may be regulated in the nervous system, at least in part by injury-induced regulation of the expression of TRAIL and its receptors.
Both TRAIL-R3 and TRAIL-R4 have been called decoy receptors because their ligation fails to induce apoptosis. One hypothesis is that these non-functional “decoy” receptors bind the available TRAIL, thereby protecting TRAIL-sensitive cells from the ligand.40,41 However, studies examining expression of mRNA and protein for TRAIL-R3/DcR1 and TRAIL-R4/DcR2 in a variety of cell types have not found a significant correlation with TRAIL sensitivity.29,42,43 Thus, the function of TRAIL-R3 and TRAIL-R4 has not been conclusively demonstrated.
p75NTR
Recent studies have provided evidence that the low affinity neurotrophin receptor, p75NTR, has significant homology to other members of the TNFR superfamily, and that it may play a role as a death receptor under some circumstances. In addition to its well-established role as a component of neurotrophin receptors, p75NTR may induce the death of selected neuronal populations when expressed in the absence of Trkreceptors. For example, embryonic or neonatal motor neurons derived from wild-type, but not p75NTR-deficient mice undergo apoptosis after NGF exposure.44,45 These cells also show increased survival after axotomy when compared to wild-type motor neurons, suggesting that p75NTR may promote neuronal cell death after injury.46 p75NTR knockout mice also show decreased cell death of a subset of spinal cord interneurons, retinal neurons, and sympathetic neurons.47,48
The mechanism by which p75NTR signals cell death is poorly understood. It has a different death domain structure than that of other TNFRs, and there is conflicting evidence as to whether it signals apoptosis when occupied or unoccupied.11 Studies have implicated NF-κB activation, ceramide production and caspase activation in the p75NTR signaling pathway (for review, see Ref. 49). In immortalized striatal neurons containing an inducible p75NTR, expression of the receptor was found to activate caspases -9, -6 and -3 in an NGF-independent manner.50 Cell death was inhibited by Bcl-XL, by a dominant-negative form of caspase-9 and by a viral FLIP, E8. The protection by viral FLIP suggests that DEDs are involved in the signaling cascade. However, FADD, TRADD and caspase-8 were not involved. Other proteins identified that interact with p75NTR and promote apoptosis include NRIF (neurotrophin receptor interacting factor, Ref. 51), NADE (p75NTR-associated cell death executor, Ref. 52), and NRAGE (neurotrophin receptor-interacting MAGE homolog, Ref. 53), while RIP254 was shown to suppress apoptosis.
Signal Transduction Pathways
Signaling Kinases Pro- and Anti-Apoptotic Effectors in the Nervous System
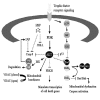
The mitogen-activated protein (MAP) kinases and phosphatidylinositol-3 kinase (PI3K) are serine/threonine protein kinases that play critical roles in neuronal growth, differentiation and survival (Figs. 1 and 2). In general, activation of the extracellular signal-regulated kinase members of the MAP kinase family (ERK or p42/p44 MAP kinase) and the PI3KAkt signaling pathway promote cell survival, while members of the MAP kinase family known as the stress-activated protein kinases (SAPK's), c-Jun N-terminal kinases (JNK's) and the p38 MAP kinase (p38 MAPK), promote cell death. In this section, we will review the molecular pathways and contributions of these kinases based upon evidence from animal studies, as well as neuronal cell lines and various cultured neuronal populations.
PI3K-Akt Signaling
Observations over the past decade have identified the PI3K-Akt pathway's importance in mediating survival in PC12 cells55 and cultured neurons from the peripheral56 and central nervous systems.57 A recent study by Kuruvilla et al.58 demonstrated the importance of PI3K activity under conditions that may more accurately simulate in vivo function. By applying NGF exclusively onto the distal axon compartment, the authors were able to demonstrate that PI3K activation in the axons and its retrograde signaling play a critical role in survival of both the axons and cell body.58
Neurotrophic factors such as NGF, brain-derived neurotrophic factor (BDNF), glial cell line-derived neurotrophic factor (GDNF), and insulin-like growth factor I (IGFI), activate the PI3-Akt signaling cascade through corresponding receptor tyrosine kinases such as the high affinity neurotrophin receptors (Trk's) (reviewed in Ref. 59). After receptor dimerization, PI3K is recruited to the plasma membrane where its catalytic subunit generates lipid second messengers, phosphoinositide phosphates (PIP2, PIP3), at the inner surface of the plasma membrane. Phosphoinositide-dependent protein kinase-1 (PDK1) then acts in concert with PIP2 and PIP3 to phosphorylate and activate Akt (a.k.a. protein kinase B, PKB), a protein kinase first identified in the AKT virus (reviewed in Ref. 60). Alternately, Trk receptors may stimulate PI3K via the Ras G-protein, insulin receptor substrate (IRS) signaling, and Gab-1, an adaptor protein, which binds to Trk and directly stimulates PI3K.61,62
Dominant-negative and constitutively active forms of Ras,62 PI3K,63,64 and Akt65 have been used to study signaling through the PI3K-Akt pathway. These and other studies have demonstrated downstream signaling effects that regulate cellular survival, proliferation, and metabolism. For example, Akt phosphorylates and inactivates FKHRL1, a member of the family of Forkhead transcriptional regulators. Inactivated FKHRL1 is unable to induce the expression of death genes in cerebellar granule neurons.66 In primary hippocampal neurons subjected to hypoxia or nitric oxide, p53 activation and p53-mediated Bax upregulation are also blocked by Akt signaling.67 Akt activates the cAMP-responsive element binding protein (CREB) and nuclear factor-κB (NF-κB), additional transcriptional regulators that may promote neuronal survival.68,69In addition, Akt can directly inhibit the apoptotic machinery by phosphorylation at sites both upstream (BAD70) and downstream (Caspase-971) of mitochondrial cytochrome c release. Finally, there is evidence to support the role of Akt in promoting neuronal survival through metabolic effects, by regulating glucose metabolism in neurons.72
ERK/MAP Kinase Pathways
The ERK cascade represents one of the evolutionarily conserved MAP kinase pathways, first found to be important in the regulation of neuronal survival in response to neurotrophin withdrawal.73 A wide body of evidence has also been collected to demonstrate the role of ERK in neuronal plasticity and memory formation.74,75 For the purposes of this discussion, we will focus on the data supporting ERK's role in promoting survival although ERK's can be activated by toxic stimuli as well and have been implicated in cell death.76-78
At least six ERK isoforms have been identified.79 In general, the common pathway leading to ERK activation by trophic factor signaling involves engagement of the membrane receptor at the cell surface, activation of small (p21ras, rap) GTPases, which in turn activate the protein kinase Raf. Raf, in turn, phosphorylates and activates MAP kinase kinase 1/2 (MKK 1/2), which phosphorylates and activates ERK (reviewed in Ref. 80). The ERK pathway can also be stimulated and modulated by intracellular Ca2+ levels, protein kinase A, diacylglycerol, and cAMP (reviewed in Refs. 74,75,81).
Constitutively activated or dominant-negative mutants directed at Ras, Raf, and MEK 1/2 (a.k.a. MKK1/2) have been used to determine the effects of the ERK pathway in the setting of neurotrophin withdrawal. Current evidence indicates a direct role for the ERK pathway in protecting neurons after injury. Activation of the ERK pathway is essential for BDNF-mediated protection of cortical neurons from DNA damage,84 glutamate-induced excitotoxicity and ischemic preconditioning.85 The role of ERK in suppressing neuronal apoptosis, however, has been controversial. The importance of this pathway as a neuroprotective mechanism appears related to the nature of the cell type and exposure. For example, a MKK1 protein kinase inhibitor protects against damage resulting from focal cerebral ischemia.82 These conflicting findings may stem from the use of different experimental systems and the possibility that the regulation of ERK is complex and might also be controlled by MKK1-independent mechanisms.83 While PI3K-Akt is thought to be more important in protecting cells from trophic factor withdrawal, ERK is thought to protect against injury-induced apoptosis.84
The mechanisms by which ERK mediates its protective effect are poorly understood. Downstream signaling may be directed through the serine/threonine kinase p90/rsk2 and CREB phosphorylation,68 but a direct link between CREB and ERK has not yet been established in neurons. Alternately, ERK can phosphorylate Bad resulting in its sequestration by 14-3-3 and prevents Bad-mediated induction of apoptosis.68
JNKs
As their name implies, stress-activated protein kinases, such as the c-Jun N-terminal kinase (JNK) are activated by numerous noxious stimuli including trophic factor withdrawal73, excitotoxicity,86,87 seizures,88 irradiation,86 hypoxia,89 exposure to β-amyloid (Abeta),90,91 arsenite toxicity,92 and axotomy93 (reviewed in Ref. 94). At least ten isoforms of JNK are expressed in the human adult brain, encoded by three separate genes through alternative splicing mechanisms.95
The relevance of JNK signaling to neuronal cell death has been demonstrated using JNK-deficient mice, JNK inhibitors and dominant-negative forms of upstream activating kinases. Transfecting cortical neurons with various dominant-negative forms of components comprising the JNK signaling pathway significantly decreased the number of neurons undergoing apoptosis in response to the β-amyloid peptide.90 Similarly, treatment with an inhibitor of JNK activation, CEP-1347, effectively blocked increases in cellular JNK activity and protected PC12 cells, sympathetic neurons,91 and cortical neurons96 from β-amyloid-induced death. These results are consistent with the demonstration that JNK is activated and redistributed in the brains of patients with Alzheimer's disease.97 Moreover, CEP-1347 also inhibited MPTP-mediated MKK4 and JNK signaling and attenuated MPTP-induced dopaminergic cell loss,98 suggesting that the JNK pathway may be activated in the degenerative process in Parkinson's disease. Cultured rat sympathetic neurons and neuronally differentiated PC12 cells were protected from nerve growth factor withdrawal, exposure to ultraviolet irradiation, and oxidative stress following treatment with the JNK inhibitor CEP-134798. Interestingly, CEP-1347 failed to protect undifferentiated PC12 cells induced to die by serum withdrawal or Jurkat T cells from Fas activation, even though each injury stimulus activated JNK and was inhibited by CEP-1347. Blocking JNK activation also protects neurons from cell death induced by the withdrawal of survival signals.73,99,100 These results are consistent with the data obtained applying NGF withdrawal to JNK3-deficient sympathetic neurons.101 Moreover, the deficiency in JNK3 activity87 and the expression of a mutant form of jun that is resistant to phosphorylation by JNK102 confers resistance to kainic acid-induced seizures and subsequent neuronal damage. Collectively, these results illustrate the importance of JNK signaling to the regulation of neuronal injury responses.
Upstream regulation of JNK activation is poorly understood. As in the ERK system, the immediate upstream activators of JNK are MKK's. While the ERK pathway is stimulated by MKK's 1/2, JNK appears to be activated by MKK4103 or MKK7,104 depending upon the cell type, developmental stage,105 and stressful stimulus.106 Other kinases such as MLK3107, MEKK1,108 and ASK1109,110 lie further upstream of the MKKs and probably activate both MKK's and JNK in a similar cell type- and stimulus-dependent manner.106 ASK1 also activates p38 MAPK and is up-regulated in PC12 cells following NGF withdrawal.111 Overexpression of a constitutively active mutant of ASK1 activates JNK and induces apoptosis in differentiated PC12 cells and sympathetic neurons.111 JNK activity is further modified by JNK interacting proteins (JIP's), which are scaffolding proteins thought to sequester all of the necessary kinases for JNK activation.112
The downstream effectors of the JNK pathway are numerous and include both cytoplasmic and nuclear targets. In the nucleus, JNK's phosphorylate and activate the transcription factor c-jun,113 a component of the AP-1 transcription factor, which regulates genes involved in apoptosis. For example, in PC12 cells, JNK activation is followed by induction of Fas ligand (FasL) expression100 (see Fas/CD95/Apo1 in the previous section). Cytoplasmic targets include p53114 (see Nuclear Signaling Pathways in the following section), which is stabilized and activated by JNK,115 and a cell death domain protein MADD, which co-translocates with JNK to the nucleus after hypoxic stress.116
p38 MAP Kinases
p38 MAPK's, like JNK's, are stress-activated kinases that are highly conserved in evolution, exist in multiple isoforms, and are activated in response to numerous cellular stresses (reviewed in Refs. 94,106). Like other members of the MAP kinase family, upstream regulation involves phosphorylation by MKK's, specifically MKK3, MKK4, and MKK6.117-119 The downstream targets of the p38 MAPK pathway are similar to the targets of JNKs and involve transcription factors, kinases, and pathways influencing pro-inflammatory cytokines (reviewed in Ref. 106). However, little is known about the downstream targets of p38 MAPK signaling in the brain.
Inhibition of p38 MAPK has been shown to attenuate neuronal cell death in response to a variety of different cellular stresses. The p38 MAPK inhibitor SB203580 significantly reduced apoptosis in potassium-deprived cerebellar granule cells120,121 and prevented the activation of downstream effectors such as caspase-3.120 p38 MAPK inhibition also conferred protection against neuronal cell death induced by C2-ceramide,122 oxidative damage,123 and trophic factor deprivation.73,124 Focal ischemia induces p38 MAPK activity and treatment with a second-generation p38 MAPK inhibitor, SB 239063, significantly reduced infarct size and behavioral deficits.125 Inhibiting p38 MAPK activity in the focal ischemia model was also associated with decreased expression of IL-1beta and TNFa, cytokines known to contribute to stroke-induced brain injury. Glutamate-mediated cell death, an important contributor to stroke-induced changes in viability, is also attenuated in cerebellar granule cells by p38 MAPK inhibition.126
Mechanistic aspects of the contribution of p38 MAPK activation to cell death were further revealed in a study from our laboratory.127 We showed that nitric oxide (NO)-induced cell death of human neuroblastoma cells and murine cortical neurons in culture was dependent upon p38 MAPK activity and Bax. We further demonstrated that inhibition of p38 MAPK activity blocked Bax translocation from the cytosol to the mitochondria and thereby conferred protection from cell death. Cheng et al. recently reported a similar result.128 Thus, p38 MAPK represents a potential target for interrupting Bax translocation and preserving neuronal viability following neuronal injury involving NO toxicity. Collectively, the stress-activated kinases, p38 MAPK and JNK, play important roles in the neuronal response to injury and newly developed inhibitors for these enzymes hold promise for therapeutic intervention.
Glycogen Synthase Kinase-3β
Another emerging kinase that has been implicated in neuronal cell death is glycogen synthase kinase-3β (GSK-3β). GSK-3β regulates a diverse array of proteins including many transcription factors (for review, see Ref. 129) and is inhibited by the PI3K pathway.130,131 Its function in the nervous system has been linked to neuropathology, such as Alzheimer's disease, based on its ability to promote tau hyperphosphorylation,132,133 although the significance of this finding is still controversial.134 Several cellular stresses including serum deprivation stimulate GSK-3β activity in cortical neurons prior to the induction of apoptosis.72 Expression of an inhibitory GSK-3β binding protein or a dominant interfering form of GSK-3β reduced neuronal apoptosis, suggesting that GSK-3β contributes to trophic factor deprivation-induced apoptosis. Moreover, overexpressing GSK-3β in neurons was sufficient to trigger neuronal cell death. Elucidating the intriguing role played by this kinase in neuronal cell survival/death is certainly warranted and will require the identification of specific substrates and their functions in the context of a specific survival or death-promoting stimulus.
Nuclear Signaling Pathways
Cyclins, Cyclin-Dependent Kinases, Rb and E2F1
What is seemingly a contradiction in our understanding of cell death in post-mitotic neurons is the demonstration that neuronal damage results in the elaboration of events that are normally associated with proliferating cells. Neurons express a range of proteins after injury that normally function to control cell cycle progression (for review, see Ref. 135). Alterations in the levels of cyclin-dependent kinases (CDK's) and their respective cyclin partners have been observed in neurons in response to trophic factor deprivation,136 ischemia and seizures,137 stroke,138 exposure to the β-amyloid peptide,139,140 and DNA damage.141 In addition, abnormal expression of these cell cycle regulators has also been implicated in neurodegenerative diseases such as Alzheimer's disease142-148 and amyotrophic lateral sclerosis (ALS).149 Pharmacological and molecular manipulation of CDK activity has been used to evaluate the relationship between cell cycle regulatory proteins and neuronal cell death.
The use of CDK inhibitors or dominant-negative forms of CDK 4/6 protects cultured postmitotic sympathetic neurons from death evoked by NGF withdrawal.150 Pharmacological inhibitors of cyclin-dependent kinases, such as flavopiridol, olomoucine, and roscovitine, prevent cerebellar granule neuron apoptosis induced by non-depolarizing KCl.151 Moreover, camptothecin-induced cell death of PC12 cells, sympathetic neurons, and cerebral cortical neurons is also suppressed by the CDK-inhibitors flavopiridol and olomoucine.152,153 However, these cell cycle inhibitors did not prevent cell death induced by a strong oxidative stress (SOD1 depletion),154 suggesting that cell cycle proteins are not involved in all forms of neuronal cell death.
The mechanism by which the CDK's facilitate neuronal cell death is under active investigation. Increased activity of cyclin-dependent kinase 5 (Cdk5) may contribute to neuronal death and cytoskeletal abnormalities in Alzheimer's disease148 and ALS149 through its ability to hyperphosphorylate tau, although the finding that cdk5 is a major protein tau kinase has been disputed.155 However, Cdk5 may alter neuronal viability through its effects on other signaling cascades, consistent with the finding that it directly phosphorylates β-catenin and regulates the latter's binding to presenilin-1.156
Cdk4/6 activation results in the phosphorylation of the Rb tumor suppressor protein in neurons following DNA damage.141 One consequence of Rb phosphorylation is the activation of E2F family members. Overexpression of E2F1 is sufficient to induce neuronal cell death,157 159 while E2F1 deficiency protects neurons from multiple cellular stresses including DNA damage,160 ischemia,161,162 staurosporine,157 reduced potassium158 and β-amyloid toxicity.163 In addition, overexpression of dominant-negative versions of DP-1, a binding partner for E2F family members, also protects neurons from cell death induced by DNA damage.139,160 Interestingly, overexpression of E2F activity in dissociated sensory neurons from adult rats stimulated their entry into S-phase, although the authors found no evidence for subsequent mitotic events in the E2F-overexpressing cells.159 These results suggest that other molecular signals are responsible for sister chromatid separation and continued cycling in neurons. The absence of these additional mitotic factors in damaged or degenerating neurons might provide an explanation for why the inappropriate activation of cell cycle regulatory genes promotes neuronal cell death.
