

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Enveloped viruses rely on fusion proteins in their envelope to fuse the viral membrane to the host cell membrane. This key step in viral entry delivers the viral genome into the cytoplasm for replication. Although class II fusion proteins are genetically and structurally unrelated to class I fusion proteins, they use the same physical principles and topology as other fusion proteins to drive membrane fusion. Exposure of a fusion loop first allows it to insert into the host cell membrane. Conserved hydrophobic residues in the fusion loop act as an anchor, which penetrates only partway into the outer bilayer leaflet of the host cell membrane. Subsequent folding back of the fusion protein on itself directs the C terminal viral transmembrane anchor towards the fusion loop. This fold back forces the host cell membrane (held by the fusion loop) and the viral membrane (held by the C terminal transmembrane anchor) against each other, resulting in membrane fusion. In class II fusion proteins, the fold back is triggered by the reduced pH of an endosome, and is accompanied by the assembly of fusion protein monomers into trimers. The fold back occurs by domain rearrangement rather than by an extensive refolding of secondary structure, but this domain rearrangement and the assembly of monomers into trimers together bury a large surface area. The energy that is thus released exerts a bending force on the apposed viral and cellular membranes, causing them to bend towards each other and, eventually, to fuse.
Introduction
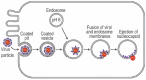
Enveloped viruses acquire a lipid bilayer membrane when they bud across the plasma membrane or the membrane of the endoplasmic reticulum (ER) during assembly of the virion.1,2 During infection, the viral membrane must be fused to the host cell membrane to deliver the viral genome into the cytoplasm for replication (Fig. 1). The fusion of the viral and host cell membranes is therefore the central molecular event during the entry of enveloped viruses into cells. Adjacent membranes do not fuse spontaneously, membrane fusion requires considerable energy (on the order of 100 kJ mol1 or 40 kT).3,4 Envelope proteins anchored in the viral membrane provide this energy in the form of a conformational rearrangement that bends the apposed membranes towards each other, inducing them to fuse.5-7 Most ‘fusion proteins’ (or their cleavage products) also effect cellular attachment of the virus prior to the membrane fusion event by binding to a receptor on the cell surface, except the paramyxo and alphaviruses, in which a second envelope protein binds the receptor.
Fusion proteins of enveloped viruses fall into two structural classes. The influenza virus haemagglutinin (HA) is the prototype of class I fusion proteins,8 which encompass those of other orthomyxo and paramyxoviruses such as measles virus, retroviruses such as human immunodeficiency virus (HIV), filoviruses such as Ebola virus, and coronaviruses such as SARS (see Chapters 4-6). Class II fusion proteins are a structurally and evolutionarily distinct class of proteins found in Flaviviridae, such as dengue, yellow fever, and West Nile viruses, and on alphaviruses, such as Semliki Forest and Sindbis viruses. Hepatitis C has a similar genomic organization to the flaviviruses, and therefore most likely relies on a Class II fusion protein as well. Crystal structures of several class I and class II fusion proteins before9-15 and after5,16-29 their fusogenic conformational rearrangements have provided us with a detailed molecular understanding of the fusion mechanism (Table 1). The structures show that, despite the absence of similarities in the protein folds of the two classes, fusion proteins from both classes use the same physical principles and general topology to drive membrane fusion. First the fusion protein inserts a hydrophobic fusion anchor partway into the outer bilayer leaflet of the host cell membrane. The fusion anchor is either an N terminal peptide,30 as in influenza and HIV,31 or an internal loop, as in SARS coronavirus,32 avian sarcoma leucosis virus33 and all class II enveloped viruses.34 Second, the fusion protein folds back on itself, directing the (C terminal) viral transmembrane anchor towards the fusion anchor. This fold back forces the host cell membrane (held by the fusion anchor) and the viral membrane (held by the C terminal transmembrane anchor) against each other, resulting in fusion of the two membranes. In this chapter, I describe our current picture of how class II fusion proteins drive viral membrane fusion, based on the structural and biochemical data available to date.
Table 1.
Class II fusion protein crystal structures and corresponding electron cryomicroscopy structures.
Overall Architecture
Three dimensional structures of eight class II fusion proteins in their native, or prefusion states,10,12-14,35,93-95,97 that is, the conformation that they adopt on the surface of a mature virus particle, have been determined at near atomic resolution. Figure 2 shows the three domain structures of E13 and E1,12 the fusion proteins of dengue virus (a representative flavivirus) and of Semliki Forest Virus (an alphavirus), respectively. The two proteins share a common molecular architecture, despite a lack of significant sequence similarity. Domain I, an eight stranded β barrel, organizes the structure. Two long insertions between pairs of consecutive β strands in domain I form the elongated domain II, which bears the fusion anchor, a fusion loop in class II proteins, at its tip (Figs. 2, 4). Domain II contains twelve β strands and two α helices. Domain III is an IgC like module, with ten β strands. Domain III contains most of the antigenic sites on e, as well as most of the structural determinants of virulence and tropism.10 This observation, and the widespread occurrence of immunoglobulin modules in cell adhesion proteins, suggest that domain III participates in attachment to a cellular receptor.10 Indeed, positively charged patches on the surface of domain III in dengue virus have been suggested to promote attachment by binding heparan sulfate on the cell surface.36 Both e1 and e have one or more glycosylation sites. These glycans can aid viral attachment to the cell surface, in the case of dengue virus by binding to the lectin DC SIGN.37,38 As expected from their sequence identities (= 37%), flaviviral E proteins have very similar overall structures, and differ only in the length and structure of surface exposed loops, some of which have been implicated in receptor binding.10,39,40 Despite these hints on the basis of cellular attachment, however, a cellular receptor that specifically recognizes an envelope protein on a class II enveloped virus has yet to be conclusively identified, although candidate receptors for dengue virus type 141 and West Nile virus42 were recently suggested.