The CDK-dependent inactivation of Rb is likely to have many other effects beyond the activation E2F family members. Rb has been shown to repress NF-κB transcriptional activity164 as well as JNK activity,165 both important neuronal cell death mediators. The relevance of Rb phosphorylation to the process of neuronal cell death is illustrated by the finding that overexpressing a mutant form of Rb lacking critical phosphorylation sites protects neurons from cell death induced by DNA damage.160,166 Moreover, deregulation of the Rb cell cycle pathway during development promotes ectopic cell cycle entry and elevated apoptosis which is, in part, p53-dependent.167,168 The latter finding is consistent with a role for the Rb-regulated E2F1 protein as a specific inducer of apoptosis and p53 accumulation.169
p53 Tumor Suppressor Gene
The p53 Gene
The p53 tumor suppressor gene encodes a nuclear phosphoprotein that functions as a key regulator of DNA repair, cell cycle progression and apoptosis. The p53 protein is up-regulated in response to a diverse array of cellular stresses, including DNA damage, hypoxia, oxidative stress, ribonucleotide depletion and oncogene activation.170,171 p53 protein levels are primarily regulated by changes in protein degradation in response to injury. In response to cellular stress, p53 induces its biological response largely through the transactivation of specific target genes. These downstream effectors have been characterized with respect to p53-mediated growth arrest,172 but the pathways associated with p53-mediated apoptosis have not been completely elucidated.173 In addition to its transactivating activity, p53 may also promote apoptosis by repressing the expression of select genes.174,175 Moreover, p53-mediated apoptosis may also occur through transcription-independent pathways requiring direct protein-protein interactions.176,177
p53 Expression and Neuronal Injury
Alterations in p53 mRNA and protein expression have been associated with neuronal damage in a variety of model systems (for review, see Ref. 178). Neuronal damage resulting from stimulation by excitatory amino acids (excitotoxicity) has been strongly associated with p53 accumulation. The systemic injection of kainic acid, an excitotoxin that produces seizures, induced p53 expression in neurons exhibiting morphological evidence of damage.179,180 Activation of glutamate receptors by intrastriatal infusion of either N-methyl-D-aspartate (NMDA), the NMDA receptor agonist quinolinic acid (QA) or kainic acid produced a significant elevation in p53 levels in striatal neurons.181-183 Elevated expression of the p53 gene has also been observed in models of experimental traumatic brain injury.184-186 As early as 6 h post-injury, p53 mRNA is induced predominantly in neurons that are vulnerable to traumatic brain injury. Transient or permanent occlusion of the middle cerebral artery causes ischemia-induced cell death in striatal and cerebral cortical neurons, which is associated with a significant increase in the expression of p53 mRNA187 and protein.188
Elevated p53 immunoreactivity has also been detected in brain tissue derived from animal models of human neurodegenerative disease or from patients that have been diagnosed with a neurodegenerative disorder. Patients with Alzheimer's disease189,190 show increased p53 immunoreactivity in morphologically damaged neurons, consistent with the presence of increased p53 immunoreactivity in neurons from mice overexpressing the β-amyloid peptide (Ab 142).191 Mutation of the E6AP ubiquitin ligase in a mouse model of Angelman syndrome results in increased cytoplasmic abundance of the p53 protein in hippocampal pyramidal neurons and cerebellar Purkinje neurons.192 Animals expressing the Angelman mutation display motor dysfunction, inducible seizures and a deficiency in contextual learning. Thus, increased levels of the p53 protein in Angelman syndrome resulting from abnormalities in the ubiquitination process may contribute to neuronal dysfunction.
The brains of patients with Down's syndrome, a genetic disorder manifesting a similar pathology to Alzheimer's disease, have also been shown to express elevated levels of apoptosis effectors, including the p53 protein.190,193,194Motor neuron degeneration observed in amyotrophic lateral sclerosis has been associated with increased levels of p53 in motor neurons of the spinal cord and motor cortex.195 Recent evidence also suggests that p53 could be involved in the pathogenesis of Huntington's disease. The Huntington's disease protein, huntingtin, was found to interact with p53 and the CREB-binding protein,196 and to repress transcription of several p53-regulated promoters. These results raise the possibility that the mutant huntingtin protein may cause neuronal dysfunction and cell death by interacting with transcription factors and altering gene transcription.
The results obtained with in vitro models of neuronal injury are consistent with the data described above for the in vivo models. Excitotoxicity, which figured so prominently in the whole animal studies, is a potent inducer of p53 protein in cultured cerebellar granule neurons.197,198 Another potent stimulus for elevating p53 expression in cultured neurons is DNA damage induced by cytosine arabinoside,199,200 ionizing radiation,166 camptothecin,153 and the absence of essential DNA repair proteins.201 Hypoxia in culture, which models the ischemia produced by middle cerebral artery occlusion, increases p53 protein expression in rat embryonic cortical neurons.202
p53 Expression and Neuronal Cell Death
The physiological relevance of p53 expression to neuronal cell death has been evaluated in numerous models of injury and disease. p53-deficient mice or neurons derived from these mice have been used most often, but inhibitors of p53 expression or p53 function have also been used to evaluate the role of p53 in the context of neuronal injury. The absence of p53 has been shown to protect neurons from a wide variety of toxic insults including focal ischemia,203 ionizing radiation,204-208 kainate-induced seizures,209 MPTP-induced neurotoxicity,210 methamphetamine-induced neurotoxicity,211 adrenalectomy,212 traumatic brain injury,186 DNA-damaging agents,199,200,206,213-215 glutamate,197,198 hypoxia,202,216 and NGF withdrawal.217,218 A role for p53 has also been demonstrated for apoptosis associated with developmental neuronal death in certain subpopulations of neurons217 as well as cell death occurring during abnormal development.167,219 In addition to the use of p53-deficient mice, the application of antisense oligonucleotides197,200,202,220,221 or a synthetic inhibitor of p53222 have been successfully used to protect neurons against cell death induced by a variety of cellular stresses.
However, the absence of p53 does not protect neurons against all forms of cellular stress. Cerebellar neurons lacking p53 die when transferred to a low potassium medium,213 and postnatal cortical and hippocampal neurons also die after staurosporine exposure in a p53-independent manner.204 Cerebellar granule neuron death induced by methylazoxymethanol is not alleviated in p53-null mice. Moreover, two separate reports failed to demonstrate that p53 is involved in the death of neurons in a mutant SOD1 transgenic mouse model of familial ALS.223,224 Despite evidence that p53 plays an important role in mediating cell death after acute neuronal injury, additional studies will be required to fully evaluate the role of p53 in late-onset neurodegenerative diseases.
p53-Mediated Cell Death Signaling Pathways
The mechanism by which p53 specifies the neuronal response to injury is poorly understood. However, currently available data suggest that the Bcl2 family member, Bax, is involved in p53-mediated neuronal death. Bax-deficient neurons are protected from cell death induced by DNA-damaging agents204,215,225 and adenovirus-mediated p53 overexpression.215,226 Moreover, various forms of neuronal injury are associated with Bax translocation from the cytosol to the mitochondria.127,153,227,228 The redistribution of Bax to the mitochondria has been associated with a reduction in mitochondrial membrane potential, mitochondrial release of cytochrome c, and activation of caspases,229-233 suggesting that caspases may also be a component of a p53-induced cell death pathway. Recent studies indeed demonstrated that p53 is required for caspase activation in response to genotoxic stress.215,225,226,234 These findings suggest that some forms of neuronal injury invoke a common pathway involving signal transduction through p53, Bax, mitochondrial dysfunction, cytochrome c release, and caspase activation.
Clearly, additional studies are required to elucidate the downstream effectors mediating neuronal cell death in response to p53 activation. The glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene has been identified as a p53-inducible gene in cultured cerebellar granule neurons subjected to DNA damage.200 Apaf1, a key element of the apoptosome, which regulates initiation of the caspase-9 cascade, is induced in neurons in response to p53 activation or DNA damage.235 Other genes, such as DR5, Fas, Fas ligand,173 PERP,236 Noxa and PUMA237 have been shown to be induced by apoptotic stimuli as a result of p53 activation in a variety of non-neuronal cell types, but the involvement of these genes in p53-dependent neuronal apoptosis is not known.
It is also critical that the pathways responsible for activating and suppressing p53 activity be identified. In this regard it is interesting to note that the important survival-promoting protein, Akt, can protect neurons from cell death by inhibiting p53-dependent transcriptional activity.67 These results demonstrate the interconnection that exists between pathways that govern cell death and viability and serve to remind us that the response and the outcome of neurons to stress are exceedingly complex.
DNA Damage-Activated Enzymes
Excessive DNA damage may result from exposure to free radicals generated by oxidative stress and thereby may be a common initiating stimulus for neuronal apoptosis. Oxidative stress is associated with a variety of neuropathological conditions including acute excitotoxicity (stroke, traumatic brain injury) and progressive neurodegeneration (Alzheimer's, Parkinson's, Huntington's, ALS).238,239 Several enzymes are activated in response to DNA damage (strand breaks) and are associated with the control of cell cycle checkpoints, DNA repair programs and the regulation of apoptosis. These include the gene product for ataxia telangiectasia (ataxia telangiectasia mutated, ATM), DNA-dependent protein kinase (DNA-PK), and poly(ADP-ribose) polymerase-1 (PARP1). Interestingly, all three of these enzymes are substrates for caspase-3,240,244 implicating them in the regulation of apoptosis.
ATM
ATM (ataxia telangiectasia mutated), a protein kinase belonging to the PI3 kinase family, is activated by DNA double-strand breaks secondary to ionizing radiation (IR) and radiomimetics. Multiple phosphorylation substrates have been identified including p53, MDM2, and BRCA1, proteins involved in DNA repair and cell cycle checkpoint control (for a review, see Ref. 245). The generation of an ATM-deficient mouse verified the relationship of ATM function to DNA damage-induced responses. For example, ATM−/− mice display extreme sensitivity to IR due to its toxic effects on the gastrointestinal tract, and fibroblasts derived from the mice show compromised G1/S checkpoint function.246 In contrast to these results from non-neuronal and proliferative cell types, developing CNS neurons in ATM−/− mice turn out to be radioresistant compared to those in wild-type mice which show massive cell death, indicating that IR-induced neuronal apoptosis is ATM-dependent.208 IR-induced, ATM-mediated neuronal cell death also depends on p53, Bax and caspase-3.225 ATM is known to phosphorylate p53 at Ser-15 directly247,248 and Ser-20 through the activation of CHK2.249 ATM also phosphorylates MDM2 at Ser-395.250 These phosphorylation events are thought to collectively contribute to p53 stabilization and transcriptional activation.251,252 However, it is still not known precisely how these ATM-dependent modifications of p53 result in the induction of apoptotic mediators.
Interestingly, IR-induced death of neural progenitor cells is not dependent on ATM, while p53 is required for apoptosis in both neural progenitor and post-mitotic neuronal populations.225,253 Thus, the linkage between ATM and p53-dependent cell death is not observed in undifferentiated, multipotent precursors but appears to develop in newly formed post-mitotic neurons; it still remains to be established if ATM is also required for IR-induced apoptosis in fully differentiated neurons in adults. Indeed, adult neurons, irrespective of the ATM genotype, don't seem to be sensitive to IR.246 This implies that a critical function of ATM in neuronal apoptotic signaling may be the elimination of neurons born with a substantial amount of DNA damage accumulated during development.253 Consistent with this idea is the observation that ATM deficiency in Ataxia Telangiectasia (AT) patients causes early-onset progressive neurodegeneration, which is apparently due to compromised genomic integrity based on the phenotypic similarities of A-T to pathological conditions caused by genetic defects in DNA repair and cell cycle checkpoints.254 ATR, an ATM-related protein primarily involved in the response to UV-induced DNA damage,255 has not been evaluated in the process of neuronal apoptosis and remains to be examined.
DNA-Dependent Protein Kinase
DNA-dependent protein kinase (DNA-PK) is a member of the PI3 kinase family and is activated in response to DNA strand breaks,256 and is essential for DNA repair.257 DNA-PK phosphorylates p53,258 MDM2,259 and other factors implicated in the regulation of cell death including c-abl260 and IkB.261 However, several recent reports suggest that DNA-PK is not required for neuronal apoptosis but may instead be important for neuronal survival after genotoxic insult. Neurons derived from scid mice, which express a truncated form of DNA-PK with impaired kinase activity, show higher rates of spontaneous cell death and increased vulnerability to apoptotic insults in culture as well as in vivo.262-264 Although an inhibitor of DNA-PK reportedly reduces the manifestation of apoptotic nuclear changes in staurosporine-treated neuroblastoma cells,265 this finding may be due to the tumorigenic and proliferative properties of neuroblastoma cells. These results suggest that the compromise in DNA repair activity associated with the scid mutation facilitates the accumulation of unrepaired DNA strand breaks, which eventually culminates in neuronal apoptosis.
Poly(ADP-Ribose) Polymerase
Poly(ADP-ribose) polymerase-1 (PARP-1) is the principal member of an expanding family of PARP enzymes,266 which are activated in response to single-strand DNA breaks. In response to DNA damage, PARP-1 modifies a variety of nuclear proteins, including itself, by attaching poly(ADP-ribose) chains. The activated enzyme plays an important role in base excision repair and transcriptional regulation, accounting for its contribution to genomic stability.267-269 Apart from its involvement in DNA repair, PARP1 has been proposed to have a major impact on cell viability as excessive activation of the enzyme depletes cellular NAD and ATP, causing necrotic cell death270 (for a review, see Ref. 271). In agreement with this idea, PARP-1 inhibitors attenuate neuronal cell death caused by excitotoxicity in culture.272,273 More compelling results were obtained from studies involving PARP-1 knockout mice, in which PARP-1 deficiency resulted in substantially reduced areas of infarct in an animal model of stroke.274,275 In addition, the absence of PARP-1 also dramatically reduces the size of lesions induced by intrastriatal NMDA injection276 and the extent of MPTP-induced loss of dopaminergic neurons.277 The protection conferred by genetic or pharmacological suppression of PARP-1 activity in models of stroke,278 NMDA excitotoxicity276 and traumatic brain injury279 appears to be long-lasting based on anatomical and behavioral analyses.
The cleavage of PARP-1 by caspase-3 during the process of apoptosis is thought to serve as a switch between necrotic vs. apoptotic cell death, ensuring that cells maintain sufficient ATP levels to die by apoptosis. Consistent with this idea, PARP-1 inhibitors attenuate necrotic, but not apoptotic, neuronal death in an in vitro model of cerebral ischemia.280 What is not clear in the stroke model, however, is whether subsequent to PARP1 inhibition, neurons in the core region of an infarct that would otherwise die by necrosis are rescued completely or die by apoptosis. Even though ATP depletion is prevented by PARP-1 inhibition, neurons in the core region still suffer from extensive oxidative damage, which should eventually activate apoptotic pathways. Apparently, however, even the apoptotic process is efficiently blocked since the protection afforded by PARP-1 inhibition is sustained for at least 2-3 weeks,276,278,279 beyond the typical time course of apoptosis. Thus, these results suggest that PARP-1 inhibition may also work to suppress apoptotic neuronal cell death. This would be consistent with the finding that PARP-1 inhibition can partially suppress p53-dependent neuronal cell death234 as well as p53 induction.281
A number of important issues remain to be examined with respect to the role of PARP in neuronal injury. For example, it is not clear whether NAD/ATP depletion can account for the entire cascade of PARP-mediated changes in neuronal viability. In addition, it is not clear if other members of the PARP family are involved in the neuronal response toxic insults. Nevertheless, data obtained thus far, suggests that PARP-1 plays an important role in several forms of neuronal apoptosis.
Bcl-2 Family Members and Mitochondrial Integrity
Properties of the Bcl-2 Family
Proteins belonging to the Bcl-2 family are key regulators of neuronal cell death and survival, and individual family members can serve to inhibit or promote apoptosis.282 The prototypical anti-apoptotic member, Bcl-2, was first discovered because of its involvement in 95% of follicular B-cell lymphomas as a result of a chromosomal translocation t(14;18). High levels of bcl-2 did not act to increase proliferation but rather increased cell survival, thus defining a new class of oncogenes.283-285 Proteins in this family share a number of Bcl-2 homology domains (BH domains). In general, the anti-apoptotic members, Bcl-2 and Bcl-XL, contain BH1-4, whereas the pro-apoptotic members, Bax and Bak, contain BH1-3. A third subset of family members including Bad, Bik, Blk, Hrk, Bim, Bid286 and Noxa287 contain only the BH3 domain. Many of the family members contain a hydrophobic C-terminal tail, which may serve to anchor these proteins into intracellular membranes.
Bcl-2 proteins regulate the apoptotic cascade mainly at the level of the mitochondria. Upon activation by an apoptotic signal, pro-apoptotic Bcl-2 proteins accumulate at the mitochondrial membrane.288-291 This is associated with alterations in mitochondrial membrane potential and the release of several pro-apoptotic factors including cytochrome c, which, in conjunction with Apaf-1, facilitates the activation of the caspase cascade (Fig. 1).292,293
The impact of pro-apoptotic Bcl-2 proteins on mitochondria can be inhibited by Bcl-2 and/or Bcl-XL.294,295 Although the mechanism underlying the regulation of mitochondrial membrane permeability is not completely understood, current hypotheses suggest that Bcl-2 family members regulate the outer mitochondrial membrane by interaction with the voltage-dependent anion channel (VDAC),296 and other components of the mitochondrial permeability transition pore (PTP) complex.297 Furthermore, there is evidence indicating that some Bcl-2 family members can form hetero- and homodimers with inherent ion channel-forming abilities.298-300
The ability of Bcl-2 family members to form dimers is essential to their function and regulation. Enforced homodimerization of Bax stimulates Bax translocation, caspase activation and cell death.301 On the other hand, heterodimerization of anti-apoptotic members such as Bcl-2 or Bcl-XL with pro-apoptotic members such as Bax can inhibit or activate apoptosis depending on the relative levels of each protein (Rheostat model302). Mutational analyses have shown that the BH domains are important for the dimerization of Bcl-2 family members. The BH1 and BH2 domains in Bcl-2 and Bcl-XL are essential for their dimerization to Bax.303 NMR and X-ray crystallographic analyses of Bcl-XL show that the α-helical BH1-3 domains are closely juxtaposed to form a hydrophobic binding pocket.304,305 Mutation of the BH3 domain in Bax, Bad and Bid have shown that this domain is essential for both the binding ability of these proteins and their pro-apoptotic function.306,307 These findings support the rheostat model for Bcl-2 proteins in apoptotic regulation. This hypothesis proposes that anti-apoptotic and pro-apoptotic Bcl-2 proteins exist in a delicate balance at the mitochondrial membrane. A shift in one direction or the other may determine the fate of the cell.
However, the rheostat model is perhaps too simple, as more recent evidence has shown the regulation of these proteins to be much more complex. Pro-apoptotic proteins such as Bad and Bid contain the critical amphipathic BH3 domain but lack a C-terminal hydrophobic tail present in many of the other membrane-anchored family members. These proteins can move between the cytosol and membranes in a regulated fashion. For example, the pro-apoptotic activity of Bad is regulated by phosphorylation. When Bad is phosphorylated on two specific serine residues, it is sequestered by binding to 14-3-3 and can no longer bind Bcl-2 or Bcl-XL.308 Phosphorylation also appears to play an important role in regulating other Bcl-2 family proteins. For example, Bcl-2 can be phosphorylated at serine 70 to activate Bcl-2's anti-apoptotic activity or it can be hyperphosphorylated on multiple sites to inhibit Bcl-2 activity.309,310 Furthermore, we have shown that p38 MAP kinase activity is involved in regulating Bax function.127 These changes in phosphorylation state may very well affect the protein-protein interactions of these Bcl-2 family members.