It is important to note that all the crystal structures of fusion proteins determined so far, from both classes and regardless of their conformational state, lack the C terminal viral membrane anchor. This anchor consists of one or two transmembrane helices, and has been intentionally omitted in constructs targeted for crystallization to facilitate expression and handling, and to promote crystallization. The crystallized species are therefore referred to as soluble fragments of the ectodomains of the full length fusion protein. Furthermore, all available crystal structures of class II fusion proteins lack the ‘stem’ region,43 a 30-55 amino acid linker between Domain III and the C terminal transmembrane anchor (Figs. 2A–B, 3). As I will discuss below, the stem region plays a key role in the final stages of membrane fusion. Its function is analogous to that of the ‘outer helix’ in class I fusion proteins.8
Maturation and Priming
Both class I and class II fusion proteins rely on a proteolytic cleavage event to become primed to respond to the environmental conditions appropriate for fusion. These conditions are usually the acidic pH of an endosome (Fig. 1), but for some class I enveloped viruses, such as HIV, coreceptor binding is required instead. In contrast to class I fusion proteins, however, class II fusion proteins rely on a priming proteolytic cleavage that does not cleave the fusion protein itself. Instead, class II proteins associate with a second, ‘protector’ protein, called M (for membrane protein) in flaviviruses or E2 in alphaviruses. The protector protein is cleaved by furin when immature virus particles assembled in the ER reach the trans Golgi network.44 The cleavage produces mature virus particles, which are then released from the host cell by exocytosis. The cleavage of the protector protein releases a conformational constraint on the fusion protein, which allows it to adopt its mature conformation (described above) in a large rearrangement on the viral surface. In the mature conformation, the fusion protein is primed to respond to acidic pH and induce membrane fusion with a further conformational rearrangement (described below).
Structures from electron cryomicroscopy of both immature45,46 and mature47-50 flavivirus and alphavirus particles, provide a detailed picture of the rearrangement that accompanies maturation in these viruses. Alphaviruses retain the T = 4 icosahedral packing of their envelope proteins, but domains that form spikes on the immature virion swing in towards the threefold symmetry axis, during maturation.46,50 The rearrangement is more dramatic in flaviviruses, in which the fusion protein e breaks the T = 3 icosahedral symmetry of the immature virion45 to adopt an unusual icosahedral herringbone pattern in the mature virion.47,51 In both alphaviruses and flaviviruses, the fusion proteins form dimers in the mature virion albeit in different configurations.10,12 The key feature of the maturation process in both genera, however, is that cleavage of the protector protein allows the fusion loop to reposition itself so that it is poised to insert into the host cell membrane in response to acidification of the solute in the endosome. Mature virus particles are therefore infectious,44,52 unlike immature virions,53,54 which are insensitive to pH. the fusion loop is shielded from the viral surface in mature virions by E E dimer contacts in flaviviruses, or by protein E2 in alphaviruses (Figs . 3A,B,5A).
The Fusogenic Conformational Rearrangement
The three dimensional structures of four class II fusion proteins in their postfusion states29,55,56,94 reveal striking differences from the prefusion forms (Fig. 3), and suggest a molecular mechanism for membrane fusion (see below and Fig. 5). Like class I fusion proteins, flaviviral e proteins and alphaviral e1 proteins are both homotrimers in their postfusion conformations. Class II proteins form trimers from monomers on the viral surface, while class I proteins are trimeric in their prefusion state.8 however, a comparison of the pre and postfusion states of influenza ha the only example in its class where both structures are known for the same protein shows that, as in class II fusion proteins, nearly all of the trimer contacts in the postfusion state are formed during the fusogenic conformational rearrangement.
Unlike influenza HA, which undergoes extensive refolding during membrane fusion, the three domains of class II fusion proteins retain most of their folded structures (Fig. 3). Instead, the domains undergo major rearrangements in their relative orientations, through flexion of the interdomain linkers. Domain III undergoes the most significant displacement in the fusion transition. It rotates by about 70, and its center of mass shifts by 30-40Å towards domain II. This folding over brings the C terminus of domain III about 40Å closer to the fusion loop, at tip of domain II (Fig. 3). Domain II rotates 15-30 with respect to domain I about a hinge region13 in which mutations affect the pH threshold of fusion in various flaviviruses.57-62 These conformational rearrangements position the end of domain III and the beginning of the stem region that links domain III to the C terminal viral transmembrane anchor towards the fusion loop (Fig. 3E F).29,55 A deep channel extends from the C terminus of the crystallized fragment along the intersubunit contact between domains II to the fusion loops, in both the dengue and Semliki Forest virus postfusion trimer structures. In the full length fusion proteins, it is thought that the stem binds in this channel in an extended, but mainly α helical conformation.29,48 This proposed stem conformation places the viral transmembrane anchor in the immediate vicinity of the fusion loop, just as in the postfusion conformation of class I viral fusion proteins.
The fusion transition in class II viral proteins is irreversible. The refoldings just described may impart irreversibility by contributing a high barrier to initiation of trimerization and an even higher barrier to dissociation of postfusion trimers once they have formed. Moreover, many new polar and nonpolar contacts are formed during the fusion transition, in several different areas along the threefold axis of the trimer. The total surface buried is 13,000-15,000Å2,29,55 nearly four times more than is buried in the prefusion dimer. The stem, which is missing from currently available crystal structures, most likely forms additional contacts with the core trimer structure. The stem does indeed promote trimer assembly even in the absence of liposomes.43
The Fusion Loop
The process of viral membrane fusion in both class I and class II enveloped viruses begins with the exposure of a fusion anchor, and its subsequent insertion into the host cell membrane. Fusion anchors from both viral classes vary in length but are in general rich in glycines and hydrophobic residues, particularly aromatic residues such as Trp or Phe. Sequence conservation is poor between fusion proteins of both classes. The fusion anchor in class I fusion proteins the ‘fusion peptide’ is a region of approximately 20 residues at or near the N terminus of the envelope protein. The crystal structure of the parainfluenza virus 5 fusion (F) protein in its prefusion form reveals the fusion peptide wedged between two subunits of the protein, in a partly extended, partly β sheet and partly α helical conformation.15 Structural studies on influenza HA in its postfusion conformation using NMR and other spectroscopic techniques show that the fusion peptide is mostly α helical in character and that its structure changes only subtly as it inserts partway into the outer leaflet of the host cell lipid bilayer.63,64 None of the currently available postfusion class I protein crystal structures contain information on the fusion peptide.