The BH3 domain-only proteins function by modulating the activity of other Bcl-2 family members. Bcl-2 proteins such as Bad, Bim, Bid, Bik, Blk and Hrk can bind to the hydrophobic groove of the anti-apoptotic family members as well as pro-apoptotic family members such as Bax and Bak.311,312 For example, Bid is a pro-apoptotic Bcl-2 homolog that, once cleaved by caspase-8, is targeted to mitochondria by cardiolipin.313 Truncated Bid binds and activates pro-apoptotic members, Bax and Bak.314-316 Other members, such as Bim, Bad and Noxa bind and inhibit the anti-apoptotic members, Bcl-2 and Bcl-XL.287,317,318 These interactions suggest that the BH3 domain-only proteins play an important role regulating the function of other Bcl-2 family members.
Bcl-2 Family Members in the Nervous System
Members of the Bcl-2 family are expressed in the nervous system during development and in the adult. Bcl-2 is widely expressed during development, whereas in the adult, its expression is low in the CNS but remains high in the PNS. Conversely, Bcl-XL remains highly expressed in the nervous system throughout both development and adult life.319 Bax is down-regulated in the adult CNS but highly expressed in the nervous system during the period of natural cell death when the number of neurons is reduced by 20-80%, presumably to match the number of innervating neurons with the size of the target tissue.320
Studies with transgenic mice have demonstrated the importance of Bcl-2 family members in regulating neuronal cell death. Animals overexpressing Bcl-2 contained 30–40% more neurons than wild-type animals following the period of developmental neuronal death. Furthermore, neurons overexpressing Bcl-2 are protected from apoptosis after facial and sciatic nerve axotomy as well as optic nerve transection. Although the exact mechanism is not known, it may be related to the effects of Bcl-2 on intracellular Ca2+ homeostasis.321 High levels of Bcl-2 expression have also proven protective in other models of neuronal cell death, for example, following ischemia and excitotoxic injury.322
Conversely, the absence of pro-apoptotic family members such as Bax tends to be protective. For example, Bax deficiency protects neurons from ionizing radiation, changes in extracellular ionic conditions, excitotoxicity and ischemia.204,215,323 One of the important initiators of cell death following stroke is thought to be DNA damage. DNA damage can independently activate cyclin-dependent kinases and p53 pathways, and both mediate Bax activation in neurons.153,215 Recently, a novel splice variant of Bax, Bax k, has been shown to promote death following ischemia, and its mRNA was distributed in selectively vulnerable hippocampal CA1 neurons that are destined to die following global ischemia.324
Recent evidence has shown Bim to be important in regulating neuronal cell death. Upon withdrawal of nerve growth factor (NGF), neurons activate c-Jun and induce BIMEL expression, and subsequently die in a Bax-dependent manner.325 Furthermore, overexpression of BIMEL induces cytochrome c release and apoptosis even in the presence of NGF. Finally, neurons treated with Bim antisense oligonucleotides and neurons from Bim−/− mice die more slowly following NGF withdrawal.326,327 These studies suggest that Bim and perhaps other BH3 domain-only proteins play redundant roles upstream of Bax in the apoptotic pathway following growth factor withdrawal, and that these proteins may be regulated by kinases such as JNK.328
On the other hand, there are some Bcl-2 family members that act quite different in neuronal versus non-neuronal cells. Bid is an example of a Bcl-2 family member that seems to have cell type-specific functions. One report suggests that, unlike in other cell types, Bid does not play an essential role in either naturally occurring or AraC-induced cell death in neurons.329 Conversely, another report suggests that cleavage of Bid may amplify caspase-8-induced neuronal death in a seizure model.330 Interestingly, another BH3 domain-only protein, N-Bak, plays opposite roles in neuronal versus non-neuronal cells. N-Bak is a neuron-specific, BH3-only isoform of Bak that is anti-apoptotic in neurons, but pro-apoptotic in non-neuronal cells.331 This was the first example of a neuron-specific Bcl-2 family member.
Bcl-2 family members have also been implicated in various neurodegenerative diseases such as Parkinson's Disease (PD), Alzheimer's Disease (AD) and motor neuron diseases such as amyotrophic lateral sclerosis (ALS) and spinal muscular atrophy (SMA). Overexpression of Bcl-2 and ablating Bax expression protected dopaminergic neurons from MPTP toxicity, a model of PD.332,333 Presenilin-1 and -2, genes involved in AD, have also been shown to interact with Bcl-XL.334 Mutations in the superoxide dismutase-1 (SOD-1) gene are linked to familial ALS, and spinal cord motor neuron death in mutant SOD-1 mice is associated with a decrease in Bcl-2 expression and an increase in Bax expression.335-337 Furthermore, in the spinal cords of these mice, Bax translocates from the cytosol to the mitochondria during the progression of the disease.338 Overexpression of Bcl-2 delays caspase expression, increases motor neuron survival, and improves muscle function in mutant SOD-1 mice.335,339 However, the disease eventually progresses, and duration of the disease is not altered.340 Survival of motor neuron protein (SMN), which has been implicated in SMA, has also been shown to interact with Bcl-2.341 In fact, Bcl-2 acts synergistically with SMN to inhibit Bax-induced or Fas-mediated toxicity.342 These studies suggest a possible role for Bcl-2 family members in a variety of neuropathological processes. Moreover, manipulating the expression and/or activity of Bcl-2 family members may prove instrumental in maintaining neuronal survival after injury and in response to disease.
Mitochondrial Pro-Apoptotic Factors
Mitochondria play a pivotal role in the amplification of an apoptotic signal since cytochrome c is normally sequestered in the mitochondrial intra-membrane space. In many cell types, apoptosis is associated with a loss in the normal electrochemical gradient employed by mitochondria to generate ATP (the proton motive force).343 Loss of mitochondrial membrane potential leads to opening of a protein complex known as the permeability transition pore.344 When this channel is opened, cytochrome c is released into the cytoplasm, leading to the formation of the apoptosome and activation of caspase-9.343 In neurons, mitochondrial cytochrome c release can be independent of permeability transition.345 Cytochrome c release in this situation may be mediated by the action of pro-apoptotic bcl-2 family members300 (discussed elsewhere).
In addition to cytochrome c, several other factors are released from mitochondria in response to apoptotic stimuli. One pro-apoptotic protein released by mitochondria is the apoptosis-inducing factor (AIF). AIF is released from mitochondria after permeability transition and translocates to the nucleus where it promotes chromatin condensation and DNA fragmentation in a caspase-independent manner.346 A second factor normally sequestered in mitochondria is known by the dual name of Smac/DIABLO. When released into the cytoplasm, this small protein up-regulates the proteolytic activity of caspases by interacting with members of the IAP family. A third pro-apoptotic factor normally resident in mitochondria, endonuclease G, was recently identified as an important apoptosis initiator.347 At this point, little is known about the role of the mitochondrial proteins Smac/DIABLO, AIF or endonuclease G in excitotoxicity or death receptor-mediated neuronal apoptosis. Caspase-2-deficient neurons demonstrate increased levels of smac, which is thought to allow developmental neuronal death to proceed in the absence of caspase-2 by releasing caspase-9 from IAP inhibition.348 A mutation in the C. elegans homologue for endonuclease G also prevents normal progression to neuronal apoptosis during development.349
Proteolytic Enzymes
Caspases
Pioneering studies aimed at identifying genes required for programmed neuronal death during the course of C. elegans development led to the discovery that one of these genes shared strong molecular homology with interlukin-1 converting enzyme (ICE).350 ICE turned out to be a member of a family of proteases later designated caspase enzymes.351 Caspase stands for cysteine-dependent, aspartate-specific protease. To date, fourteen family members have been identified. Some family members (caspases -4 and -5) are thus far unique to humans, and others, caspases -12 and -13, have not yet been identified in the human genome. All caspases are synthesized as inactive proenzymes and activated by proteolytic cleavage. The pro-enzyme is cleaved into an N-terminal peptide, a large subunit containing the catalytic portion of the enzyme and a small subunit at the C-terminal end of the peptide. The active enzyme consists of a tetrameric complex of two large subunits and two small subunits.352 Caspase enzymes have been divided into several distinct groups based on protein structure and putative function. Caspases -1, -4, -5, -11 and -13 are classified as cytokine activators, caspases -2, -8, -9, -10 and 12 are believed to act as initiators of the apoptotic cascade, and caspases -3, -6 and -7 are designated executioners of apoptosis. Caspase-14 appears to have a unique role in supporting the terminal differentiation of keratinocytes in the epidermis and cornea without promoting the typical features of apoptosis.353,354 Not all studies have supported these designations, and the biologic function associated with some of the less well-studied members of this enzyme family have not been evaluated in neurons or neural tissues. In addition, some debate remains about the possibility that caspases known as cytokine activators, primarily caspase-1, may also participate as apoptotic initiators in neurons355 or oligodendrocytes.356,357
The initiator caspases can be further subdivided into two distinct groups. One group is activated following ligand binding to members of the tumor necrosis factor α (TNF-α) receptor (TNF-R) family including FAS, TNF-R and TRAIL receptors. These caspases (-8 and -10) contain death effector domains (DED) in the N-terminal region of the pro-enzyme. The DED confers homomeric binding ability to adaptor proteins in the death-inducing signaling complex (DISC) at the cytoplasmic region of TNF-R family members known as death receptors. Ligand binding of death receptor family members promotes receptor clustering. The predominant theory of caspase activation is that death receptor clustering brings molecules of pro-caspase-8 or pro-caspase-10 in such close proximity that they can be activated by autocleavage.352
A second group of caspases (-2 and -9) contain a domain known as the caspase recruitment domain (CARD). The CARD domain appears to confer homomeric binding capabilities between caspases and their specific regulatory complexes. Caspase-2 is recruited to the DISC through interactions between its CARD and the CARD on an adapter protein called RAIDD. RAIDD contains both a CARD domain and a DED domain allowing it to interact with caspase-2 on one end and a DED containing serine/threonine kinase, RIP, that is associated with TNF-R1.358 Thus, despite lacking a DED domain, caspase-2 may still be activated by proximity-induced auto-processing. Pro-caspase-9 binds apoptotic protease activating factor-1 (Apaf-1) by CARD domain interaction, and Apaf-1 oligomerization promotes proximity-induced caspase-9 activation.359,360 Apaf-1 is a required peptide co-factor for caspase-9 activation, but caspase-9 activation also requires two additional factors, cytochrome c and dATP.361,362 The complex of these factors, designated the apoptosome, is under intense investigation and additional regulatory proteins continue to be identified.363 In addition, there is some evidence that human caspase activity is regulated by phosphorylation71 although the putative phosphorylation sites are not conserved in several other species.364
Once initiator caspases are activated, they can proteolytically process executioner caspases (caspases -3, -6 and -7) into their active form. This group of caspases contains truncated N-terminal sequences without known regulatory function. Once activated, their substrates include nuclear and cytoskeletal components that must be proteolyzed for a cell to develop the phenotypic characteristics of apoptosis. The activation of executioner caspases has been demonstrated in variety of neuronal death paradigms. A large number of studies have demonstrated that caspase-3 is activated in the CNS in response to a variety of injurious stimuli including ischemia, excitotoxic insult, trauma and neurodegenerative diseases.365,371 Caspase-3 is also required for normal developmental cell death in the CNS.372 Caspase-6 may play a role in the pathogenesis of Alzheimer's and Huntington's disease.373,374 Caspase-7 is activated during motoneuron degeneration in a mouse model of amyotrophic lateral sclerosis.338 However, the inhibition or ablation of executioner caspases may not improve neuronal survival, even though the morphologic changes associated with apoptosis do not develop.375,376
Endogenous Caspase Inhibitors
Apoptosis appears to be a tightly regulated cellular process. In addition to specific patterns of caspase activation, several endogenous inhibitors of caspase enzyme activity have also been described. One class of caspase inhibitor acts by forming proteolytically inactive heterodimeric complexes with active caspases. Examples of this strategy include splice variants of caspase-2377 and caspase-14.354 Very little is known about the presence of caspase-inhibitory splice variants in the central or peripheral nervous system. The FLICE (caspase-8) inhibitory protein (FLIP) shares a high degree of sequence homology with caspase-8 and inhibits caspase-8 activity by promoting the formation of a caspase-8/FLIP heterodimers devoid of proteolytic activity.378 Motoneurons expressing high levels of FLIP are resistant to FasL-induced apoptosis,4 and FLIP is expressed in neuroblasts in rat cortex during the early postnatal period.379 The localization of FLIP in developing neural tissue suggests that FLIP may play a regulatory role in cell survival during nervous system development. However, FLIP knockout mice have an embryonic lethal phenotype similar to that observed in caspase-8-deficient mice,380 suggesting that endogenous caspase inhibition is also required for normal caspase function, perhaps by preventing proteolytic inactivation of other important apoptosis regulators.
The peptide family of inhibitor of apoptosis (IAP) genes381 employs a second strategy for endogenous caspase inhibition. This family of proteins consists of at least seven mammalian members including X-chromosome-linked inhibitor of apoptosis (XIAP). All members of this family contain at least one BIR (baculovirus inhibitor of apoptosis repeat) domain that binds and inhibits the proteolytic activity of caspases -3 and -7. Some of the IAP proteins also inhibit caspase-9 activity (for review, see Ref. 382). Inhibition by IAP family members can be overcome by several different mechanisms. The C-terminal region of some IAPs contains a domain referred to as the RING (Really Interesting New Gene) domain, which contains ubiquitin E3 ligase activity that targets them for rapid proteasomal degradation.383 Two other proteins that bind IAPs and prevent caspase inhibition have also been identified. The XIAP-associated factor 1 (XAF1) binds XIAP in the nucleus, potentially promoting release of caspase inhibition once the enzymes have been able to translocate into the nuclear compartment.384 The second mitochondria-derived activator of caspase (Smac) or DIABLO protein is normally sequestered in mitochondria but will bind and inhibit the anti-apoptotic activities of IAPs when it is released into the cytoplasm.385,386
Calpains
In addition to caspases, there are additional classes of proteases associated with neuronal cell death. The calpains are calcium-activated cysteine proteases implicated in neuronal cell death following acute neurological insults. Administration of calpain inhibitors during the initial 24-h period following injury can attenuate injury-induced derangements of neuronal structure and function following traumatic brain injury and excitotoxicity.387-393 At least one substrate, fodrin (spectrin), appears to be shared between the calpains and caspases, but it is not presently known if the caspases and calpains activate one another. In addition, a novel serine protease has been identified which is expressed at ten-fold higher concentrations in the CNS than in peripheral tissues of the rat and human.394 The mRNA for this protease was found to be significantly elevated by excitotoxic injury. It seems clear that proteases can have profound effects on neuronal viability following injury although additional studies will be required to determine if they represent a rate-limiting step in the apoptotic process.
Abbreviations
AIF, apoptosis-inducing factor; ALS, amyotrophic lateral sclerosis; Apaf-1, apoptotic protease activating factor-1; ATM, ataxia telangiectasia mutated; BH domain, Bcl-2 homology domain; CARD, caspase recruitment domain; CDK, cyclin-dependent kinase; DD, death domain; DISC, death-inducing signaling complex; DNA-PK, DNA-dependent protein kinase; ERK, extracellular signal-regulated kinase; FADD, Fas-associated death domain; FasL, Fas ligand; FLIP, FLICE (caspase-8) inhibitory protein; IAP, inhibitor of apoptosis; IR, ionizing radiation; JNK, c-Jun N-terminal kinase; MKK, MAP kinase kinase; NMDA, N-methyl-D-aspartate; PARP, poly(ADP-ribose) polymerase; PI3K, phosphatidylinositol-3 kinase; RIP, receptor-interacting protein; TNFR, tumor necrosis factor receptor; TRADD, TNFR-associated death domain; TRAF2, TNF receptor-associated factor 2
References
- 1.
- Walczak H, Krammer PH. The CD95 (APO-1/Fas) and the TRAIL (APO-2L) apoptosis systems. Exp Cell Res. 2000;256:58–66. [PubMed: 10739652]
- 2.
- Bechmann I, Mor G, Nilsen J, Eliza M, Nitsch R, Naftolin F. FasL (CD95L, Apo1L) is expressed in the normal rat and human brain: Evidence for the existence of an immunological brain barrier. Glia. 1999;27:62–74. [PubMed: 10401633]
- 3.
- Felderhoff-Mueser U, Taylor DL, Greenwood K, Kozma M, Stibenz D, Joashi UC. et al. Fas/CD95/APO-1 can function as a death receptor for neuronal cells in vitro and in vivo and is upregulated following cerebral hypoxic- ischemic injury to the developing rat brain. Brain Pathol. 2000;10:17–29. [PMC free article: PMC8098164] [PubMed: 10668892]
- 4.
- Raoul C, Henderson CE, Pettmann B. Programmed cell death of embryonic motoneurons triggered through the Fas death receptor. J Cell Biol. 1999; 147:1049–1062. [PMC free article: PMC2169347] [PubMed: 10579724]
- 5.
- Matsushita K, Wu Y, Qiu J, Lang-Lazdunski L, Hirt L, Waeber C. et al. Fas receptor and neuronal cell death after spinal cord ischemia. J Neurosci. 2000;20:6879–6887. [PMC free article: PMC6772830] [PubMed: 10995832]
- 6.
- Martin-Villalba A, Herr I, Jeremias I, Hahne M, Brandt R, Vogel J. et al. CD95 ligand (Fas-L/APO-1L) and tumor necrosis factor-related apoptosis-inducing ligand mediate ischemia-induced apoptosis in neurons. J Neurosci. 1999;19:3809–3817. [PMC free article: PMC6782733] [PubMed: 10234013]
- 7.
- Rosenbaum DM, Gupta G, D'Amore J, Singh M, Weidenheim K, Zhang H. et al. Fas (CD95/APO-1) plays a role in the pathophysiology of focal cerebral ischemia. J Neurosci Res. 2000;61:686–692. [PubMed: 10972965]
- 8.
- Elovaara I, Sabri F, Gray F, Alafuzoff I, Chiodi F. Upregulated expression of Fas and Fas ligand in brain through the spectrum of HIV-1 infection. Acta Neuropathol (Berl). 1999;98:355–362. [PubMed: 10502040]
- 9.
- Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410:988–994. [PubMed: 11309629]
- 10.
- Sharma K, Wang RX, Zhang LY, Yin DL, Luo XY, Solomon JC. et al. Death the Fas way: Regulation and pathophysiology of CD95 and its ligand. Pharmacol Ther. 2000;88:333–347. [PubMed: 11337030]
- 11.