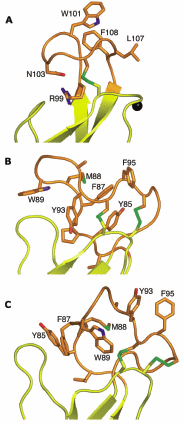
The recently determined crystal structures of class II fusion proteins in pre10,12-14 and postfusion29,55,56 conformations offer the first direct views of fusion anchors in this case, the fusion loops as they insert into a target membrane (Fig. 4). Like the class I fusion peptide, the class II fusion loop penetrates only partway into the hydrocarbon layer of the target membrane. Exposed carbonyls and charged residues prevent the fusion loop from penetrating further than 6Å.29,56 In flaviviruses, the fusion loop adopts a tightly folded conformation, which is stabilized by a disulfide bond (Fig. 4A). The structure of the fusion loop is essentially identical in the pre and postfusion conformations of the protein, suggesting that membrane insertion has no effect on the structure of the fusion loop. During the fusion transition, three hydrophobic residues in the fusion loop (Trp, Leu, and Phe) become exposed on the molecular surface. Three fusion loops end up in close proximity at the tip of the trimer in the postfusion conformation, where they form a crater like surface with a hydrophobic rim (Fig. 3E). Electron cryomicroscopy29 and mutagenesis studies34 confirm that these hydrophobic, mostly aromatic residues on the crater rim insert into the host cell membrane, acting as an ‘aromatic anchor’ for the fusion protein. The concave shape of the crater is thought to be important in generating distortions or perturbations in the host cell membrane,29 which are required for fusion.65
In alphaviruses, the fusion loop is also rich in aromatic and other hydrophobic residues. Unlike flaviviral fusion loops, however, alphaviviral fusion loops do not form trimer contacts (Fig. 3F). Indeed, in the postfusion structure of the Semliki Forest virus e1 trimer, the fusion loops have high temperature factors and exhibit a high degree of flexibility despite the presence of two disulfide bonds. Thus, the structures of the fusion loops are poorly defined, but each fusion loop seems to adopt a very different conformation (Fig. 4B,C).55 The fusion loops in the postfusion Semliki Forest virus e1 structure form quite polar surfaces, with many mainchain carbonyls and some polar or charged sidechains exposed on the surface. This suggests that, in contrast to flaviviral fusion loops, alphaviral fusion loops either change their conformation upon membrane insertion to shield polar groups from the membrane, or the fusion loops only interact with the polar headgroups of the lipids, and do not penetrate into the hydrocarbon layer.
Semliki Forest virus E1 trimers form irregular clusters, or ‘rosettes’ of about 40-60 trimers through contacts between fusion loops in adjacent trimers.55 This is reminiscent of influenza virus HA, which aggregates into rosettes through interactions between the fusion peptide, at low pH and after proteolytic activation.66 This fusion loop/peptide clustering may provide a mechanism for the direct coupling of several E1/HA trimers to work in concert around a single fusion site (see below).
Mechanism of Membrane Fusion
Combined with previous knowledge, the structures of the fusion proteins from class II viruses in their postfusion states29,55,56 have led to a much better understanding of how conformational changes in the proteins drive membrane fusion. The structures confirm two major principles of membrane fusion machineries: (1) the fusion protein must insert an anchor into each of the two membranes to be fused, and (2) the protein folds back on itself in a thermodynamically favorable conformational rearrangement that drives membrane fusion by forcing the two anchors into close proximity.
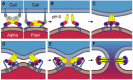
In the current model, viral membrane fusion proceeds as follows (Fig. 5). First, receptor binding by an envelope protein, which in flaviviruses is also the fusion protein, leads to clathrin mediated endocytosis of the virus (Figs. 1, 5A). When the virus reaches endosomal compartments the low pH of the lumen (pH 6) causes an initial conformational rearrangement that leads to the exposure of the previously buried fusion loop48,50 at the tip of domain II. In flaviviruses, domains I and II flex relative to each other by 30.29 This hinge motion causes domain II, and therefore the fusion loop, to swing away from the viral surface and towards the host cell membrane (Fig. 5B). Indeed, mutations at the domain I domain II interface in various flaviviruses alter the pH threshold of fusion.13,57-62 As domain II swings away from the viral surface, constraints imposed by the tight packing of E on the viral surface are released, allowing E to diffuse freely in the plane of the viral membrane. The stem may also be able to extend away from the membrane at this stage. In alphaviruses, constraints are released in response to low pH by the dissociation of the protector (and receptor binding) protein E2. This exposes the fusion loop and allows domain II of E1 to swing towards the nearest threefold symmetry axis in the virus particle in a 15 hinge motion relative to domain I, leading to the formation of trimer contacts with adjacent E1 molecules.46
The second key step in the fusion process is insertion of the exposed fusion loop into the host cell membrane (Fig. 5C). Alphaviral E1 has already formed some trimer contacts at this stage, but flaviviral E proteins probably insert their fusion loops as monomers. Membrane insertion probably catalyzes trimerization of the fusion loops,67 by lateral rearrangement of e monomers. This trimeric prefusion intermediate (Fig. 5C) bridges host cell and viral membranes, anchored by its fusion loops in the former and by the viral transmembrane anchors in the latter. This proposed intermediate is analogous to the ‘prehairpin’ intermediate postulated for class I viral fusion mechanisms.68
Upon insertion of the fusion loops into the host cell membrane, formation of trimer contacts spreads from the fusion loops at the trimer tip to domain I at the trimer base. Domain II shifts and rotates, folding the stem and C terminal anchor back towards the fusion loop (Fig. 5D), and burying additional protein surfaces. Free energy released by this refolding drives the two membranes to bend towards each other,5-7 as the C terminal anchor is forced closer to the fusion loop, forming apposing nipples in the membranes (Fig. 5D).3 Fusion loop insertion may induce positive bilayer curvature, which would stabilize the lateral surfaces of the nipples. The concave shape of the crater like surface formed by the fusion loops at the trimer tip may also have a destabilizing effect on the membrane, as has been postulated for fusion peptides in class I fusion proteins.65 Based on the energy required to deform lipid bilayers, it seems likely that a ring of trimers refolding in concert is needed to provide sufficient energy to form nipples in the membranes.3,4 It is unclear exactly how many trimers are needed to drive membrane fusionin class II viruses, norhow their conformational changes are coupled. In the case of influenza, fusion requires the concerted action of at least three HA trimers,69 and is more likely driven by rings of 6-8 trimers.70 The clustering of fusion loops may provide a mechanism for the direct coupling of several E1 trimers to work in concert around a single fusion site in alphaviruses, but such clustering has not been observed in flaviviruses. It is possible that coupling occurs via the membrane: only when several trimers fold back in concert can they overcome the resistance of the membrane to deformation and reach their final, most stable postfusion conformation (Figs. 5D–F).
As the fusion transition proceeds, the stem zippers up onto the core of the trimer, along a channel that spans domain II, at the intersubunit contact regions (Figs. 3, 5D–F). The zippering up of the stem onto the domain II forces the fusion loop and the viral transmembrane anchor closer and closer, until the proximal leaflets of the two membranes fuse to form a ‘hemifusion stalk’ (Fig. 5E). Hemifusion is thought to be an essential intermediate of membrane fusion.3,4,71 (Fig. 5E) illustrates the need for shallow penetration of the viral fusion anchor into the host cell membrane: assuming several trimers do in fact act in concert around a single fusion site, fusion anchors from different trimers would collide if they inserted beyond the outer (proximal) lipid bilayer leaflet. This constraint on the length of the fusion anchor holds true for both class I fusion peptides and class II fusion loops.
Hemifusion stalks can ‘flicker’ open into narrow fusion pores.71 In order to prevent the transient fusion pores from closing, the stem must complete its zippering up onto the core of the trimer, and the C terminal transmembrane anchor must migrate into the pore (Fig. 5F). Indeed, the transition from hemifusion stalk to full fusion pore appears to require that the viral transmembrane anchor span the membrane completely, in all biological membrane fusion systems. Thus, the replacement of the C terminal transmembrane anchor of influenza HA with a glycosylphophatidylinositol (GPI) lipid anchor,72-74 or with a half length protein α helical anchor,75 stalls the fusion reaction at the stage of hemifusion. Other viral fusion proteins and cellular SNARE fusion proteins also require at least one transmembrane anchor.76-83 Upon completion of fusion, the trimer has reached the conformation seen in the postfusion crystal structures.29,55,56 The stems (not present in the structures) are docked along the surface of domains II, and the fusion loops and transmembrane anchors lie next to each other in the fused membrane (Fig. 5F).
Some class II fusion proteins, including those of alphaviruses, can only fuse membranes containing cholesterol and sphingolipids.84 The structural basis for this requirement is still not well understood. Several mutations in different regions of the Semliki Forest virus fusion protein e1 lower its dependence on cholesterol and/or sphingolipids for membrane fusion.85,86 It is unclear, however, whether the lower dependence on cholesterol of these mutants is due to an apparent destabilization of the E1 homotrimer,87 or to the different physical properties of membranes lacking cholesterol and sphingolipids. In flaviviruses, cholesterol facilitates fusion, but neither cholesterol nor sphingolipids are essential for fusion.88
Strategies for Fusion Inhibition
Many class II viruses, especially the flaviviruses, represent important human pathogens such as dengue, hepatitis C, yellow fever, West Nile, Japanese encephalitis and tick borne encephalitis viruses.89 For most of these viruses, there are no specific treatments for infection, their control by vaccination has proved elusive,89 and the number of infections is on the rise. Recently determined three dimensional structures of class II fusion proteins suggest new strategies for inhibiting viral entry by blocking membrane fusion. One such strategy stems for the discovery in dengue virus E of a long, tapering channel lined with hydrophobic side chains.13 In the crystal structure, the channel is occupied by a molecule of the detergent n octyl β d glucoside. In the absence of detergent, a β hairpin covering the channel swings towards the protein, and closes up the channel.13 The location of this ‘ligand binding pocket’ at the domain I domain II interface coincides with that of mutations affecting the pH threshold of fusion in various flaviviruses.57-62 Most of these mutations involve side chains lining the ligand binding pocket. The postfusion structure of dengue virus E shows that this region acts as a hinge between domains I and II during the fusogenic conformational rearrangement (see above).29 The opening up of a ligand binding pocket just at the locus of a hinge suggests that compounds tightly inserted at this position might hinder the conformational changes required for membrane fusion (Fig. 6A). The mechanism of action of such compounds might resemble that of some of the well studied antipicornaviral compounds, which block a concerted structural transition in the icosahedral assembly.90 alternatively, small molecules that pry open the β hairpin on binding in the pocket may inhibit infection by facilitating the low pH conformational change, causing premature triggering. Knowledge of the structure of the binding pocket with a bound ligand will guide efforts to design derivative ligands with higher affinities for use as inhibitors of flaviviral membrane fusion.
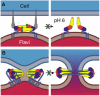
The postfusion structures of dengue29 and Semliki Forest55 viruses suggest a second possible strategy for fusion inhibition, related to an approach successful in developing an HIV antiviral compound.91 Peptides corresponding to the stem region of the gp41 fusion protein inhibit HIV entry by binding to the trimeric, N terminal ‘inner core’ of the protein and interfering with the folding back against it of the stem and C terminal viral transmembrane anchor. The way in which the stem is likely to fold back in class II viral fusion proteins (Figs. 3, 5D–F) suggests that an analogous strategy may be successful with class II viruses. Peptides derived from stem sequences could block completion of the fusogenic conformational change, by competing with the stem for interaction with surfaces on domain II, at the trimer interface (Fig. 6B). Stem like peptides or peptidomimetic compounds could thus inhibit viral membrane fusion in class II enveloped viruses by preventing the final folding back of the fusion protein that is required to drive the viral and host cell membranes together to fuse.
Conclusion
All viral membrane fusion proteins use the same physical principles and topology to drive membrane fusion. Class II fusion proteins are structurally and evolutionarily distinct class of proteins found in Flaviviridae, such as dengue, yellow fever, and West Nile viruses, and on alphaviruses, such as Semliki Forest and Sindbis viruses. Unlike class I fusion proteins such as influenza HA, which undergoes extensive refolding during membrane fusion, the three domains of class II fusion proteins retain most of their folded structures. Instead, the domains undergo major rearrangements in their relative orientations, through flexion of the interdomain linkers. Class II fusion proteins rely on a hydrophobic fusion loop to anchor themselves in the target cellular membrane. Like the class I fusion peptide, the class II fusion loop penetrates only partway into the hydrocarbon layer of the target membrane. Class II fusion proteins drive membrane fusion in a foldback rearrangement of a trimeric protein assembly. Crystal structures of class II envelope proteins have suggested two specific strategies for fusion inhibition, with hydrophobic small molecules and "stem" like peptides or peptidomimetics, respectively.
Note Added After Proofs
This chapter was originally written in 2006 and was updated in 2012 to add recent relevant advances.
References
- 1.
- Lindenbach BD, Rice CM. Flaviviridae: The viruses and their replication. In: Knipe DM, Howley PM, eds. Fields Virolo. 4th ed. Philadelphia: Lippincott Williams and Wilkins. 2001:991–1041.