- Raoul C, Pettmann B, Henderson CE. Active killing of neurons during development and following stress: a role for p75(NTR) and Fas? Curr Opin Neurobiol. 2000;10:111–117. [PubMed: 10679436]
- 12.
- Varfolomeev EE, Schuchmann M, Luria V, Chiannilkulchai N, Beckmann JS, Mett IL. et al. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity. 1998;9:267–276. [PubMed: 9729047]
- 13.
- Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. [PubMed: 9727492]
- 14.
- Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli KJ. et al. Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 1998;17:1675–1687. [PMC free article: PMC1170515] [PubMed: 9501089]
- 15.
- Bratton SB, MacFarlane M, Cain K, Cohen GM. Protein complexes activate distinct caspase cascades in death receptor and stress-induced apoptosis. Exp Cell Res. 2000;256:27–33. [PubMed: 10739648]
- 16.
- Wang J, Zheng L, Lobito A, Chan FK, Dale J, Sneller M. et al. Inherited human Caspase 10 mutations underlie defective lymphocyte and dendritic cell apoptosis in autoimmune lymphoproliferative syndrome type II. Cell. 1999;98:47–58. [PubMed: 10412980]
- 17.
- Reimann-Philipp U, Ovase R, Weigel PH, Grammas P. Mechanisms of cell death in primary cortical neurons and PC12 cells. J Neurosci Res. 2001;64:654–660. [PubMed: 11398190]
- 18.
- Barker V, Middleton G, Davey F, Davies AM. TNFalpha contributes to the death of NGF-dependent neurons during development. Nat Neurosci. 2001;4:1194–1198. [PubMed: 11685224]
- 19.
- Kim GM, Xu J, Song SK, Yan P, Ku G, Xu XM. et al. Tumor necrosis factor receptor deletion reduces nuclear factor-kappaB activation, cellular inhibitor of apoptosis protein 2 expression, and functional recovery after traumatic spinal cord injury. J Neurosci. 2001;21:6617–6625. [PMC free article: PMC6763083] [PubMed: 11517251]
- 20.
- Lin Y, Devin A, Cook A, Keane MM, Kelliher M, Lipkowitz S. et al. The death domain kinase RIP is essential for TRAIL (Apo2L)-induced activation of IkappaB kinase and c-Jun N-terminal kinase. Mol Cell Biol. 2000;20: 6638–6645. [PMC free article: PMC86162] [PubMed: 10958661]
- 21.
- Wajant H, Henkler F, Scheurich P. The TNF-receptor-associated factor family: Scaffold molecules for cytokine receptors, kinases and their regulators. Cell Signal. 2001;13:389–400. [PubMed: 11384837]
- 22.
- Baud V, Karin M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 2001;11:372–377. [PubMed: 11514191]
- 23.
- Grell M, Zimmermann G, Gottfried E, Chen CM, Grunwald U, Huang DC. et al. Induction of cell death by tumour necrosis factor (TNF) receptor 2, CD40 and CD30: A role for TNF-R1 activation by endogenous membrane-anchored TNF. EMBO J. 1999;18:3034–3043. [PMC free article: PMC1171385] [PubMed: 10357816]
- 24.
- Griffith TS, Lynch DH. TRAIL: A molecule with multiple receptors and control mechanisms. Curr Opin Immunol. 1998;10:559–563. [PubMed: 9794836]
- 25.
- Wiley SR, Schooley K, Smolak PJ, Din WS, Huang CP, Nicholl JK. et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. 1995;3:673–682. [PubMed: 8777713]
- 26.
- Sheikh MS, Fornace A J Jr. Death and decoy receptors and p53-mediated apoptosis. Leukemia. 2000;14:1509–1513. [PubMed: 10942251]
- 27.
- Degli-Esposti MA, Dougall WC, Smolak PJ, Waugh JY, Smith CA, Goodwin RG. The novel receptor TRAIL-R4 induces NF-kappaB and protects against TRAIL-mediated apoptosis, yet retains an incomplete death domain. Immunity. 1997;7:813–820. [PubMed: 9430226]
- 28.
- Ashkenazi A, Pai RC, Fong S, Leung S, Lawrence DA, Marsters SA. et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest. 1999;104:155–162. [PMC free article: PMC408479] [PubMed: 10411544]
- 29.
- Zhang XD, Nguyen T, Thomas WD, Sanders JE, Hersey P. Mechanisms of resistance of normal cells to TRAIL induced apoptosis vary between different cell types. FEBS Lett. 2000;482:193–199. [PubMed: 11024459]
- 30.
- Jo M, Kim TH, Seol DW, Esplen JE, Dorko K, Billiar TR. et al. Apoptosis induced in normal human hepatocytes by tumor necrosis factor- related apoptosis-inducing ligand. Nat Med. 2000;6:564–567. [PubMed: 10802713]
- 31.
- Frank S, Kohler U, Schackert G, Schackert HK. Expression of TRAIL and its receptors in human brain tumors. Biochem Biophys Res Commun. 1999;257:454–459. [PubMed: 10198234]
- 32.
- Nitsch R, Bechmann I, Deisz RA, Haas D, Lehmann TN, Wendling U. et al. Human brain-cell death induced by tumour-necrosis-factor-related apoptosis-inducing ligand (TRAIL). Lancet. 2000;356:827–828. [PubMed: 11022932]
- 33.
- Eggert A, Grotzer MA, Zuzak TJ, Wiewrodt BR, Ikegaki N, Brodeur GM. Resistance to TRAIL-induced apoptosis in neuroblastoma cells correlates with a loss of caspase-8 expression. Med Pediatr Oncol. 2000;35:603–607. [PubMed: 11107127]
- 34.
- Sprick MR, Weigand MA, Rieser E, Rauch CT, Juo P, Blenis J. et al. FADD/MORT1 and caspase-8 are recruited to TRAIL receptors 1 and 2 and are essential for apoptosis mediated by TRAIL receptor 2. Immunity. 2000;12:599–609. [PubMed: 10894160]
- 35.
- Mariani SM, Matiba B, Armandola EA, Krammer PH. Interleukin 1 beta-converting enzyme related proteases/caspases are involved in TRAIL-induced apoptosis of myeloma and leukemia cells. J Cell Biol. 1997;137:221–229. [PMC free article: PMC2139852] [PubMed: 9105050]
- 36.
- Kuang AA, Diehl GE, Zhang J, Winoto A. FADD is required for DR4- and DR5-mediated apoptosis: lack of trail- induced apoptosis in FADD-deficient mouse embryonic fibroblasts. J Biol Chem. 2000;275:25065–25068. [PubMed: 10862756]
- 37.
- Meng RD, McDonald E R 3rd ,, Sheikh MS, Fornace A J Jr., El-Deiry WS. The TRAIL decoy receptor TRUNDD (DcR2, TRAIL-R4) is induced by adenovirus-p53 overexpression and can delay TRAIL-, p53-, and KILLER/DR5-dependent colon cancer apoptosis. Mol Ther. 2000;1:130–144. [PubMed: 10933923]
- 38.
- Meng RD, El-Deiry WS. p53-independent upregulation of KILLER/DR5 TRAIL receptor expression by glucocorticoids and interferon-gamma. Exp Cell Res. 2001;262:154–169. [PubMed: 11139340]
- 39.
- Sheikh MS, Huang Y, Fernandez-Salas EA, El-Deiry WS, Friess H, Amundson S. et al. The antiapoptotic decoy receptor TRID/TRAIL-R3 is a p53-regulated DNA damage-inducible gene that is overexpressed in primary tumors of the gastrointestinal tract. Oncogene. 1999;18:4153–4159. [PubMed: 10435597]
- 40.
- Pan G, Ni J, Wei YF, Yu G, Gentz R, Dixit VM. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science. 1997;277:815–818. [PubMed: 9242610]
- 41.
- Sheridan JP, Marsters SA, Pitti RM, Gurney A, Skubatch M, Baldwin D. et al. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science. 1997;277:818–821. [PubMed: 9242611]
- 42.
- Griffith TS, Rauch CT, Smolak PJ, Waugh JY, Boiani N, Lynch DH. et al. Functional analysis of TRAIL receptors using monoclonal antibodies. J Immunol. 1999;162:2597–2605. [PubMed: 10072501]
- 43.
- Zhang XD, Franco A, Myers K, Gray C, Nguyen T, Hersey P. Relation of TNF-related apoptosis-inducing ligand (TRAIL) receptor and FLICE-inhibitory protein expression to TRAIL-induced apoptosis of melanoma. Cancer Res. 1999;59:2747–2753. [PubMed: 10364001]
- 44.
- Sedel F, Bechade C, Triller A. Nerve growth factor (NGF) induces motoneuron apoptosis in rat embryonic spinal cord in vitro. Eur J Neurosci. 1999;11:3904–3912. [PubMed: 10583479]
- 45.
- Wiese S, Metzger F, Holtmann B, Sendtner M. The role of p75NTR in modulating neurotrophin survival effects in developing motoneurons. Eur J Neurosci. 1999;11:1668–1676. [PubMed: 10215920]
- 46.
- Ferri CC, Moore FA, Bisby MA. Effects of facial nerve injury on mouse motoneurons lacking the p75 low- affinity neurotrophin receptor. J Neurobiol. 1998;34:1–9. [PubMed: 9469614]
- 47.
- Bamji SX, Majdan M, Pozniak CD, Belliveau DJ, Aloyz R, Kohn et al. The p75 neurotrophin receptor mediates neuronal apoptosis and is essential for naturally occurring sympathetic neuron death. J Cell Biol. 1998;140:911–923. [PMC free article: PMC2141754] [PubMed: 9472042]
- 48.
- Frade JM, Barde YA. Genetic evidence for cell death mediated by nerve growth factor and the neurotrophin receptor p75 in the developing mouse retina and spinal cord. Development. 1999;126:683–690. [PubMed: 9895316]
- 49.
- Casaccia-Bonnefil P, Kong H, Chao MV. Neurotrophins: the biological paradox of survival factors eliciting apoptosis. Cell Death Differ. 1998;5:357–364. [PubMed: 10200484]
- 50.
- Wang X, Bauer JH, Li Y, Shao Z, Zetoune FS, Cattaneo E. et al. Characterization of a p75(NTR) apoptotic signaling pathway using a novel cellular model. J Biol Chem. 2001;276:33812–33820. [PubMed: 11451944]
- 51.
- Casademunt E, Carter BD, Benzel I, Frade JM, Dechant G, Barde YA. The zinc finger protein NRIF interacts with the neurotrophin receptor p75(NTR) and participates in programmed cell death. EMBO J. 1999;18:6050–6061. [PMC free article: PMC1171670] [PubMed: 10545116]
- 52.
- Mukai J, Hachiya T, Shoji-Hoshino S, Kimura MT, Nadano D, Suvanto P. et al. NADE, a p75NTR-associated cell death executor, is involved in signal transduction mediated by the common neurotrophin receptor p75NTR. J Biol Chem. 2000;275:17566–17570. [PubMed: 10764727]
- 53.
- Salehi AH, Roux PP, Kubu CJ, Zeindler C, Bhakar A, Tannis LL. et al. NRAGE, a novel MAGE protein, interacts with the p75 neurotrophin receptor and facilitates nerve growth factor-dependent apoptosis. Neuron. 2000;27:279–288. [PubMed: 10985348]
- 54.
- Khursigara G, Bertin J, Yano H, Moffett H, DiStefano PS, Chao MV. A prosurvival function for the p75 receptor death domain mediated via the caspase recruitment domain receptor-interacting protein 2. J Neurosci. 2001;21:5854–5863. [PMC free article: PMC6763175] [PubMed: 11487608]
- 55.
- Yao R, Cooper GM. Requirement for phosphatidylinositol-3 kinase in the prevention of apoptosis by nerve growth factor. Science. 1995;267:2003–2006. [PubMed: 7701324]
- 56.
- Crowder RJ, Freeman RS. Phosphatidylinositol 3-kinase and Akt protein kinase are necessary and sufficient for the survival of nerve growth factor-dependent sympathetic neurons. J Neurosci. 1998;18:2933–2943. [PMC free article: PMC6792598] [PubMed: 9526010]
- 57.
- Ghosh A, Greenberg ME. Distinct roles for bFGF and NT-3 in the regulation of cortical neurogenesis. Neuron. 1995;15:89–103. [PubMed: 7619533]
- 58.
- Kuruvilla R, Ye H, Ginty DD. Spatially and functionally distinct roles of the PI3-K effector pathway during NGF signaling in sympathetic neurons. Neuron. 2000;27:499–512. [PubMed: 11055433]
- 59.
- Segal RA, Greenberg ME. Intracellular signaling pathways activated by neurotrophic factors. Annu Rev Neurosci. 1996;19:463–489. [PubMed: 8833451]
- 60.
- Brunet A, Datta SR, Greenberg ME. Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr Opin Neurobiol. 2001;11:297–305. [PubMed: 11399427]
- 61.
- Holgado-Madruga M, Moscatello DK, Emlet DR, Dieterich R, Wong AJ. Grb2-associated binder-1 mediates phosphatidylinositol 3-kinase activation and the promotion of cell survival by nerve growth factor. Proc Natl Acad Sci USA. 1997;94:12419–12424. [PMC free article: PMC24976] [PubMed: 9356464]
- 62.
- Kauffmann-Zeh A, Rodriguez-Viciana P, Ulrich E, Gilbert C, Coffer P, Downward J. et al. Suppression of c-Myc-induced apoptosis by Ras signalling through PI(3)K and PKB. Nature. 1997;385:544–548. [PubMed: 9020362]
- 63.
- Dhand R, Hara K, Hiles I, Bax B, Gout I, Panayotou G. et al. PI 3-kinase: structural and functional analysis of intersubunit interactions. EMBO J. 1994;13:511–521. [PMC free article: PMC394840] [PubMed: 8313896]
- 64.
- Hu Q, Klippel A, Muslin AJ, Fantl WJ, Williams LT. Ras-dependent induction of cellular responses by constitutively active phosphatidylinositol-3 kinase. Science. 1995;268:100–102. [PubMed: 7701328]
- 65.
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM. et al. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. [PubMed: 9005851]
- 66.
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS. et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. [PubMed: 10102273]
- 67.
- Yamaguchi A, Tamatani M, Matsuzaki H, Namikawa K, Kiyama H, Vitek MP. et al. Akt activation protects hippocampal neurons from apoptosis by inhibiting transcriptional activity of p53. J Biol Chem. 2001;276:5256–5264. [PubMed: 11054421]
- 68.
- Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286:1358–1362. [PubMed: 10558990]
- 69.
- Maggirwar SB, Sarmiere PD, Dewhurst S, Freeman RS. Nerve growth factor-dependent activation of NF-kappaB contributes to survival of sympathetic neurons. J Neurosci. 1998;18:10356–10365. [PMC free article: PMC6793355] [PubMed: 9852573]
- 70.
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y. Akt phosphorylation of BAD couples survival signals to the cell- intrinsic death machinery. Cell. 1997;91:231–241. [PubMed: 9346240]
- 71.
- Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E. et al. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–1321. [PubMed: 9812896]
- 72.
- Hetman M, Cavanaugh JE, Kimelman D, Xia Z. Role of glycogen synthase kinase-3beta in neuronal apoptosis induced by trophic withdrawal. J Neurosci. 2000;20:2567–2574. [PMC free article: PMC6772233] [PubMed: 10729337]
- 73.
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. [PubMed: 7481820]
- 74.
- Impey S, Obrietan K, Storm DR. Making new connections: role of ERK/MAP kinase signaling in neuronal plasticity. Neuron. 1999;23:11–14. [PubMed: 10402188]
- 75.
- Sweatt JD. The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J Neurochem. 2001;76:1–10. [PubMed: 11145972]
- 76.
- Murray B, Alessandrini A, Cole AJ, Yee AG, Furshpan EJ. Inhibition of the p44/42 MAP kinase pathway protects hippocampal neurons in a cell-culture model of seizure activity. Proc Natl Acad Sci USA. 1998;95:11975–11980. [PMC free article: PMC21750] [PubMed: 9751775]
- 77.
- Kulich SM, Chu CT. Sustained extracellular signal-regulated kinase activation by 6- hydroxydopamine: Implications for Parkinson's disease. J Neurochem. 2001;77:1058–1066. [PMC free article: PMC1868550] [PubMed: 11359871]
- 78.
- Stanciu M, DeFranco DB. Prolonged nuclear retention of activated ERK promotes cell death generated by oxidative toxicity or proteasome inhibition in a neuronal cell line. J Biol Chem. 2002;277:4010–4017. [PubMed: 11726647]
- 79.
- Abe MK, Kahle KT, Saelzler MP, Orth K, Dixon JE, Rosner MR. ERK7 is an autoactivated member of the MAPK family. J Biol Chem. 2001;276:21272–21279. [PubMed: 11287416]
- 80.
- Hetman M, Xia Z. Signaling pathways mediating anti-apoptotic action of neurotrophins. Acta Neurobiol Exp (Wars). 2000;60:531–545. [PubMed: 11200182]
- 81.
- Grewal SS, York RD, Stork PJ. Extracellular-signal-regulated kinase signalling in neurons. Curr Opin Neurobiol. 1999;9:544–553. [PubMed: 10508738]
- 82.
- Alessandrini A, Namura S, Moskowitz MA, Bonventre JV. MEK1 protein kinase inhibition protects against damage resulting from focal cerebral ischemia. Proc Natl Acad Sci USA. 1999;96:12866–12869. [PMC free article: PMC23136] [PubMed: 10536014]
- 83.
- Grammer TC, Blenis J. Evidence for MEK-independent pathways regulating the prolonged activation of the ERK-MAP kinases. Oncogene. 1997;14:1635–1642. [PubMed: 9135064]
- 84.
- Hetman M, Kanning K, Cavanaugh JE, Xia Z. Neuroprotection by brain-derived neurotrophic factor is mediated by extracellular signal-regulated kinase and phosphatidylinositol 3-kinase. J Biol Chem. 1999;274:22569–22580. [PubMed: 10428835]
- 85.
- Gonzalez-Zulueta M, Feldman AB, Klesse LJ, Kalb RG, Dillman JF, Parada LF. et al. Requirement for nitric oxide activation of p21(ras)/extracellular regulated kinase in neuronal ischemic preconditioning. Proc Natl Acad Sci USA. 2000;97:436–441. [PMC free article: PMC26681] [PubMed: 10618436]
- 86.
- Ferrer I, Ballabriga J, Pozas E. Transient forebrain ischemia in the adult gerbil is associated with a complex c-Jun response. Neuroreport. 1997;8:2483–2487. [PubMed: 9261813]
- 87.
- Yang DD, Kuan CY, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ. et al. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 1997;389:865–870. [PubMed: 9349820]
- 88.
- Jeon SH, Kim YS, Bae CD, Park JB. Activation of JNK and p38 in rat hippocampus after kainic acid induced seizure. Exp Mol Med. 2000;32:227–230. [PubMed: 11190275]
- 89.