- 2.
- Schlesinger S, Schlesinger MJ. Togaviridae: The viruses and their replication. In: Knipe DM, Howley PM, eds. Fields Virology. 4th ed. Philadelphia: Lippincott Williams and Wilkins. 2001:895–916.
- 3.
- Kuzmin PI, Zimmerberg J, Chizmadzhev YA, et al. A quantitative model for membrane fusion based on low-energy intermediates. Proc Natl Acad Sci USA. 2001;98(13):7235–7240. [PMC free article: PMC34652] [PubMed: 11404463]
- 4.
- Kozlov MM, Chernomordik LV. A mechanism of protein-mediated fusion: Coupling between refolding of the influenza hemagglutinin and lipid rearrangements. Biophys J. 1998;75(3):1384–1396. [PMC free article: PMC1299812] [PubMed: 9726939]
- 5.
- Baker KA, Dutch RE, Lamb RA, et al. Structural basis for paramyxovirus-mediated membrane fusion. Mol Cell. 1999;3(3):309–319. [PubMed: 10198633]
- 6.
- Melikyan GB, Markosyan RM, Hemmati H, et al. Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J Cell Biol. 2000;151(2):413–423. [PMC free article: PMC2192659] [PubMed: 11038187]
- 7.
- Russell CJ, Jardetzky TS, Lamb RA. Membrane fusion machines of paramyxoviruses: Capture of intermediates of fusion. EMBO J. 2001;20(15):4024–4034. [PMC free article: PMC149161] [PubMed: 11483506]
- 8.
- Skehel JJ, Wiley DC. Receptor binding and membrane fusion in virus entry: The influenza hemagglutinin. Annu Rev Biochem. 2000;69:531–569. [PubMed: 10966468]
- 9.
- Wilson IA, Skehel JJ, Wiley DC. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature. 1981;289(5796):366–373. [PubMed: 7464906]
- 10.
- Rey FA, Heinz FX, Mandl C, et al. The envelope glycoprotein from tick-borne encephalitis virus at 2 A resolution. Nature. 1995;375(6529):291–298. [PubMed: 7753193]
- 11.
- Rosenthal PB, Zhang X, Formanowski F, et al. Structure of the haemagglutinin-esterase-fusion glycoprotein of influenza C virus. Nature. 1998;396(6706):92–96. [PMC free article: PMC7095117] [PubMed: 9817207]
- 12.
- Lescar J, Roussel A, Wien MW, et al. The Fusion glycoprotein shell of Semliki Forest virus: An icosahedral assembly primed for fusogenic activation at endosomal pH. Cell. 2001;105(1):137–148. [PubMed: 11301009]
- 13.
- Modis Y, Harrison SC. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc Natl Acad Sci USA. 2003;100:6986–6991. [PMC free article: PMC165817] [PubMed: 12759475]
- 14.
- Modis Y, Ogata S, Clements D, et al. Variable surface epitopes in the crystal structure of dengue virus type 3 envelope glycoprotein. J Virol. 2005;79(2):1223–1231. [PMC free article: PMC538574] [PubMed: 15613349]
- 15.
- Yin HS, Wen X, Paterson RG, et al. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature. 2006;439(7072):38–44. [PMC free article: PMC7095149] [PubMed: 16397490]
- 16.
- Bullough PA, Hughson FM, Skehel JJ, et al. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature. 1994;371(6492):37–43. [PubMed: 8072525]
- 17.
- Fass D, Harrison SC, Kim PS. Retrovirus envelope domain at 1.7 angstrom resolution. Nat Struct Biol. 1996;3(5):465–469. [PubMed: 8612078]
- 18.
- Chan DC, Fass D, Berger JM, et al. Core structure of gp41 from the HIV envelope glycoprotein. Cell. 1997;89(2):263–273. [PubMed: 9108481]
- 19.
- Tan K, Liu J, Wang J, et al. Atomic structure of a thermostable subdomain of HIV-1 gp41. Proc Natl Acad Sci USA. 1997;94(23):12303–12308. [PMC free article: PMC24915] [PubMed: 9356444]
- 20.
- Weissenhorn W, Dessen A, Harrison SC, et al. Atomic structure of the ectodomain from HIV-1 gp41. Nature. 1997;387(6631):426–430. [PubMed: 9163431]
- 21.
- Malashkevich VN, Chan DC, Chutkowski CT, et al. Crystal structure of the simian immunodeficiency virus (SIV) gp41 core: Conserved helical interactions underlie the broad inhibitory activity of gp41 peptides. Proc Natl Acad Sci USA. 1998;95(16):9134–9139. [PMC free article: PMC21304] [PubMed: 9689046]
- 22.
- Caffrey M, Cai M, Kaufman J, et al. Three-dimensional solution structure of the 44 kDa ectodomain of SIV gp41. EMBO J. 1998;17(16):4572–4584. [PMC free article: PMC1170787] [PubMed: 9707417]
- 23.
- Weissenhorn W, Carfi A, Lee KH, et al. Crystal structure of the Ebola virus membrane fusion subunit, GP2, from the envelope glycoprotein ectodomain. Mol Cell. 1998;2(5):605–616. [PubMed: 9844633]
- 24.
- Kobe B, Center RJ, Kemp BE, et al. Crystal structure of human T cell leukemia virus type 1 gp21 ectodomain crystallized as a maltose-binding protein chimera reveals structural evolution of retroviral transmembrane proteins. Proc Natl Acad Sci USA. 1999;96(8):4319–4324. [PMC free article: PMC16330] [PubMed: 10200260]
- 25.
- Chen J, Skehel JJ, Wiley DC. N- and C-terminal residues combine in the fusion-pH influenza hemagglutinin HA(2) subunit to form an N cap that terminates the triple-stranded coiled coil. Proc Natl Acad Sci USA. 1999;96(16):8967–8972. [PMC free article: PMC17716] [PubMed: 10430879]
- 26.
- Zhao X, Singh M, Malashkevich VN, et al. Structural characterization of the human respiratory syncytial virus fusion protein core. Proc Natl Acad Sci USA. 2000;97(26):14172–14177. [PMC free article: PMC18890] [PubMed: 11106388]
- 27.
- Xu Y, Lou Z, Liu Y, et al. Crystal structure of severe acute respiratory syndrome coronavirus spike protein fusion core. J Biol Chem. 2004;279(47):49414–49419. [PMC free article: PMC8008698] [PubMed: 15345712]
- 28.