- Chihab R, Ferry C, Koziel V, Monin P, Daval JL. Sequential activation of activator protein-1-related transcription factors and JNK protein kinases may contribute to apoptotic death induced by transient hypoxia in developing brain neurons. Brain Res Mol Brain Res. 1998;63:105–120. [PubMed: 9838068]
- 90.
- Morishima Y, Gotoh Y, Zieg J, Barrett T, Takano H, Flavell R. et al. Beta-amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand. J Neurosci. 2001;21:7551–7560. [PMC free article: PMC6762892] [PubMed: 11567045]
- 91.
- Troy CM, Rabacchi SA, Xu Z, Maroney AC, Connors TJ, Shelanski ML. et al. beta-Amyloid-induced neuronal apoptosis requires c-Jun N-terminal kinase activation. J Neurochem. 2001;77:157–164. [PubMed: 11279271]
- 92.
- Namgung U, Xia Z. Arsenite-induced apoptosis in cortical neurons is mediated by c-Jun N- terminal protein kinase 3 and p38 mitogen-activated protein kinase. J Neurosci. 2000;20:6442–6451. [PMC free article: PMC6772983] [PubMed: 10964950]
- 93.
- Herdegen T, Claret FX, Kallunki T, Martin-Villalba A, Winter C, Hunter T. et al. Lasting N-terminal phosphorylation of c-Jun and activation of c-Jun N-terminal kinases after neuronal injury. J Neurosci. 1998;18:5124–5135. [PMC free article: PMC6793486] [PubMed: 9651196]
- 94.
- Mielke K, Herdegen T. JNK and p38 stresskinasesDegenerative effectors of signal-transduction-cascades in the nervous system. Prog Neurobiol. 2000;61:45–60. [PubMed: 10759064]
- 95.
- Gupta S, Barrett T, Whitmarsh AJ, Cavanagh J, Sluss HK, Derijard B. et al. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996;15:2760–2770. [PMC free article: PMC450211] [PubMed: 8654373]
- 96.
- Bozyczko-Coyne D, O'Kane TM, Wu ZL, Dobrzanski P, Murthy S, Vaught JL. et al. CEP-1347/KT-7515, an inhibitor of SAPK/JNK pathway activation, promotes survival and blocks multiple events associated with Abeta-induced cortical neuron apoptosis. J Neurochem. 2001;77:849–863. [PubMed: 11331414]
- 97.
- Zhu X, Raina AK, Rottkamp CA, Aliev G, Perry G, Boux H. et al. Activation and redistribution of c-Jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer's disease. J Neurochem. 2001;76:435–441. [PubMed: 11208906]
- 98.
- Saporito MS, Thomas BA, Scott RW. MPTP activates c-Jun NH(2)-terminal kinase (JNK) and its upstream regulatory kinase MKK4 in nigrostriatal neurons in vivo. J Neurochem. 2000;75:1200–1208. [PubMed: 10936203]
- 99.
- Harding TC, Xue L, Bienemann A, Haywood D, Dickens M, Tolkovsky AM. et al. Inhibition of JNK by overexpression of the JNK binding domain of JIP-1 prevents apoptosis in sympathetic neurons. J Biol Chem. 2001;276:4531–4534. [PubMed: 11121395]
- 100.
- Le-Niculescu H, Bonfoco E, Kasuya Y, Claret FX, Green DR, Karin M. Withdrawal of survival factors results in activation of the JNK pathway in neuronal cells leading to Fas ligand induction and cell death. Mol Cell Biol. 1999;19:751–763. [PMC free article: PMC83932] [PubMed: 9858598]
- 101.
- Bruckner SR, Tammariello SP, Kuan CY, Flavell RA, Rakic P, Estus S. JNK3 contributes to c-Jun activation and apoptosis but not oxidative stress in nerve growth factor-deprived sympathetic neurons. J Neurochem. 2001;78:298–303. [PubMed: 11461965]
- 102.
- Behrens A, Sibilia M, Wagner EF. Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat Genet. 1999;21:326–329. [PubMed: 10080190]
- 103.
- Yang D, Tournier C, Wysk M, Lu HT, Xu J, Davis RJ. et al. Targeted disruption of the MKK4 gene causes embryonic death, inhibition of c-Jun NH2-terminal kinase activation, and defects in AP-1 transcriptional activity. Proc Natl Acad Sci USA. 1997;94:3004–3009. [PMC free article: PMC20312] [PubMed: 9096336]
- 104.
- Holland PM, Suzanne M, Campbell JS, Noselli S, Cooper JA. MKK7 is a stress-activated mitogen-activated protein kinase kinase functionally related to hemipterous. J Biol Chem. 1997;272:24994–24998. [PubMed: 9312105]
- 105.
- Ham J, Eilers A, Whitfield J, Neame SJ, Shah B. c-Jun and the transcriptional control of neuronal apoptosis. Biochem Pharmacol. 2000;60:1015–1021. [PubMed: 11007936]
- 106.
- Harper SJ, LoGrasso P. Signalling for survival and death in neurones: The role of stress- activated kinases, JNK and p38. Cell Signal. 2001;13:299–310. [PubMed: 11369511]
- 107.
- Tibbles LA, Ing YL, Kiefer F, Chan J, Iscove N, Woodgett JR. et al. MLK-3 activates the SAPK/JNK and p38/RK pathways via SEK1 and MKK3/6. EMBO J. 1996;15:7026–7035. [PMC free article: PMC452528] [PubMed: 9003778]
- 108.
- Lin A, Minden A, Martinetto H, Claret FX, Lange-Carter C, Mercurio F. et al. Identification of a dual specificity kinase that activates the Jun kinases and p38-Mpk2. Science. 1995;268:286–290. [PubMed: 7716521]
- 109.
- Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T. et al. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–94. [PubMed: 8974401]
- 110.
- Wang XS, Diener K, Jannuzzi D, Trollinger D, Tan TH, Lichenstein H. et al. Molecular cloning and characterization of a novel protein kinase with a catalytic domain homologous to mitogen-activated protein kinase kinase kinase. J Biol Chem. 1996;271:31607–31611. [PubMed: 8940179]
- 111.
- Kanamoto T, Mota M, Takeda K, Rubin LL, Miyazono K, Ichijo H. et al. Role of apoptosis signal-regulating kinase in regulation of the c-Jun N-terminal kinase pathway and apoptosis in sympathetic neurons. Mol Cell Biol. 2000;20:196–204. [PMC free article: PMC85075] [PubMed: 10594022]
- 112.
- Whitmarsh AJ, Cavanagh J, Tournier C, Yasuda J, Davis RJ. A mammalian scaffold complex that selectively mediates MAP kinase activation. Science. 1998;281:1671–1674. [PubMed: 9733513]
- 113.
- Derijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T. et al. JNK1: A protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–1037. [PubMed: 8137421]
- 114.
- Hu MC, Qiu WR, Wang YP. JNK1, JNK2 and JNK3 are p53 N-terminal serine 34 kinases. Oncogene. 1997;15:2277–2287. [PubMed: 9393873]
- 115.
- Fuchs SY, Adler V, Pincus MR, Ronai Z. MEKK1/JNK signaling stabilizes and activates p53. Proc Natl Acad Sci USA. 1997;95:10541–10546. [PMC free article: PMC27930] [PubMed: 9724739]
- 116.
- Zhang Y, Zhou L, Miller CA. A splicing variant of a death domain protein that is regulated by a mitogen-activated kinase is a substrate for c-Jun N-terminal kinase in the human central nervous system. Proc Natl Acad Sci USA. 1998;95:2586–2591. [PMC free article: PMC19423] [PubMed: 9482930]
- 117.
- Derijard B, Raingeaud J, Barrett T, Wu IH, Han J, Ulevitch RJ. et al. Independent human MAP-kinase signal traduction pathways defined by MEK and MKK isoforms. Science. 1995;267:682–685. [PubMed: 7839144]
- 118.
- Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ. et al. Pro-inflammatory cytokines and environmental stress cause p38 mitogen- activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem. 1995;270:7420–7426. [PubMed: 7535770]
- 119.
- Han J, Lee JD, Jiang Y, Li Z, Feng L, Ulevitch RJ. Characterization of the structure and function of a novel MAP kinase kinase (MKK6). J Biol Chem. 1996;271:2886–2891. [PubMed: 8621675]
- 120.
- Nath R, McGinnis K, Dutta S, Shivers B, Wang KK. Inhibition of p38 kinase mimics survival signal-linked protection against apoptosis in rat cerebellar granule neurons. Cell Mol Biol Lett. 2001;6:173–184. [PubMed: 11544639]
- 121.
- Yamagishi S, Yamada M, Ishikawa Y, Matsumoto T, Ikeuchi T, Hatanaka H. p38 mitogen-activated protein kinase regulates low potassium-induced c-Jun phosphorylation and apoptosis in cultured cerebellar granule neurons. J Biol Chem. 2001;276:5129–5133. [PubMed: 11083864]
- 122.
- Willaime S, Vanhoutte P, Caboche J, Lemaigre-Dubreuil Y, Mariani J, Brugg B. Ceramide-induced apoptosis in cortical neurons is mediated by an increase in p38 phosphorylation and not by the decrease in ERK phosphorylation. Eur J Neurosci. 2011;13:2037–2046. [PubMed: 11422444]
- 123.
- McLaughlin B, Pal S, Tran MP, Parsons AA, Barone FC, Erhardt JA. et al. p38 activation is required upstream of potassium current enhancement and caspase cleavage in thiol oxidant-induced neuronal apoptosis. J Neurosci. 2001;21:3303–3311. [PMC free article: PMC3746747] [PubMed: 11331359]
- 124.
- De Zutter GS, Davis RJ. Pro-apoptotic gene expression mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Proc Natl Acad Sci USA. 2001;98:6168–6173. [PMC free article: PMC33440] [PubMed: 11344273]
- 125.
- Barone FC, Irving EA, Ray AM, Lee JC, Kassis S, Kumar S. et al. Inhibition of p38 mitogen-activated protein kinase provides neuroprotection in cerebral focal ischemia. Med Res Rev. 2001;21:129–145. [PubMed: 11223862]
- 126.
- Kawasaki H, Morooka T, Shimohama S, Kimura J, Hirano T, Gotoh Y. et al. Activation and involvement of p38 mitogen-activated protein kinase in glutamate-induced apoptosis in rat cerebellar granule cells. J Biol Chem. 1997;272:18518–18521. [PubMed: 9228012]
- 127.
- Ghatan S, Larner S, Kinoshita Y, Hetman M, Patel L, Xia Z. et al. p38 MAP kinase mediates bax translocation in nitric oxide-induced apoptosis in neurons. J Cell Biol. 2000;150:335–347. [PMC free article: PMC2180235] [PubMed: 10908576]
- 128.
- Cheng A, Chan SL, Milhavet O, Wang S, Mattson MP. p38 MAP kinase mediates nitric oxide-induced apoptosis of neural progenitor cells. J Biol Chem. 2001;276:43320–43327. [PubMed: 11555660]
- 129.
- Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol. 2001;65:391–426. [PubMed: 11527574]
- 130.
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. [PubMed: 8524413]
- 131.
- Delcommenne M, Tan C, Gray V, Rue L, Woodgett J, Dedhar S. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc Natl Acad Sci USA. 1998;95:11211–11216. [PMC free article: PMC21621] [PubMed: 9736715]
- 132.
- Spittaels K, Van den Haute C, Van Dorpe J, Geerts H, Mercken M, Bruynseels K. et al. Glycogen synthase kinase-3beta phosphorylates protein tau and rescues the axonopathy in the central nervous system of human four-repeat tau transgenic mice. J Biol Chem. 2000;275:41340–41349. [PubMed: 11007782]
- 133.
- Lucas JJ, Hernandez F, Gomez-Ramos P, Moran MA, Hen R, Avila J. Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J. 2001;20:27–39. [PMC free article: PMC140191] [PubMed: 11226152]
- 134.
- Mattson MP. Neuronal death and GSK-3beta: A tau fetish? Trends Neurosci. 2001;24:255–256. [PubMed: 11311365]
- 135.
- Copani A, Uberti D, Sortino MA, Bruno V, Nicoletti F, Memo M. Activation of cell-cycle-associated proteins in neuronal death: a mandatory or dispensable path? Trends Neurosci. 2001;24:25–31. [PubMed: 11163884]
- 136.
- Gao CY, Zelenka PS. Induction of cyclin B and H1 kinase activity in apoptotic PC12 cells. Exp Cell Res. 1995;219:612–618. [PubMed: 7641812]
- 137.
- Timsit S, Rivera S, Ouaghi P, Guischard F, Tremblay E, Ben-Ari Y. et al. Increased cyclin D1 in vulnerable neurons in the hippocampus after ischaemia and epilepsy: a modulator of in vivo programmed cell death? Eur J Neurosci. 1999;11:263–278. [PubMed: 9987030]
- 138.
- Osuga H, Osuga S, Wang F, Fetni R, Hogan MJ, Slack RS. et al. Cyclin-dependent kinases as a therapeutic target for stroke. Proc Natl Acad Sci USA. 2000;97:10254–10259. [PMC free article: PMC27851] [PubMed: 10944192]
- 139.
- Giovanni A, Wirtz-Brugger F, Keramaris E, Slack R, Park DS. Involvement of cell cycle elements, cyclin-dependent kinases, pRb, and E2F x DP, in B-amyloid-induced neuronal death. J Biol Chem. 1999;274:19011–19016. [PubMed: 10383401]
- 140.
- Copani A, Condorelli F, Caruso A, Vancheri C, Sala A, Giuffrida Stella AM. et al. Mitotic signaling by beta-amyloid causes neuronal death. FASEB J. 1999;13:2225–2234. [PubMed: 10593870]
- 141.
- Park DS, Morris EJ, Padmanabhan J, Shelanski ML, Geller HM, Greene LA. Cyclin-dependent kinases participate in death of neurons evoked by DNA- damaging agents. J Cell Biol. 1998;143:457–467. [PMC free article: PMC2132832] [PubMed: 9786955]
- 142.
- McShea A, Harris PL, Webster KR, Wahl AF, Smith MA. Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer's disease. Am J Pathol. 1997;150:1933–1939. [PMC free article: PMC1858317] [PubMed: 9176387]
- 143.
- Vincent I, Jicha G, Rosado M, Dickson DW. Aberrant expression of mitotic cdc2/cyclin B1 kinase in degenerating neurons of Alzheimer's disease brain. J Neurosci. 1997;17:3588–3598. [PMC free article: PMC6573674] [PubMed: 9133382]
- 144.
- Busser J, Geldmacher DS, Herrup K. Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer's disease brain. J Neurosci. 1998;18:2801–2807. [PMC free article: PMC6792587] [PubMed: 9525997]
- 145.
- Ding XL, Husseman J, Tomashevski A, Nochlin D, Jin LW, Vincent I. The cell cycle Cdc25A tyrosine phosphatase is activated in degenerating postmitotic neurons in Alzheimer's disease. Am J Pathol. 2000;157:1983–1990. [PMC free article: PMC1885767] [PubMed: 11106571]
- 146.
- Maccioni RB, Otth C, Concha, II, Munoz JP. The protein kinase Cdk5. Structural aspects, roles in neurogenesis and involvement in Alzheimer's pathology. Eur J Biochem. 2001;268:1518–1527. [PubMed: 11248668]
- 147.
- Vincent I, Bu B, Hudson K, Husseman J, Nochlin D, Jin L. Constitutive Cdc25B tyrosine phosphatase activity in adult brain neurons with M phase-type alterations in Alzheimer's disease. Neuroscience. 2001;105:639–650. [PubMed: 11516829]
- 148.
- Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402:615–622. [PubMed: 10604467]
- 149.
- Nguyen MD, Lariviere RC, Julien JP. Deregulation of Cdk5 in a mouse model of ALS: Toxicity alleviated by perikaryal neurofilament inclusions. Neuron. 2001;30:135–147. [PubMed: 11343650]
- 150.
- Park DS, Levine B, Ferrari G, Greene LA. Cyclin dependent kinase inhibitors and dominant negative cyclin dependent kinase 4 and 6 promote survival of NGF-deprived sympathetic neurons. J Neurosci. 1997;17:8975–8983. [PMC free article: PMC6573623] [PubMed: 9364045]
- 151.
- Padmanabhan J, Park DS, Greene LA, Shelanski ML. Role of cell cycle regulatory proteins in cerebellar granule neuron apoptosis. J Neurosci. 1999;19:8747–8756. [PMC free article: PMC6782785] [PubMed: 10516294]
- 152.
- Park DS, Morris EJ, Greene LA, Geller HM. G1/S cell cycle blockers and inhibitors of cyclin-dependent kinases suppress camptothecin-induced neuronal apoptosis. J Neurosci. 1997;17:1256–1270. [PMC free article: PMC6793728] [PubMed: 9006970]
- 153.
- Morris EJ, Keramaris E, Rideout HJ, Slack RS, Dyson NJ, Stefanis L. et al. Cyclin-dependent kinases and P53 pathways are activated independently and mediate Bax activation in neurons after DNA damage. J Neurosci. 2001;21:5017–5026. [PMC free article: PMC6762857] [PubMed: 11438577]
- 154.
- Park DS, Morris EJ, Stefanis L, Troy CM, Shelanski ML, Geller HM. Multiple pathways of neuronal death induced by DNA-damaging agents, NGF deprivation, and oxidative stress. J Neurosci. 1998;18:830–840. [PMC free article: PMC6792759] [PubMed: 9437005]
- 155.
- Van den Haute C, Spittaels K, Van Dorpe J, Lasrado R, Vandezande K, Laenen I. et al. Coexpression of human cdk5 and its activator p35 with human protein tau in neurons in brain of triple transgenic mice. Neurobiol Dis. 2001;8:32–44. [PubMed: 11162238]
- 156.
- Kesavapany S, Lau KF, McLoughlin DM, Brownlees J, Ackerley S, Leigh PN. et al. p35/cdk5 binds and phosphorylates beta-catenin and regulates beta-catenin/presenilin-1 interaction. Eur J Neurosci. 2001;13:241–247. [PubMed: 11168528]
- 157.
- Hou ST, Callaghan D, Fournier MC, Hill I, Kang L, Massie B. et al. The transcription factor E2F1 modulates apoptosis of neurons. J Neurochem. 2000;75:91–100. [PubMed: 10854251]
- 158.
- O'Hare MJ, Hou ST, Morris EJ, Cregan SP, Xu Q, Slack RS. et al. Induction and modulation of cerebellar granule neuron death by E2F-1. J Biol Chem. 2000;275:25358–25364. [PubMed: 10851232]
- 159.
- Smith DS, Leone G, DeGregori J, Ahmed MN, Qumsiyeh MB, Nevins JR. Induction of DNA replication in adult rat neurons by deregulation of the retinoblastoma/E2F G1 cell cycle pathway. Cell Growth Differ. 2000;11:625–633. [PubMed: 11149597]
- 160.