- Xu Y, Liu Y, Lou Z, et al. Structural basis for coronavirus-mediated membrane fusion. Crystal structure of mouse hepatitis virus spike protein fusion core. J Biol Chem. 2004;279(29):30514–30522. [PMC free article: PMC7982547] [PubMed: 15123674]
- 29.
- Modis Y, Ogata S, Clements D, et al. Structure of the dengue virus envelope protein after membrane fusion. Nature. 2004;427(6972):313–319. [PubMed: 14737159]
- 30.
- Gething MJ, White JM, Waterfield MD. Purification of the fusion protein of Sendai virus: Analysis of the NH2-terminal sequence generated during precursor activation. Proc Natl Acad Sci USA. 1978;75(6):2737–2740. [PMC free article: PMC392638] [PubMed: 208074]
- 31.
- Gallaher WR. Detection of a fusion peptide sequence in the transmembrane protein of human immunodeficiency virus. Cell. 1987;50(3):327–328. [PubMed: 3496970]
- 32.
- Supekar VM, Bruckmann C, Ingallinella P, et al. Structure of a proteolytically resistant core from the severe acute respiratory syndrome coronavirus S2 fusion protein. Proc Natl Acad Sci USA. 2004;101(52):17958–17963. [PMC free article: PMC539766] [PubMed: 15604146]
- 33.
- Cheng SF, Wu CW, Kantchev EA, et al. Structure and membrane interaction of the internal fusion peptide of avian sarcoma leukosis virus. Eur J Biochem. 2004;271(23-24):4725–4736. [PubMed: 15606759]
- 34.
- Allison SL, Schalich J, Stiasny K, et al. Mutational evidence for an internal fusion peptide in flavivirus envelope protein E. J Virol. 2001;75(9):4268–4275. [PMC free article: PMC114172] [PubMed: 11287576]
- 35.
- Zhang Y, Zhang W, Ogata S, et al. Conformational changes of the flavivirus E glycoprotein. Structure (Camb). 2004;12(9):1607–1618. [PMC free article: PMC4152830] [PubMed: 15341726]
- 36.
- Chen Y, Maguire T, Hileman RE, et al. Dengue virus infectivity depends on envelope protein binding to target cell heparan sulfate. Nat Med. 1997;3(8):866–871. [PubMed: 9256277]
- 37.
- Navarro-Sanchez E, Altmeyer R, Amara A, et al. Dendritic-cell-specific ICAM3-grabbing nonintegrin is essential for the productive infection of human dendritic cells by mosquito-cell-derived dengue viruses. EMBO Rep. 2003;4(7 Suppl):1–6. [PMC free article: PMC1326316] [PubMed: 12783086]
- 38.
- Tassaneetrithep B, Burgess TH, Granelli-piperno A, et al. DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J Exp Med. 2003;197(7):823–829. [PMC free article: PMC2193896] [PubMed: 12682107]
- 39.
- Crill WD, Roehrig JT. Monoclonal antibodies that bind to domain III of dengue virus E glycoprotein are the most efficient blockers of virus adsorption to Vero cells. J Virol. 2001;75(16):7769–7773. [PMC free article: PMC115016] [PubMed: 11462053]
- 40.
- Hung JJ, Hsieh MT, Young MJ, et al. An external loop region of domain III of dengue virus type 2 envelope protein is involved in serotype-specific binding to mosquito but not mammalian cells. J Virol. 2004;78(1):378–388. [PMC free article: PMC303388] [PubMed: 14671119]
- 41.
- Thepparit C, Smith DR. Serotype-specific entry of dengue virus into liver cells: Identification of the 37-kilodalton/67-kilodalton high-affinity laminin receptor as a dengue virus serotype 1 receptor. J Virol. 2004;78(22):12647–12656. [PMC free article: PMC525075] [PubMed: 15507651]
- 42.
- Chu JJ, Ng ML. Interaction of West Nile virus with alpha v beta 3 integrin mediates virus entry into cells. J Biol Chem. 2004;279(52):54533–54541. [PubMed: 15475343]
- 43.
- Allison SL, Stiasny K, Stadler K, et al. Mapping of functional elements in the stem-anchor region of tick-borne encephalitis virus envelope protein E. J Virol. 1999;73(7):5605–5612. [PMC free article: PMC112618] [PubMed: 10364309]
- 44.
- Stadler K, Allison SL, Schalich J, et al. Proteolytic activation of tick-borne encephalitis virus by furin. J Virol. 1997;71(11):8475–8481. [PMC free article: PMC192310] [PubMed: 9343204]
- 45.
- Zhang Y, Corver J, Chipman PR, et al. Structures of immature flavivirus particles. EMBO J. 2003;22(11):2604–2613. [PMC free article: PMC156766] [PubMed: 12773377]
- 46.
- Ferlenghi I, Gowen B, de Haas F, et al. The first step: Activation of the Semliki Forest virus spike protein precursor causes a localized conformational change in the trimeric spike. J Mol Biol. 1998;283(1):71–81. [PubMed: 9761674]
- 47.
- Kuhn RJ, Zhang W, Rossmann MG, et al. Structure of dengue virus: Implications for flavivirus organization, maturation, and fusion. Cell. 2002;108(5):717–725. [PMC free article: PMC4152842] [PubMed: 11893341]
- 48.
- Zhang W, Chipman PR, Corver J, et al. Visualization of membrane protein domains by cryo-electron microscopy of dengue virus. Nat Struct Biol. 2003;10(11):907–912. [PMC free article: PMC4148076] [PubMed: 14528291]
- 49.
- Mukhopadhyay S, Kim BS, Chipman PR, et al. Structure of West Nile virus. Science. 2003;302(5643):248. [PubMed: 14551429]
- 50.
- Mancini EJ, Clarke M, Gowen Be, et al. Cryo-electron microscopy reveals the functional organization of an enveloped virus, Semliki Forest virus. Mol Cell. 2000;5(2):255–266. [PubMed: 10882067]
- 51.
- Zhang W, Mukhopadhyay S, pletnev SV, et al. placement of the structural proteins in Sindbis virus. J Virol. 2002;76(22):11645–11658. [PMC free article: PMC136788] [PubMed: 12388725]
- 52.
- Elshuber S, Allison SL, Heinz FX, et al. Cleavage of protein prM is necessary for infection of BHK-21 cells by tick-borne encephalitis virus. J Gen Virol. 2003;84(Pt 1):183–191. [PubMed: 12533715]
- 53.