- Park DS, Morris EJ, Bremner R, Keramaris E, Padmanabhan J, Rosenbaum M. et al. Involvement of retinoblastoma family members and E2F/DP complexes in the death of neurons evoked by DNA damage. J Neurosci. 2000;20:3104–3114. [PMC free article: PMC6773109] [PubMed: 10777774]
- 161.
- MacManus JP, Koch CJ, Jian M, Walker T, Zurakowski B. Decreased brain infarct following focal ischemia in mice lacking the transcription factor E2F1. Neuroreport. 1999;10:2711–2714. [PubMed: 10511428]
- 162.
- Gendron TF, Mealing GA, Paris J, Lou A, Edwards A, Hou ST. et al. Attenuation of neurotoxicity in cortical cultures and hippocampal slices from E2F1 knockout mice. J Neurochem. 2001;78:316–324. [PubMed: 11461967]
- 163.
- Giovanni A, Keramaris E, Morris EJ, Hou ST, O'Hare M, Dyson N. et al. E2F1 mediates death of B-amyloid-treated cortical neurons in a manner independent of p53 and dependent on Bax and caspase 3. J Biol Chem. 2000;275:11553–11560. [PubMed: 10766769]
- 164.
- Tamami M, Lindholm PF, Brady JN. The retinoblastoma gene product (Rb) induces binding of a conformationally inactive nuclear factor-kappaB. J Biol Chem. 1996;271:24551–24556. [PubMed: 8798717]
- 165.
- Shim J, Park HS, Kim MJ, Park J, Park E, Cho SG. et al. Rb protein down-regulates the stress-activated signals through inhibiting c-Jun N-terminal kinase/stress-activated protein kinase. J Biol Chem. 2000;275:14107–14111. [PubMed: 10799486]
- 166.
- Jordan J, Galindo MF, Prehn JH, Weichselbaum RR, Beckett M, Ghadge GD. et al. p53 expression induces apoptosis in hippocampal pyramidal neuron cultures. J Neurosci. 1997;17:1397–1405. [PMC free article: PMC6793734] [PubMed: 9006981]
- 167.
- Macleod KF, Hu Y, Jacks T. Loss of Rb activates both p53-dependent and independent cell death pathways in the developing mouse nervous system. EMBO J. 1996;15:6178–6188. [PMC free article: PMC452439] [PubMed: 8947040]
- 168.
- Lipinski MM, Macleod KF, Williams BO, Mullaney TL, Crowley D, Jacks T. Cell-autonomous and non-cell-autonomous functions of the Rb tumor suppressor in developing central nervous system. EMBO J. 2001;20:3402–3413. [PMC free article: PMC125524] [PubMed: 11432828]
- 169.
- Nip J, Strom DK, Eischen CM, Cleveland JL, Zambetti GP, Hiebert SW. E2F-1 induces the stabilization of p53 but blocks p53-mediated transactivation. Oncogene. 2001;20:910–920. [PubMed: 11314026]
- 170.
- Ko LJ, Prives C. p53: Puzzle and paradigm. Genes Dev. 1996;10:1054–1072. [PubMed: 8654922]
- 171.
- Giaccia AJ, Kastan MB. The complexity of p53 modulation: Emerging patterns from divergent signals. Genes Dev. 1998;12:2973–2983. [PubMed: 9765199]
- 172.
- Bates S, Vousden KH. p53 in signaling checkpoint arrest or apoptosis. Curr Opin Genet Dev. 1996;6:12–18. [PubMed: 8791489]
- 173.
- Asker C, Wiman KG, Selivanova G. p53-induced apoptosis as a safeguard against cancer. Biochem Biophys Res Commun. 1999;265:1–6. [PubMed: 10548481]
- 174.
- Miyashita T, Harigai M, Hanada M, Reed JC. Identification of a p53-dependent negative response element in the bcl- 2 gene. Cancer Res. 1994;54:3131–3135. [PubMed: 8205530]
- 175.
- Roperch JP, Alvaro V, Prieur S, Tuynder M, Nemani M, Lethrosne F. et al. Inhibition of presenilin 1 expression is promoted by p53 and p21WAF-1 and results in apoptosis and tumor suppression. Nat Med. 1998;4:835–838. [PubMed: 9662377]
- 176.
- Ding HF, McGill G, Rowan S, Schmaltz C, Shimamura A, Fisher DE. Oncogene-dependent regulation of caspase activation by p53 protein in a cell-free system. J Biol Chem. 273:28378–28383. [PubMed: 9774464]
- 177.
- Gottlieb E, Oren M.(1998)p53 facilitates pRb cleavage in IL-3-deprived cells: Novel pro- apoptotic activity of p53 EMBO J 1998173587–3596. [PMC free article: PMC1170695] [PubMed: 9649429]
- 178.
- Morrison RS, Kinoshita Y. The role of p53 in neuronal cell death. Cell Death Differ. 2000;7:868–879. [PubMed: 11279532]
- 179.
- Sakhi S, Bruce A, Sun N, Tocco G, Baudry M, Schreiber SS. p53 induction is associated with neuronal damage in the central nervous system. Proc Natl Acad Sci USA. 1994;91:7525–7529. [PMC free article: PMC44434] [PubMed: 8052613]
- 180.
- Sakhi S, Sun N, Wing LL, Mehta P, Schreiber SS. Nuclear accumulation of p53 protein following kainic acid-induced seizures. Neuroreport. 1996;7:493–496. [PubMed: 8730813]
- 181.
- Nakai M, Qin ZH, Chen JF, Wang Y, Chase TN. Kainic acid-induced apoptosis in rat striatum is associated with nuclear factor-kappaB activation. J Neurochem. 2000;74:647–658. [PubMed: 10646516]
- 182.
- Qin ZH, Chen RW, Wang Y, Nakai M, Chuang DM, Chase TN. Nuclear factor kappaB nuclear translocation upregulates c-Myc and p53 expression during NMDA receptor-mediated apoptosis in rat striatum. J Neurosci. 1999;19:4023–4033. [PMC free article: PMC6782699] [PubMed: 10234031]
- 183.
- Wang Y, Qin ZH, Nakai M, Chen RW, Chuang DM, Chase TN. Co-stimulation of cyclic-AMP-linked metabotropic glutamate receptors in rat striatum attenuates excitotoxin-induced nuclear factor-kappaB activation and apoptosis. Neuroscience. 1999;94:1153–1162. [PubMed: 10625054]
- 184.
- Napieralski JA, Raghupathi R, McIntosh TK. The tumor-suppressor gene, p53, is induced in injured brain regions following experimental traumatic brain injury. Brain Res Mol Brain Res. 1999;71:78–86. [PubMed: 10407189]
- 185.
- Lu J, Moochhala S, Kaur C, Ling E. Changes in apoptosis-related protein (p53, Bax, Bcl-2 and Fos) expression with DNA fragmentation in the central nervous system in rats after closed head injury. Neurosci Lett. 2000;290:89–92. [PubMed: 10936684]
- 186.
- Martin LJ, Kaiser A, Yu JW, Natale JE, Al-Abdulla NA. Injury-induced apoptosis of neurons in adult brain is mediated by p53-dependent and p53-independent pathways and requires Bax. J Comp Neurol. 2001;433:299–311. [PubMed: 11298357]
- 187.
- Chopp M, Li Y, Zhang ZG, Freytag SO. p53 expression in brain after middle cerebral artery occlusion in the rat. Biochem Biophys Res Commun. 1992;182:1201–1207. [PubMed: 1540165]
- 188.
- Watanabe H, Ohta S, Kumon Y, Sakaki S, Sakanaka M. Increase in p53 protein expression following cortical infarction in the spontaneously hypertensive rat. Brain Res. 1999;837:38–45. [PubMed: 10433986]
- 189.
- de la Monte SM, Sohn YK, Wands JR. Correlates of p53- and Fas (CD95)-mediated apoptosis in Alzheimer's disease. J Neurol Sci. 1997;152:73–83. [PubMed: 9395128]
- 190.
- de la Monte SM, Sohn YK, Ganju N, Wands JR. P53- and CD95-associated apoptosis in neurodegenerative diseases. Lab Invest. 1998;78:401–411. [PubMed: 9564885]
- 191.
- LaFerla FM, Hall CK, Ngo L, Jay G. Extracellular deposition of beta-amyloid upon p53-dependent neuronal cell death in transgenic mice. J Clin Invest. 1996;98:1626–1632. [PMC free article: PMC507596] [PubMed: 8833912]
- 192.
- Jiang YH, Armstrong D, Albrecht U, Atkins CM, Noebels JL, Eichele G. et al. Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation [see comments] Neuron. 1998;21:799–811. [PubMed: 9808466]
- 193.
- Sawa A. Neuronal cell death in Down's syndrome. J Neural Transm Suppl. 1999;57:87–97. [PubMed: 10666670]
- 194.
- Seidl R, Fang-Kircher S, Bidmon B, Cairns N, Lubec G. Apoptosis-associated proteins p53 and APO-1/Fas (CD95) in brains of adult patients with Down syndrome. Neurosci Lett. 1999;260:9–12. [PubMed: 10027687]
- 195.
- Martin LJ. p53 is abnormally elevated and active in the CNS of patients with amyotrophic lateral sclerosis. Neurobiol Dis. 2000;7:613–622. [PubMed: 11114260]
- 196.
- Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H. et al. The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci USA. 2000;97:6763–6768. [PMC free article: PMC18731] [PubMed: 10823891]
- 197.
- Uberti D, Belloni M, Grilli M, Spano P, Memo M. Induction of tumour-suppressor phosphoprotein p53 in the apoptosis of cultured rat cerebellar neurones triggered by excitatory amino acids. Eur J Neurosci. 1998;10:246–254. [PubMed: 9753133]
- 198.
- Chen RW, Chuang DM. Long term lithium treatment suppresses p53 and Bax expression but increases Bcl-2 expression. A prominent role in neuroprotection against excitotoxicity. J Biol Chem. 1999;274:6039–6042. [PubMed: 10037682]
- 199.
- Anderson C N G, Tolkovsky AM. A role for MAPK/ERK in sympathetic neuron survival: Protection against a p53-dependent, JNK-independent induction of apoptosis by cytosine arabinoside. J Neurosci. 1999;19:664–673. [PMC free article: PMC6782192] [PubMed: 9880587]
- 200.
- Chen RW, Saunders PA, Wei H, Li Z, Seth P, Chuang DM. Involvement of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and p53 in neuronal apoptosis: evidence that GAPDH is upregulated by p53. J Neurosci. 1999;19:9654–9662. [PMC free article: PMC6782921] [PubMed: 10531467]
- 201.
- Frank KM, Sharpless NE, Gao Y, Sekiguchi JM, Ferguson DO, Zhu C. et al. DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol Cell. 2000;5:993–1002. [PubMed: 10911993]
- 202.
- Banasiak KJ, Haddad GG. Hypoxia-induced apoptosis: Effect of hypoxic severity and role of p53 in neuronal cell death. Brain Res. 1998;797:295–304. [PubMed: 9666152]
- 203.
- Crumrine RC, Thomas AL, Morgan PF. Attenuation of p53 expression protects against focal ischemic damage in transgenic mice. J Cereb Blood Flow Metab. 1994;14:887–891. [PubMed: 7929653]
- 204.
- Johnson MD, Xiang H, London S, Kinoshita Y, Knudson M, Mayberg M. et al. Evidence for involvement of Bax and p53, but not caspases, in radiation-induced cell death of cultured postnatal hippocampal neurons. J Neurosci Res. 1998;54:721–733. [PubMed: 9856857]
- 205.
- Wood KA, Youle RJ. The role of free radicals and p53 in neuron apoptosis in vivo. J Neurosci. 1995;15:5851–5857. [PMC free article: PMC6577632] [PubMed: 7643225]
- 206.
- Enokido Y, Araki T, Tanaka K, Aizawa S, Hatanaka H. Involvement of p53 in DNA strand break-induced apoptosis in postmitotic CNS neurons. Eur J Neurosci. 1996;8:1812–1821. [PubMed: 8921272]
- 207.
- D'Sa-Eipper C, Leonard JR, Putcha G, Zheng TS, Flavell RA, Rakic P. et al. DNA damage-induced neural precursor cell apoptosis requires p53 and caspase 9 but neither Bax nor caspase 3. Development. 2001;128:137–146. [PubMed: 11092819]
- 208.
- Herzog KH, Chong MJ, Kapsetaki M, Morgan JI, McKinnon PJ. Requirement for Atm in ionizing radiation-induced cell death in the developing central nervous system. Science. 1998;280:1089–1091. [PubMed: 9582124]
- 209.
- Morrison RS, Wenzel HJ, Kinoshita Y, Robbins CA, Donehower LA, Schwartzkroin PA. Loss of the p53 tumor suppressor gene protects neurons from kainate- induced cell death. J Neurosci. 1996;16:1337–1345. [PMC free article: PMC6578556] [PubMed: 8778285]
- 210.
- Trimmer PA, Smith TS, Jung AB, Bennett J P Jr. Dopamine neurons from transgenic mice with a knockout of the p53 gene resist MPTP neurotoxicity. Neurodegeneration. 1996;5:233–239. [PubMed: 8910901]
- 211.
- Hirata H, Cadet JL. p53-knockout mice are protected against the long-term effects of methamphetamine on dopaminergic terminals and cell bodies. J Neurochem. 1997;69:780–790. [PubMed: 9231739]
- 212.
- Sakhi S, Gilmore W, Tran ND, Schreiber SS. p53-deficient mice are protected against adrenalectomy-induced apoptosis. Neuroreport. 1996;8:233–235. [PubMed: 9051787]
- 213.
- Enokido Y, Araki T, Aizawa S, Hatanaka H. p53 involves cytosine arabinoside-induced apoptosis in cultured cerebellar granule neurons. Neurosci Lett. 1996;203:1–4. [PubMed: 8742032]
- 214.
- Araki T, Enokido Y, Inamura N, Aizawa S, Reed JC, Hatanaka H. Changes in c-Jun but not Bcl-2 family proteins in p53-dependent apoptosis of mouse cerebellar granule neurons induced by DNA damaging agent bleomycin. Brain Res. 1998;794:239–247. [PubMed: 9622642]
- 215.
- Xiang H, Kinoshita Y, Knudson CM, Korsmeyer SJ, Schwartzkroin PA, Morrison RS. Bax involvement in p53-mediated neuronal cell death. J Neurosci. 1998;18:1363–1373. [PMC free article: PMC6792710] [PubMed: 9454845]
- 216.
- Halterman MW, Miller CC, Federoff HJ. Hypoxia-inducible factor-1alpha mediates hypoxia-induced delayed neuronal death that involves p53. J Neurosci. 1999;19:6818–6824. [PMC free article: PMC6782875] [PubMed: 10436039]
- 217.
- Aloyz RS, Bamji SX, Pozniak CD, Toma JG, Atwal J, Kaplan DR. et al. p53 is essential for developmental neuron death as regulated by the TrkA and p75 neurotrophin receptors. J Cell Biol. 1998;143:1691–1703. [PMC free article: PMC2132983] [PubMed: 9852160]
- 218.
- Vogel KS, Parada LF. Sympathetic neuron survival and proliferation are prolonged by loss of p53 and neurofibromin. Mol Cell Neurosci. 1998;11:19–28. [PubMed: 9608530]
- 219.
- Lee EY, Chang CY, Hu N, Wang YC, Lai CC, Herrup K. et al. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis [see comments] Nature. 1992;359:288–294. [PubMed: 1406932]
- 220.
- Yeung MC, Geertsma F, Liu J, Lau AS. Inhibition of HIV-1 gp120-induced apoptosis in neuroblastoma SK-N-SH cells by an antisense oligodeoxynucleotide against p53. Aids. 1998;12:349–354. [PubMed: 9520163]
- 221.
- Lakkaraju A, Dubinsky JM, Low WC, Rahman YE. Neurons are protected from excitotoxic death by p53 antisense oligonucleotides delivered in anionic liposomes. J Biol Chem. 2001;276:32000–32007. [PubMed: 11406618]
- 222.
- Culmsee C, Zhu X, Yu QS, Chan SL, Camandola S, Guo Z. et al. A synthetic inhibitor of p53 protects neurons against death induced by ischemic and excitotoxic insults, and amyloid beta-peptide. J Neurochem. 2001;77:220–228. [PubMed: 11279278]
- 223.
- Kuntz C, Kinoshita Y, Beal F, Donehower LA, Morrison RS. The absence of p53 does not protect SOD1 mutant mice from onset of clincial symptoms or lethality. Exp Neurol. 2000;165:184–190. [PubMed: 10964497]
- 224.
- Prudlo J, Koenig J, Graser J, Burckhardt E, Mestres P, Menger M. et al. Motor neuron cell death in a mouse model of FALS is not mediated by the p53 cell survival regulator. Brain Res. 2000;879:183–187. [PubMed: 11011020]
- 225.
- Chong MJ, Murray MR, Gosink EC, Russell HR, Srinivasan A, Kapsetaki M. et al. Atm and Bax cooperate in ionizing radiation-induced apoptosis in the central nervous system. Proc Natl Acad Sci USA. 2000;97:889–894. [PMC free article: PMC15426] [PubMed: 10639175]
- 226.
- Cregan SP, MacLaurin JG, Craig CG, Robertson GS, Nicholson DW, Park DS. et al. Bax-dependent caspase-3 activation is a key determinant in p53-induced apoptosis in neurons. J Neurosci. 1999;19:7860–7869. [PMC free article: PMC6782440] [PubMed: 10479688]
- 227.
- McGinnis KM, Gnegy ME, Wang KK. Endogenous bax translocation in SH-SY5Y human neuroblastoma cells and cerebellar granule neurons undergoing apoptosis. J Neurochem. 1999;72:1899–1906. [PubMed: 10217266]
- 228.
- Putcha GV, Deshmukh M, Johnson E M Jr. BAX translocation is a critical event in neuronal apoptosis: regulation by neuroprotectants, BCL-2, and caspases. J Neurosci. 1999;19:7476–7485. [PMC free article: PMC6782498] [PubMed: 10460254]
- 229.
- Xiang J, Chao DT, Korsmeyer SJ. BAX-induced cell death may not require interleukin 1 beta-converting enzyme-like proteases. Proc Natl Acad Sci USA. 1996;93:14559–14563. [PMC free article: PMC26172] [PubMed: 8962091]
- 230.
- Vekrellis K, McCarthy MJ, Watson A, Whitfield J, Rubin LL, Ham J. Bax promotes neuronal cell death and is downregulated during the development of the nervous system. Development. 1997;124:1239–1249. [PubMed: 9102310]
- 231.
- Martinou I, Missotten M, Fernandez PA, Sadoul R, Martinou JC. Bax and Bak proteins require caspase activity to trigger apoptosis in sympathetic neurons. Neuroreport. 1998;9:15–19. [PubMed: 9592040]
- 232.
- Marzo I, Brenner C, Zamzami N, Jurgensmeier JM, Susin SA, Vieira HL. et al. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science. 1998;281:2027–2031. [PubMed: 9748162]
- 233.