- Guirakhoo F, Heinz FX, Mandl CW, et al. Fusion activity of flaviviruses: Comparison of mature and immature (prM-containing) tick-borne encephalitis virions. J Gen Virol. 1991;72(pt 6):1323–1329. [PubMed: 1710648]
- 54.
- Guirakhoo F, Bolin RA, Roehrig JT. The Murray Valley encephalitis virus prM protein confers acid resistance to virus particles and alters the expression of epitopes within the R2 domain of E glycoprotein. Virology. 1992;191(2):921–931. [PMC free article: PMC7130970] [PubMed: 1280384]
- 55.
- Gibbons DL, Vaney MC, Roussel A, et al. Conformational change and protein-protein interactions of the fusion protein of Semliki Forest virus. Nature. 2004;427(6972):320–325. [PubMed: 14737160]
- 56.
- Bressanelli S, Stiasny K, Allison SL, et al. Structure of a flavivirus envelope glycoprotein in its low-pH-induced membrane fusion conformation. EMBO J. 2004;23(4):728–738. [PMC free article: PMC380989] [PubMed: 14963486]
- 57.
- Cecilia D, Gould EA. Nucleotide changes responsible for loss of neuroinvasiveness in Japanese encephalitis virus neutralization-resistant mutants. Virology. 1991;181(1):70–77. [PubMed: 1704661]
- 58.
- Hasegawa H, Yoshida M, Shiosaka T, et al. Mutations in the envelope protein of Japanese encephalitis virus affect entry into cultured cells and virulence in mice. Virology. 1992;191(1):158–165. [PubMed: 1329314]
- 59.
- Lee E, Weir RC, Dalgarno L. Changes in the dengue virus major envelope protein on passaging and their localization on the three-dimensional structure of the protein. Virology. 1997;232(2):281–290. [PubMed: 9191841]
- 60.
- Beasley DW, Aaskov JG. Epitopes on the dengue 1 virus envelope protein recognized by neutralizing IgM monoclonal antibodies. Virology. 2001;279(2):447–458. [PubMed: 11162801]
- 61.
- Hurrelbrink RJ, McMinn PC. Attenuation of Murray Valley encephalitis virus by site-directed mutagenesis of the hinge and putative receptor-binding regions of the envelope protein. J Virol. 2001;75(16):7692–7702. [PMC free article: PMC115004] [PubMed: 11462041]
- 62.
- Monath TP, Arroyo J, Levenbook I, et al. Single mutation in the flavivirus envelope protein hinge region increases neurovirulence for mice and monkeys but decreases viscerotropism for monkeys: Relevance to development and safety testing of live, attenuated vaccines. J Virol. 2002;76(4):1932–1943. [PMC free article: PMC135909] [PubMed: 11799188]
- 63.
- Dubovskii PV, Li H, Takahashi S, et al. Structure of an analog of fusion peptide from hemagglutinin. Protein Sci. 2000;9(4):786–798. [PMC free article: PMC2144621] [PubMed: 10794422]
- 64.
- Han X, Bushweller JH, Cafiso DS, et al. Membrane structure and fusion-triggering conformational change of the fusion domain from influenza hemagglutinin. Nat Struct Biol. 2001;8(8):715–720. [PubMed: 11473264]
- 65.
- Tamm LK, Han X, Li Y, et al. Structure and function of membrane fusion peptides. Biopolymers. 2002;66(4):249–260. [PubMed: 12491538]
- 66.
- Ruigrok RW, Aitken A, Calder LJ, et al. Studies on the structure of the influenza virus haemagglutinin at the pH of membrane fusion. J Gen Virol. 1988;69(Pt 11):2785–2795. [PubMed: 3183628]
- 67.
- Stiasny K, Allison SL, Schalich J, et al. Membrane interactions of the tick-borne encephalitis virus fusion protein E at low pH. J Virol. 2002;76(8):3784–3790. [PMC free article: PMC136097] [PubMed: 11907218]
- 68.
- Chan DC, Kim PS. HIV entry and its inhibition. Cell. 1998;93(5):681–684. [PubMed: 9630213]
- 69.
- Danieli T, Pelletier SL, Henis YI, et al. Membrane fusion mediated by the influenza virus hemagglutinin requires the concerted action of at least three hemagglutinin trimers. J Cell Biol. 1996;133(3):559–569. [PMC free article: PMC2120819] [PubMed: 8636231]
- 70.
- Blumenthal R, Sarkar DP, Durell S, et al. Dilation of the influenza hemagglutinin fusion pore revealed by the kinetics of individual cell-cell fusion events. J Cell Biol. 1996;135(1):63–71. [PMC free article: PMC2121025] [PubMed: 8858163]
- 71.
- Razinkov VI, Melikyan GB, Cohen FS. Hemifusion between cells expressing hemagglutinin of influenza virus and planar membranes can precede the formation of fusion pores that subsequently fully enlarge. Biophys J. 1999;77(6):3144–3151. [PMC free article: PMC1300584] [PubMed: 10585935]
- 72.
- Kemble GW, Danieli T, White JM. Lipid-anchored influenza hemagglutinin promotes hemifusion, not complete fusion. Cell. 1994;76(2):383–391. [PubMed: 8293471]
- 73.
- Melikyan GB, White JM, Cohen FS. GPI-anchored influenza hemagglutinin induces hemifusion to both red blood cell and planar bilayer membranes. J Cell Biol. 1995;131(3):679–691. [PMC free article: PMC2120621] [PubMed: 7593189]
- 74.
- Nussler F, Clague MJ, Herrmann A. Meta-stability of the hemifusion intermediate induced by glycosylphosphatidylinositol-anchored influenza hemagglutinin. Biophys J. 1997;73(5):2280–2291. [PMC free article: PMC1181133] [PubMed: 9370425]
- 75.
- Armstrong RT, Kushnir AS, White JM. The transmembrane domain of influenza hemagglutinin exhibits a stringent length requirement to support the hemifusion to fusion transition. J Cell Biol. 2000;151(2):425–437. [PMC free article: PMC2192652] [PubMed: 11038188]
- 76.
- Saifee O, Wei L, Nonet ML. The Caenorhabditis elegans unc-64 locus encodes a syntaxin that interacts genetically with synaptobrevin. Mol Biol Cell. 1998;9(6):1235–1252. [PMC free article: PMC25346] [PubMed: 9614171]
- 77.