- Schuler M, Bossy-Wetzel E, Goldstein JC, Fitzgerald P, Green DR. p53 induces apoptosis by caspase activation through mitochondrial cytochrome c release. J Biol Chem. 2000;275:7337–7342. [PubMed: 10702305]
- 234.
- Johnson MD, Kinoshita Y, Xiang H, Ghatan S, Morrison RS. Contribution of p53-dependent caspase activation to neuronal cell death declines with neuronal maturation. J Neurosci. 1999;19:2996–3006. [PMC free article: PMC6782293] [PubMed: 10191317]
- 235.
- Fortin A, Cregan SP, MacLaurin JG, Kushwaha N, Hickman ES, Thompson CS. et al. APAF1 is a key transcriptional target for p53 in the regulation of neuronal cell death. J Cell Biol. 2001;155:207–216. [PMC free article: PMC2198828] [PubMed: 11591730]
- 236.
- Attardi LD, Reczek EE, Cosmas C, Demicco EG, McCurrach ME, Lowe SW. et al. PERP, an apoptosis-associated target of p53, is a novel member of the PMP-22/gas3 family [In Process Citation] Genes Dev. 2000;14:704–718. [PMC free article: PMC316461] [PubMed: 10733530]
- 237.
- Ferri KF, Kroemer G. Organelle-specific initiation of cell death pathways. Nat Cell Biol. 2001;3:E255–263. [PubMed: 11715037]
- 238.
- Lewen A, Matz P, Chan PH. Free radical pathways in CNS injury. J Neurotrauma. 2000;17:871–890. [PubMed: 11063054]
- 239.
- Mattson MP. Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol. 2000;1:120–129. [PubMed: 11253364]
- 240.
- Tong X, Liu B, Dong Y, Sun Z. Cleavage of ATM during radiation-induced apoptosis: Caspase-3-like apoptotic protease as a candidate. Int J Radiat Biol. 2000;76:1387–1395. [PubMed: 11057747]
- 241.
- Song Q, Lees-Miller SP, Kumar S, Zhang Z, Chan DW, Smith GC. et al. DNA-dependent protein kinase catalytic subunit: A target for an ICE- like protease in apoptosis. EMBO J. 1996;15:3238–3246. [PMC free article: PMC451880] [PubMed: 8670824]
- 242.
- Han Z, Malik N, Carter T, Reeves WH, Wyche JH, Hendrickson EA. DNA-dependent protein kinase is a target for a CPP32-like apoptotic protease. J Biol Chem. 1996;271:25035–25040. [PubMed: 8798786]
- 243.
- Tewari M, Quan LT, O'Rourke K, Desnoyers S, Zeng Z, Beidler DR. et al. Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995;81:801–809. [PubMed: 7774019]
- 244.
- Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature. 1994;371:346–347. [PubMed: 8090205]
- 245.
- Kastan MB, Lim DS. The many substrates and functions of ATM. Nat Rev Mol Cell Biol. 2000;1:179–186. [PubMed: 11252893]
- 246.
- Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F. et al. Atm-deficient mice: A paradigm of ataxia telangiectasia. Cell. 1996;86:159–171. [PubMed: 8689683]
- 247.
- Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K. et al. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–1679. [PubMed: 9733515]
- 248.
- Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L. et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–1677. [PubMed: 9733514]
- 249.
- Caspari T. How to activate p53. Curr Biol. 2000;10:R315–317. [PubMed: 10801407]
- 250.
- Khosravi R, Maya R, Gottlieb T, Oren M, Shiloh Y, Shkedy D. Rapid ATM-dependent phosphorylation of MDM2 precedes p53 accumulation in response to DNA damage. Proc Natl Acad Sci USA. 1999;96:14973–14977. [PMC free article: PMC24757] [PubMed: 10611322]
- 251.
- Maya R, Balass M, Kim ST, Shkedy D, Leal JF, Shifman O. et al. ATM-dependent phosphorylation of Mdm2 on serine 395: Role in p53 activation by DNA damage. Genes Dev. 2001;15:1067–1077. [PMC free article: PMC312683] [PubMed: 11331603]
- 252.
- Meek DW. Mechanisms of switching on p53: A role for covalent modification? Oncogene. 1999;18:7666–7675. [PubMed: 10618706]
- 253.
- Lee Y, Chong MJ, McKinnon PJ. Ataxia telangiectasia mutated-dependent apoptosis after genotoxic stress in the developing nervous system is determined by cellular differentiation status. J Neurosci. 2001;21:6687–6693. [PMC free article: PMC6763074] [PubMed: 11517258]
- 254.
- Rolig RL, McKinnon PJ. Linking DNA damage and neurodegeneration. Trends Neurosci. 2000;23:417–424. [PubMed: 10941191]
- 255.
- Shiloh Y. ATM and ATR: networking cellular responses to DNA damage. Curr Opin Genet Dev. 2001;11:71–77. [PubMed: 11163154]
- 256.
- Hammarsten O, DeFazio LG, Chu G. Activation of DNA-dependent protein kinase by single-stranded DNA ends. J Biol Chem. 2000;275:1541–1550. [PubMed: 10636842]
- 257.
- Smith GC, Jackson SP. The DNA-dependent protein kinase. Genes Dev. 1999;13:916–934. [PubMed: 10215620]
- 258.
- Lees-Miller SP, Sakaguchi K, Ullrich SJ, Appella E, Anderson CW. Human DNA-activated protein kinase phosphorylates serines 15 and 37 in the amino-terminal transactivation domain of human p53. Mol Cell Biol. 1992;12:5041–5049. [PMC free article: PMC360437] [PubMed: 1406679]
- 259.
- Mayo LD, Turchi JJ, Berberich SJ. Mdm-2 phosphorylation by DNA-dependent protein kinase prevents interaction with p53. Cancer Res. 1997;57:5013–5016. [PubMed: 9371494]
- 260.
- Kharbanda S, Yuan ZM, Weichselbaum R, Kufe D. Determination of cell fate by c-Abl activation in the response to DNA damage. Oncogene. 1998;17:3309–3318. [PubMed: 9916993]
- 261.
- Liu L, Kwak YT, Bex F, Garcia-Martinez LF, Li XH, Meek K. et al. DNA-dependent protein kinase phosphorylation of IkappaB alpha and IkappaB beta regulates NF-kappaB DNA binding properties. Mol Cell Biol. 1998;18:4221–4234. [PMC free article: PMC109006] [PubMed: 9632806]
- 262.
- Chechlacz M, Vemuri MC, Naegele JR. Role of DNA-dependent protein kinase in neuronal survival. J Neurochem. 2001;78:141–154. [PubMed: 11432981]
- 263.
- Vemuri MC, Schiller E, Naegele JR. Elevated DNA double strand breaks and apoptosis in the CNS of scid mutant mice. Cell Death Differ. 2001;8:245–255. [PubMed: 11319607]
- 264.
- Culmsee C, Bondada S, Mattson MP. Hippocampal neurons of mice deficient in DNA-dependent protein kinase exhibit increased vulnerability to DNA damage, oxidative stress and excitotoxicity. Brain Res Mol Brain Res. 2001;87:257–262. [PubMed: 11245929]
- 265.
- Chakravarthy BR, Walker T, Rasquinha I, Hill IE, MacManus JP. Activation of DNA-dependent protein kinase may play a role in apoptosis of human neuroblastoma cells. J Neurochem. 1999;72:933–942. [PubMed: 10037464]
- 266.
- Smith S. The world according to PARP. Trends Biochem Sci. 2001;26:174–179. [PubMed: 11246023]
- 267.
- Herceg Z, Wang ZQ. Functions of poly(ADP-ribose) polymerase (PARP) in DNA repair, genomic integrity and cell death. Mutat Res. 2001;477:97–110. [PubMed: 11376691]
- 268.
- Le Rhun Y, Kirkland JB, Shah GM. Cellular responses to DNA damage in the absence of Poly(ADP-ribose) polymerase. Biochem Biophys Res Commun. 1998;245:1–10. [PubMed: 9535773]
- 269.
- Ziegler M, Oei SL. A cellular survival switch: Poly(ADP-ribosyl)ation stimulates DNA repair and silences transcription. Bioessays. 2001;23:543–548. [PubMed: 11385634]
- 270.
- Ha HC, Snyder SH. Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc Natl Acad Sci USA. 1999;96:13978–13982. [PMC free article: PMC24176] [PubMed: 10570184]
- 271.
- Ha HC, Snyder SH. Poly(ADP-ribose) polymerase-1 in the nervous system. Neurobiol Dis. 2000;7:225–239. [PubMed: 10964595]
- 272.
- Cosi C, Suzuki H, Milani D, Facci L, Menegazzi M, Vantini G. et al. Poly(ADP-ribose) polymerase: early involvement in glutamate-induced neurotoxicity in cultured cerebellar granule cells. J Neurosci Res. 1994;39:38–46. [PubMed: 7807591]
- 273.
- Zhang J, Dawson VL, Dawson TM, Snyder SH. Nitric oxide activation of poly(ADP-ribose) synthetase in neurotoxicity. Science. 1994;263:687–689. [PubMed: 8080500]
- 274.
- Eliasson MJ, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J. et al. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med. 1997;3:1089–1095. [PubMed: 9334719]
- 275.
- Endres M, Wang ZQ, Namura S, Waeber C, Moskowitz MA. Ischemic brain injury is mediated by the activation of poly(ADP- ribose)polymerase. J Cereb Blood Flow Metab. 1997;17:1143–1151. [PubMed: 9390645]
- 276.
- Mandir AS, Poitras MF, Berliner AR, Herring WJ, Guastella DB, Feldman A. et al. NMDA but not non-NMDA excitotoxicity is mediated by Poly(ADP-ribose) polymerase. J Neurosci. 2000;20:8005–8011. [PMC free article: PMC6772735] [PubMed: 11050121]
- 277.
- Mandir AS, Przedborski S, Jackson-Lewis V, Wang ZQ, Simbulan-Rosenthal CM, Smulson ME. et al. Poly(ADP-ribose) polymerase activation mediates 1-methyl-4-phenyl-1, 2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism. Proc Natl Acad Sci USA. 1999;96:5774–5779. [PMC free article: PMC21936] [PubMed: 10318960]
- 278.
- Ding Y, Zhou Y, Lai Q, Li J, Gordon V, Diaz FG. Long-term neuroprotective effect of inhibiting poly(ADP-ribose) polymerase in rats with middle cerebral artery occlusion using a behavioral assessment. Brain Res. 2001;915:210–217. [PubMed: 11595210]
- 279.
- Whalen MJ, Clark RS, Dixon CE, Robichaud P, Marion DW, Vagni V. et al. Traumatic brain injury in mice deficient in poly-ADP(ribose) polymerase: A preliminary report. Acta Neurochir Suppl. 2000;76:61–64. [PubMed: 11450092]
- 280.
- Moroni F, Meli E, Peruginelli F, Chiarugi A, Cozzi A, Picca R. et al. Poly(ADP-ribose) polymerase inhibitors attenuate necrotic but not apoptotic neuronal death in experimental models of cerebral ischemia. Cell Death Differ. 2001;8:921–932. [PubMed: 11526447]
- 281.
- Kruman II, Culmsee C, Chan SL, Kruman Y, Guo Z, Penix L. et al. Homocysteine elicits a DNA damage response in neurons that promotes apoptosis and hypersensitivity to excitotoxicity. J Neurosci. 2000;20:6920–6926. [PMC free article: PMC6772815] [PubMed: 10995836]
- 282.
- Yang E, Korsmeyer SJ. Molecular thanatopsis: A discourse on the BCL2 family and cell death. Blood. 1996;88:386–401. [PubMed: 8695785]
- 283.
- Bakhshi A, Jensen JP, Goldman P, Wright JJ, McBride OW, Epstein AL. et al. Cloning the chromosomal breakpoint of t(14;18) human lymphomas: Clustering around JH on chromosome 14 and near a transcriptional unit on 18. Cell. 1985;41:899–906. [PubMed: 3924412]
- 284.
- Cleary ML, Smith SD, Sklar J. Cloning and structural analysis of cDNAs for bcl-2 and a hybrid bcl- 2/immunoglobulin transcript resulting from the t(14;18) translocation. Cell. 1986;47:19–28. [PubMed: 2875799]
- 285.
- Tsujimoto Y, Finger LR, Yunis J, Nowell PC, Croce CM. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science. 1984;226:1097–1099. [PubMed: 6093263]
- 286.
- Huang DC, Strasser A. BH3-Only proteins-essential initiators of apoptotic cell death. Cell. 2000;103:839–842. [PubMed: 11136969]
- 287.
- Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T. et al. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;288:1053–1058. [PubMed: 10807576]
- 288.
- Nechushtan A, Smith CL, Lamensdorf I, Yoon SH, Youle RJ. Bax and Bak coalesce into novel mitochondria-associated clusters during apoptosis. J Cell Biol. 2001;153:1265–1276. [PMC free article: PMC2192024] [PubMed: 11402069]
- 289.
- Hsu YT, Wolter KG, Youle RJ. Cytosol-to-membrane redistribution of Bax and Bcl-X(L) during apoptosis. Proc Natl Acad Sci USA. 1997;94:3668–3672. [PMC free article: PMC20498] [PubMed: 9108035]
- 290.
- Wolter KG, Hsu YT, Smith CL, Nechushtan A, Xi XG. et al. Movement of Bax from the cytosol to mitochondria during apoptosis. J Cell Biol. 1997;139:1281–1292. [PMC free article: PMC2140220] [PubMed: 9382873]
- 291.
- Zha H, Fisk HA, Yaffe MP, Mahajan N, Herman B, Reed JC. Structure-function comparisons of the proapoptotic protein Bax in yeast and mammalian cells. Mol Cell Biol. 1996;16:6494–6508. [PMC free article: PMC231651] [PubMed: 8887678]
- 292.
- Thornberry NA, Lazebnik Y. Caspases: Enemies within. Science. 1998;281:1312–1316. [PubMed: 9721091]
- 293.
- Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. [PubMed: 9721092]
- 294.
- Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: A primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. [PubMed: 9027315]
- 295.
- Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X. (1997) Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked Science 2751129–1132. [PubMed: 9027314]
- 296.
- Harris MH, Thompson CB. The role of the Bcl-2 family in the regulation of outer mitochondrial membrane permeability. Cell Death Differ. 2000;7:1182–1191. [PubMed: 11175255]
- 297.
- Kowaltowski AJ, Smaili SS, Russell JT, Fiskum G. Elevation of resting mitochondrial membrane potential of neural cells by cyclosporin A, BAPTA-AM, and bcl-2. Am J Physiol Cell Physiol. 2000;279:C852–859. [PubMed: 10942734]
- 298.
- Schendel SL, Montal M, Reed JC. Bcl-2 family proteins as ion-channels. Cell Death Differ. 1998;5:372–380. [PubMed: 10200486]
- 299.
- Saito M, Korsmeyer SJ, Schlesinger PH. BAX-dependent transport of cytochrome c reconstituted in pure liposomes. Nat Cell Biol. 2000;2:553–555. [PubMed: 10934477]
- 300.
- Martinou JC, Green DR. Breaking the mitochondrial barrier. Nat Rev Mol Cell Biol. 2001;2:63–67. [PubMed: 11413467]
- 301.
- Gross A, Jockel J, Wei MC, Korsmeyer SJ. Enforced dimerization of BAX results in its translocation, mitochondrial dysfunction and apoptosis. EMBO J. 1998;17:3878–3885. [PMC free article: PMC1170723] [PubMed: 9670005]
- 302.
- Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. [PubMed: 8358790]
- 303.
- Yin XM, Oltvai ZN, Korsmeyer SJ. BH1 and BH2 domains of Bcl-2 are required for inhibition of apoptosis and heterodimerization with Bax. Nature. 1994;369:321–323. [PubMed: 8183370]
- 304.
- Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M. et al. Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science. 1997;275:983–986. [PubMed: 9020082]
- 305.
- Muchmore SW, Sattler M, Liang H, Meadows RP, Harlan JE, Yoon HS. et al. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature. 1996;381:335–341. [PubMed: 8692274]
- 306.
- Wang K, Yin XM, Chao DT, Milliman CL, Korsmeyer SJ. BID: A novel BH3 domain-only death agonist. Genes Dev. 1996;10:2859–2869. [PubMed: 8918887]
- 307.
- Zha J, Harada H, Osipov K, Jockel J, Waksman G, Korsmeyer SJ. BH3 domain of BAD is required for heterodimerization with BCL-XL and pro-apoptotic activity. J Biol Chem. 1997;272:24101–24104. [PubMed: 9305851]
- 308.
- Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell. 1996;87:619–628. [PubMed: 8929531]
- 309.
- Blagosklonny MV. Unwinding the loop of Bcl-2 phosphorylation. Leukemia. 2001;15:869–874. [PubMed: 11417471]
- 310.
- Ruvolo PP, Deng X, May WS. Phosphorylation of Bcl2 and regulation of apoptosis. Leukemia. 2001;15:515–522. [PubMed: 11368354]
- 311.
- Wei MC, Lindsten T, Mootha VK, Weiler S, Gross A, Ashiya M. et al. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 2000;14:2060–2071. [PMC free article: PMC316859] [PubMed: 10950869]
- 312.
- Fesik SW. Insights into programmed cell death through structural biology. Cell. 2000;103:273–282. [PubMed: 11057900]
- 313.
- Lutter M, Fang M, Luo X, Nishijima M, Xie X, Wang X. Cardiolipin provides specificity for targeting of tBid to mitochondria. Nat Cell Biol. 2000;2:754–761. [PubMed: 11025668]
- 314.
- Korsmeyer SJ, Wei MC, Saito M, Weiler S, Oh KJ, Schlesinger PH. Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ. 2000;7:1166–1173. [PubMed: 11175253]
- 315.
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ. et al. Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. [PMC free article: PMC3049805] [PubMed: 11326099]
- 316.
- Desagher S, Osen-Sand A, Nichols A, Eskes R, Montessuit S, Lauper S. et al. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J Cell Biol. 1999;144:891–901. [PMC free article: PMC2148190] [PubMed: 10085289]
- 317.
- O'Connor L, Strasser A, O'Reilly LA, Hausmann G, Adams JM, Cory S. et al. Bim: A novel member of the Bcl-2 family that promotes apoptosis. EMBO J. 1998;17:384–395. [PMC free article: PMC1170389] [PubMed: 9430630]
- 318.
- Ottilie S, Diaz JL, Horne W, Chang J, Wang Y, Wilson G. et al. Dimerization properties of human BAD. dentification of a BH-3 domain and analysis of its binding to mutant BCL-2 and BCL-XL proteins. J Biol Chem. 1997;272:30866–30872. [PubMed: 9388232]
- 319.
- Merry DE, Korsmeyer SJ. Bcl-2 gene family in the nervous system. Annu Rev Neurosci. 1997;20:245–267. [PubMed: 9056714]
- 320.
- Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. [PubMed: 2031577]
- 321.
- Sadoul R. Bcl-2 family members in the development and degenerative pathologies of the nervous system. Cell Death Differ. 1998;5:805–815. [PubMed: 10203696]
- 322.