- McNew JA, Weber T, parlati F, et al. Close is not enough: SNARE-dependent membrane fusion requires an active mechanism that transduces force to membrane anchors. J Cell Biol. 2000;150(1):105–117. [PMC free article: PMC2185554] [PubMed: 10893260]
- 78.
- West JT, Johnston PB, Dubay SR, et al. Mutations within the putative membrane-spanning domain of the simian immunodeficiency virus transmembrane glycoprotein define the minimal requirements for fusion, incorporation, and infectivity. J Virol. 2001;75(20):9601–9612. [PMC free article: PMC114531] [PubMed: 11559792]
- 79.
- Dutch RE, Lamb RA. Deletion of the cytoplasmic tail of the fusion protein of the paramyxovirus simian virus 5 affects fusion pore enlargement. J Virol. 2001;75(11):5363–5369. [PMC free article: PMC114942] [PubMed: 11333918]
- 80.
- Melikyan GB, Markosyan RM, Brener SA, et al. Role of the cytoplasmic tail of ecotropic moloney murine leukemia virus env protein in fusion pore formation. J Virol. 2000;74(1):447–455. [PMC free article: PMC111556] [PubMed: 10590134]
- 81.
- Melikyan GB, Jin H, Lamb RA, et al. The role of the cytoplasmic tail region of influenza virus hemagglutinin in formation and growth of fusion pores. Virology. 1997;235(1):118–128. [PubMed: 9300043]
- 82.
- Bagai S, Lamb RA. Truncation of the COOH-terminal region of the paramyxovirus SV5 fusion protein leads to hemifusion but not complete fusion. J Cell Biol. 1996;135(1):73–84. [PMC free article: PMC2121019] [PubMed: 8858164]
- 83.
- Januszeski MM, Cannon PM, Chen D, et al. Functional analysis of the cytoplasmic tail of Moloney murine leukemia virus envelope protein. J Virol. 1997;71(5):3613–3619. [PMC free article: PMC191509] [PubMed: 9094634]
- 84.
- Nieva JL, Bron R, Corver J, et al. Membrane fusion of Semliki Forest virus requires sphingolipids in the target membrane. EMBO J. 1994;13(12):2797–2804. [PMC free article: PMC395159] [PubMed: 8026464]
- 85.
- Vashishtha M, Phalen T, Marquardt Mt, et al. A single point mutation controls the cholesterol dependence of Semliki Forest virus entry and exit. J Cell Biol. 1998;140(1):91–99. [PMC free article: PMC2132589] [PubMed: 9425157]
- 86.
- Chanel-Vos C, Kielian M. A conserved histidine in the ij loop of the Semliki Forest virus E1 protein plays an important role in membrane fusion. J Virol. 2004;78(24):13543–13552. [PMC free article: PMC533937] [PubMed: 15564465]
- 87.
- Chatterjee PK, Eng CH, Kielian M. Novel mutations that control the sphingolipid and cholesterol dependence of the Semliki Forest virus fusion protein. J Virol. 2002;76(24):12712–12722. [PMC free article: PMC136714] [PubMed: 12438597]
- 88.
- Stiasny K, Koessl C, Heinz FX. Involvement of lipids in different steps of the flavivirus fusion mechanism. J Virol. 2003;77(14):7856–7862. [PMC free article: PMC161939] [PubMed: 12829825]
- 89.
- Burke DS, Monath TP. Flaviviruses. In: Knipe DM, Howley PM, eds. Fields Virology 4th ed. Philadelphia: Lippincott Williams and Wilkins. 2001:1043–1125.
- 90.
- Smith TJ, Kremer MJ, Luo M, et al. The site of attachment in human rhinovirus 14 for antiviral agents that inhibit uncoating. Science. 1986;233(4770):1286–1293. [PubMed: 3018924]
- 91.
- Baldwin CE, Sanders RW, Berkhout B. Inhibiting HIV-1 entry with fusion inhibitors. Curr Med Chem. 2003;10(17):1633–1642. [PubMed: 12871113]
- 92.
- Kilby JM, Hopkins S, Venetta TM, et al. Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat Med. 1998;4(11):1302–1307. [PubMed: 9809555]
- 93.
- Kanai R, Kar K, Anthony K, et al. Crystal structure of west nile virus envelope glycoprotein reveals viral surface epitopes. J Virol. 2006;80(22):11000–11008. [PMC free article: PMC1642136] [PubMed: 16943291]
- 94.
- Nayak V, Dessau M, Kucera K, et al. Crystal structure of dengue virus type 1 envelope protein in the postfusion conformation and its implications for membrane fusion. J Virol. 2009;83(9):4338–4344. [PMC free article: PMC2668458] [PubMed: 19244332]
- 95.
- Voss JE, Vaney MC, Duquerroy S, et al. Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature. 2010;468(7324):709–712. [PubMed: 21124458]
- 96.
- Li L, Jose J, Xiang Y, et al. Structural changes of envelope proteins during alphavirus fusion. Nature. 2010;468(7324):705–708. [PMC free article: PMC3057476] [PubMed: 21124457]
- 97.
- Cockburn JJ, Navarro Sanchez ME, et al. Structural insights into the neutralization mechanism of a higher primate antibody against dengue virus. EMBO J. 2011;31(3):767–779. [PMC free article: PMC3273384] [PubMed: 22139356]
- 98.
- Luca VC, AbiMansour J, Nelson CA, Fremont DH. Crystal structure of the Japanese encephalitis virus envelope protein. J Virol. 2012;86(4):2337–2346. [PMC free article: PMC3302414] [PubMed: 22156523]
- Class II Fusion Proteins - Madame Curie Bioscience DatabaseClass II Fusion Proteins - Madame Curie Bioscience Database
- Expression of Hox Genes in the Nervous System of Vertebrates - Madame Curie Bios...Expression of Hox Genes in the Nervous System of Vertebrates - Madame Curie Bioscience Database
- The Function of Toll-Like Receptors - Madame Curie Bioscience DatabaseThe Function of Toll-Like Receptors - Madame Curie Bioscience Database
- Gastroenterologic and Hepatic Diseases - Madame Curie Bioscience DatabaseGastroenterologic and Hepatic Diseases - Madame Curie Bioscience Database
- Human, Mouse and Yeast Telomerase - Madame Curie Bioscience DatabaseHuman, Mouse and Yeast Telomerase - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...