- Ay I, Sugimori H, Finklestein SP. Intravenous basic fibroblast growth factor (bFGF) decreases DNA fragmentation and prevents downregulation of Bcl-2 expression in the ischemic brain following middle cerebral artery occlusion in rats. Brain Res Mol Brain Res. 2001;87:71–80. [PubMed: 11223161]
- 323.
- Miller TM, Moulder KL, Knudson CM, Creedon DJ, Deshmukh M, Korsmeyer SJ. et al. Bax deletion further orders the cell death pathway in cerebellar granule cells and suggests a caspase-independent pathway to cell death. J Cell Biol. 1997;139:205–217. [PMC free article: PMC2139809] [PubMed: 9314540]
- 324.
- Jin KL, Graham SH, Mao XO, He X, Nagayama T, Simon RP. et al. Bax kappa, a novel Bax splice variant from ischemic rat brain lacking an ART domain, promotes neuronal cell death. J Neurochem. 2001;77:1508–1519. [PubMed: 11413234]
- 325.
- Deckwerth TL, Elliott JL, Knudson CM, Johnson E M Jr., Snider WD, Korsmeyer SJ. BAX is required for neuronal death after trophic factor deprivation and during development. Neuron. 1996;17:401–411. [PubMed: 8816704]
- 326.
- Whitfield J, Neame SJ, Paquet L, Bernard O, Ham J. Dominant-negative c-Jun promotes neuronal survival by reducing BIM expression and inhibiting mitochondrial cytochrome c release. Neuron. 2001;29:629–643. [PubMed: 11301023]
- 327.
- Putcha GV, Moulder KL, Golden JP, Bouillet P, Adams JA, Strasser A. et al. Induction of BIM, a proapoptotic BH3-only BCL-2 family member, is critical for neuronal apoptosis. Neuron. 2001;29:615–628. [PubMed: 11301022]
- 328.
- Harris CA, Johnson E M Jr. Bh3-only bcl-2 family members are coordinately regulated by the jnk pathway and require bax to induce apoptosis in neurons. J Biol Chem. 2001;276:37754–37760. [PubMed: 11495903]
- 329.
- Leonard JR, D'Sa C, Cahn BR, Korsmeyer SJ, Roth KA. Bid regulation of neuronal apoptosis. Brain Res Dev Brain Res. 2001;128:187–190. [PubMed: 11412905]
- 330.
- Henshall DC, Bonislawski DP, Skradski SL, Lan JQ, Meller R, Simon RP. Cleavage of bid may amplify caspase-8-induced neuronal death following focally evoked limbic seizures. Neurobiol Dis. 2001;8:568–580. [PubMed: 11493022]
- 331.
- Sun YF, Yu LY, Saarma M, Timmusk T, Arumae U. Neuron-specific Bcl-2 homology 3 domain-only splice variant of Bak is anti-apoptotic in neurons, but pro-apoptotic in non-neuronal cells. J Biol Chem. 2001;276:16240–16247. [PubMed: 11278671]
- 332.
- Vila M, Jackson-Lewis V, Vukosavic S, Djaldetti R, Liberatore G, Offen D. et al. Bax ablation prevents dopaminergic neurodegeneration in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson's disease. Proc Natl Acad Sci USA. 2001;98:2837–2842. [PMC free article: PMC30226] [PubMed: 11226327]
- 333.
- Offen D, Beart PM, Cheung NS, Pascoe CJ, Hochman A, Gorodin S. et al. Transgenic mice expressing human Bcl-2 in their neurons are resistant to 6-hydroxydopamine and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. Proc Natl Acad Sci USA. 1998;95:5789–5794. [PMC free article: PMC20458] [PubMed: 9576963]
- 334.
- Feinstein E, Kimchi A, Wallach D, Boldin M, Varfolomeev E. The death domain: A module shared by proteins with diverse cellular functions. Trends Biochem Sci. 1995;20:342–344. [PubMed: 7482697]
- 335.
- Vukosavic S, Stefanis L, Jackson-Lewis V, Guegan C, Romero N, Chen C. et al. Delaying caspase activation by Bcl-2: A clue to disease retardation in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci. 2000;20:9119–9125. [PMC free article: PMC6773037] [PubMed: 11124989]
- 336.
- Martin LJ. Neuronal death in amyotrophic lateral sclerosis is apoptosis: Possible contribution of a programmed cell death mechanism. J Neuropathol Exp Neurol. 1999;58:459–471. [PubMed: 10331434]
- 337.
- Gonzalez de Aguilar JL, Gordon JW, Rene F, de Tapia M, Lutz-Bucher B, Gaiddon C. et al. Alteration of the Bcl-x/Bax ratio in a transgenic mouse model of amyotrophic lateral sclerosis: Evidence for the implication of the p53 signaling pathway. Neurobiol Dis. 2000;7:406–415. [PubMed: 10964611]
- 338.
- Guegan C, Vila M, Rosoklija G, Hays AP, Przedborski S. Recruitment of the mitochondrial-dependent apoptotic pathway in amyotrophic lateral sclerosis. J Neurosci. 2001;21:6569–6576. [PMC free article: PMC6763092] [PubMed: 11517246]
- 339.
- Azzouz M, Hottinger A, Paterna JC, Zurn AD, Aebischer P, Bueler H. Increased motoneuron survival and improved neuromuscular function in transgenic ALS mice after intraspinal injection of an adeno-associated virus encoding Bcl-2. Hum Mol Genet. 2000;9:803–811. [PubMed: 10749988]
- 340.
- Kostic V, Jackson-Lewis V, de Bilbao F, Dubois-Dauphin M, Przedborski S. Bcl-2: Prolonging life in a transgenic mouse model of familial amyotrophic lateral sclerosis. Science. 1997;277:559–562. [PubMed: 9228005]
- 341.
- Iwahashi H, Eguchi Y, Yasuhara N, Hanafusa T, Matsuzawa Y, Tsujimoto Y. Synergistic anti-apoptotic activity between Bcl-2 and SMN implicated in spinal muscular atrophy. Nature. 1997;390:413–417. [PubMed: 9389483]
- 342.
- Coovert DD, Le TT, Morris GE, Man NT, Kralewski M, Sendtner M. et al. Does the survival motor neuron protein (SMN) interact with Bcl-2? J Med Genet. 2000;37:536–539. [PMC free article: PMC1734632] [PubMed: 10970187]
- 343.
- Bernardi P, Scorrano L, Colonna R, Petronilli V, Di Lisa F. Mitochondria and cell death. Mechanistic aspects and methodological issues. Eur J Biochem. 1999;264:687–701. [PubMed: 10491114]
- 344.
- Zamzami N, Kroemer G. The mitochondrion in apoptosis: How Pandora's box opens. Nat Rev Mol Cell Biol. 2001;2:67–71. [PubMed: 11413468]
- 345.
- Budd SL, Tenneti L, Lishnak T, Lipton SA. Mitochondrial and extramitochondrial apoptotic signaling pathways in cerebrocortical neurons. Proc Natl Acad Sci USA. 2000;97:6161–6166. [PMC free article: PMC18575] [PubMed: 10811898]
- 346.
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM. et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. [PubMed: 9989411]
- 347.
- Li LY, Luo X, Wang X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature. 2001;412:95–99. [PubMed: 11452314]
- 348.
- Troy CM, Rabacchi SA, Hohl JB, Angelastro JM, Greene LA, Shelanski ML. Death in the balance: alternative participation of the caspase-2 and -9 pathways in neuronal death induced by nerve growth factor deprivation. J Neurosci. 2001;21:5007–5016. [PMC free article: PMC6762825] [PubMed: 11438576]
- 349.
- Parrish J, Li L, Klotz K, Ledwich D, Wang X, Xue D. Mitochondrial endonuclease G is important for apoptosis in C. elegans. Nature. 2001;412:90–94. [PubMed: 11452313]
- 350.
- Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 beta-converting enzyme. Cell. 1993;75:641–652. [PubMed: 8242740]
- 351.
- Alnemri ES, Livingston DJ, Nicholson DW, Salvesen G, Thornberry NA, Wong WW. et al. Human ICE/CED-3 protease nomenclature. Cell. 1996;87:171. [PubMed: 8861900]
- 352.
- Stennicke HR, Salvesen GS. CaspasesControlling intracellular signals by protease zymogen activation. Biochim Biophys Acta. 2000;1477:299–306. [PubMed: 10708865]
- 353.
- Eckhart L, Declercq W, Ban J, Rendl M, Lengauer B, Mayer C. et al. Terminal differentiation of human keratinocytes and stratum corneum formation is associated with caspase-14 activation. J Invest Dermatol. 2000;115:1148–1151. [PubMed: 11121154]
- 354.
- Kuechle MK, Predd HM, Fleckman P, Dale BA, Presland RB. Caspase-14, a keratinocyte specific caspase: mRNA splice variants and expression pattern in embryonic and adult mouse. Cell Death Differ. 2001;8:868–870. [PubMed: 11526440]
- 355.
- Sanchez Mejia RO, Ona VO, Li M, Friedlander RM. Minocycline reduces traumatic brain injury-mediated caspase-1 activation, tissue damage, and neurological dysfunction. Neurosurgery. 2001;48:1393–1399, discussion. 1399–1401. [PubMed: 11383749]
- 356.
- Shibata M, Hisahara S, Hara H, Yamawaki T, Fukuuchi Y, Yuan J. et al. Caspases determine the vulnerability of oligodendrocytes in the ischemic brain. J Clin Invest. 2000;106:643–653. [PMC free article: PMC381288] [PubMed: 10974017]
- 357.
- Hisahara S, Yuan J, Momoi T, Okano H, Miura M. Caspase-11 mediates oligodendrocyte cell death and pathogenesis of autoimmune-mediated demyelination. J Exp Med. 2001;193:111–122. [PMC free article: PMC2195881] [PubMed: 11136825]
- 358.
- Grimm S, Stanger BZ, Leder P. RIP and FADD: two “death domain”-containing proteins can induce apoptosis by convergent, but dissociable, pathways. Proc Natl Acad Sci USA. 1996;93:10923–10927. [PMC free article: PMC38259] [PubMed: 8855284]
- 359.
- Zou H, Li Y, Liu X, Wang X. An APAF-1. cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem. 1999;274:11549–11556. [PubMed: 10206961]
- 360.
- Srinivasula SM, Ahmad M, Fernandes-Alnemri T, Alnemri ES. Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization. Mol Cell. 1998;1:949–957. [PubMed: 9651578]
- 361.
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES. et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. [PubMed: 9390557]
- 362.
- Hu Y, Benedict MA, Ding L, Nunez G. Role of cytochrome c and dATP/ATP hydrolysis in Apaf-1-mediated caspase-9 activation and apoptosis. EMBO J. 1999;18:3586–3595. [PMC free article: PMC1171437] [PubMed: 10393175]
- 363.
- Chu ZL, Pio F, Xie Z, Welsh K, Krajewska M, Krajewski S. et al. A novel enhancer of the Apaf1 apoptosome involved in cytochrome c-dependent caspase activation and apoptosis. J Biol Chem. 2001;276:9239–9245. [PubMed: 11113115]
- 364.
- Fujita E, Jinbo A, Matuzaki H, Konishi H, Kikkawa U, Momoi T. Akt phosphorylation site found in human caspase-9 is absent in mouse caspase-9. Biochem Biophys Res Commun. 1999;264:550–555. [PubMed: 10529400]
- 365.
- Namura S, Zhu J, Fink K, Endres M, Srinivasan A, Tomaselli KJ. et al. Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J Neurosci. 1998;18:3659–3668. [PMC free article: PMC6793169] [PubMed: 9570797]
- 366.
- Hartmann A, Hunot S, Michel PP, Muriel MP, Vyas S, Faucheux BA. et al. Caspase-3: A vulnerability factor and final effector in apoptotic death of dopaminergic neurons in Parkinson's disease. Proc Natl Acad Sci USA. 2000;97:2875–2880. [PMC free article: PMC16023] [PubMed: 10688892]
- 367.
- Su JH, Zhao M, Anderson AJ, Srinivasan A, Cotman CW. Activated caspase-3 expression in Alzheimer's and aged control brain: Correlation with Alzheimer pathology. Brain Res. 2001;898:350–357. [PubMed: 11306022]
- 368.
- Li M, Ona VO, Chen M, Kaul M, Tenneti L, Zhang X. et al. Functional role and therapeutic implications of neuronal caspase-1 and -3 in a mouse model of traumatic spinal cord injury. Neuroscience. 2000;99:333–342. [PubMed: 10938439]
- 369.
- Tenneti L, Lipton SA. Involvement of activated caspase-3-like proteases in N-methyl-D- aspartate-induced apoptosis in cerebrocortical neurons. J Neurochem. 2000;74:134–142. [PubMed: 10617114]
- 370.
- Benchoua A, Guegan C, Couriaud C, Hosseini H, Sampaio N, Morin D. et al. Specific caspase pathways are activated in the two stages of cerebral infarction. J Neurosci. 2001;21:7127–7134. [PMC free article: PMC6762989] [PubMed: 11549723]
- 371.
- Chen M, Ona VO, Li M, Ferrante RJ, Fink KB, Zhu S. et al. Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med. 2000;6:797–801. [PubMed: 10888929]
- 372.
- Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H. et al. Decreased apoptosis in the brain and premature lethality in CPP32- deficient mice. Nature. 2000;384:368–372. [PubMed: 8934524]
- 373.
- LeBlanc A, Liu H, Goodyer C, Bergeron C, Hammond J. Caspase-6 role in apoptosis of human neurons, amyloidogenesis, and Alzheimer's disease. J Biol Chem. 1999;274:23426–23436. [PubMed: 10438520]
- 374.
- Wellington CL, Singaraja R, Ellerby L, Savill J, Roy S, Leavitt B. et al. Inhibiting caspase cleavage of huntingtin reduces toxicity and aggregate formation in neuronal and nonneuronal cells. J Biol Chem. 2000;275:19831–19838. [PubMed: 10770929]
- 375.
- Selznick LA, Zheng TS, Flavell RA, Rakic P, Roth KA. Amyloid beta-induced neuronal death is bax-dependent but caspase- independent. J Neuropathol Exp Neurol. 2000;59:271–279. [PubMed: 10759182]
- 376.
- D'Mello SR, Kuan CY, Flavell RA, Rakic P. Caspase-3 is required for apoptosis-associated DNA fragmentation but not for cell death in neurons deprived of potassium. J Neurosci Res. 2000;59:24–31. [PubMed: 10658182]
- 377.
- Droin N, Beauchemin M, Solary E, Bertrand R. Identification of a caspase-2 isoform that behaves as an endogenous inhibitor of the caspase cascade. Cancer Res. 2000;60:7039–7047. [PubMed: 11156409]
- 378.
- Tschopp J, Irmler M, Thome M. Inhibition of fas death signals by FLIPs. Curr Opin Immunol. 1998;10:552–558. [PubMed: 9794838]
- 379.
- Cheema ZF, Wade SB, Sata M, Walsh K, Sohrabji F, Miranda RC. Fas/Apo [apoptosis]-1 and associated proteins in the differentiating cerebral cortex: induction of caspase-dependent cell death and activation of NF-kappaB. J Neurosci. 1999;19:1754–1770. [PMC free article: PMC6782175] [PubMed: 10024361]
- 380.
- Yeh WC, Itie A, Elia AJ, Ng M, Shu HB, Wakeham A. et al. Requirement for Casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity. 2000;12:633–642. [PubMed: 10894163]
- 381.
- Yang YL, Li XM. The IAP family: Endogenous caspase inhibitors with multiple biological activities. Cell Res. 2000;10:169–177. [PubMed: 11032169]
- 382.
- Holcik M, Korneluk RG. XIAP, the guardian angel. Nat Rev Mol Cell Biol. 2001;2:550–556. [PubMed: 11433370]
- 383.
- Yang Y, Fang S, Jensen JP, Weissman AM, Ashwell JD. Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science. 2000;288:874–877. [PubMed: 10797013]
- 384.
- Liston P, Fong WG, Kelly NL, Toji S, Miyazaki T, Conte D. et al. Identification of XAF1 as an antagonist of XIAP anti-Caspase activity. Nat Cell Biol. 2001;3:128–133. [PubMed: 11175744]
- 385.
- Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE. et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102:43–53. [PubMed: 10929712]
- 386.
- Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. [PubMed: 10929711]
- 387.
- Siman R, Noszek JC. Excitatory amino acids activate calpain I and induce structural protein breakdown in vivo. Neuron. 1988;1:279–287. [PubMed: 2856162]
- 388.
- Siman R, Noszek JC, Kegerise C. Calpain I activation is specifically related to excitatory amino acid induction of hippocampal damage. J Neurosci. 1989;9:1579–1590. [PMC free article: PMC6569848] [PubMed: 2542478]
- 389.
- Saatman KE, Bozyczko-Coyne D, Marcy V, Siman R, McIntosh TK. Prolonged calpain-mediated spectrin breakdown occurs regionally following experimental brain injury in the rat. J Neuropathol Exp Neurol. 1996;55:850–860. [PubMed: 8965100]
- 390.
- Saatman KE, Murai H, Bartus RT, Smith DH, Hayward NJ, Perri BR. et al. Calpain inhibitor AK295 attenuates motor and cognitive deficits following experimental brain injury in the rat. Proc Natl Acad Sci USA. 1996;93:3428–3433. [PMC free article: PMC39625] [PubMed: 8622952]
- 391.
- Faddis BT, Hasbani MJ, Goldberg MP. Calpain activation contributes to dendritic remodeling after brief excitotoxic injury in vitro. J Neurosci. 1997;17:951–959. [PMC free article: PMC6573163] [PubMed: 8994050]
- 392.
- Kampfl A, Posmantur RM, Zhao X, Schmutzhard E, Clifton GL, Hayes RL. Mechanisms of calpain proteolysis following traumatic brain injury: Implications for pathology and therapy: implications for pathology and therapy: a review and update. J Neurotrauma. 1997;14:121–134. [PubMed: 9104930]
- 393.
- Rami A, Ferger D, Krieglstein J. Blockade of calpain proteolytic activity rescues neurons from glutamate excitotoxicity. Neurosci Res. 1997;27:93–97. [PubMed: 9089703]
- 394.
- Scarisbrick IA, Towner MD, Isackson PJ. Nervous system-specific expression of a novel serine protease: Regulation in the adult rat spinal cord by excitotoxic injury. J Neurosci. 1997;17:8156–8168. [PMC free article: PMC6573769] [PubMed: 9334391]
- Neuronal Survival and Cell Death Signaling Pathways - Madame Curie Bioscience Da...Neuronal Survival and Cell Death Signaling Pathways - Madame Curie Bioscience Database
- Gossypium hirsutum strain:TM-1Gossypium hirsutum strain:TM-1Gossypium hirsutum strain:TM-1 Transcriptome or Gene expressionBioProject
Your browsing activity is empty.
Activity recording is turned off.
See more...