

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Here, we discuss the mechanisms of repression of signaling pathways that are triggered by Lipoxin (LX) and are responsible for control of pro-inflammatory response during chronic phase of Toxoplasma gondii infection. We also discuss this mechanism from the perspective of the pathogen, which pirates the host's lipoxygenase machinery to its own advantage as a probable immune-escape mechanism. Pro-inflammatory mediators such as IL-12, IFN-γ and TNF are essential in controlling parasite growth during T. gondii infection. However, it is clear that exacerbated production of these cytokines results in host tissue damage. LX, an anti-inflammatory eicosanoid, plays an important role in regulation of immune response to T. gondii.
Introduction
Toxoplasmosis is a parasitic disease, usually asymptomatic, caused by Toxoplasma gondii. Toxoplasma infection can become a serious disease in the setting of congenital infection and in immunodeficient (AIDS patients and transplant recipients1). After infection with T. gondii, the released parasites from ruptured cysts actively enter host cells2 including macrophages and dendritic cells (DCs). Once intracellular the tachyzoites quickly replicate and in immunocompetent individuals, the acute phase of infection resolves when the remaining fast-replicating parasites switch to bradyzoites form of T. gondii, probably as a response to immune attack and seclude themselves in cysts within tissues allowing the parasite escape of the immune responses and this phase is known as chronic or persistent infection.3
The main pathology resulting from toxoplasma infection occurs during chronic disease, when immune suppression caused by drugs or infections, such as HIV, can lead to reversion from the dormant encysted bradyzoite stage back to the fast replicating tachyzoites, which rupture cysts within heart or skeletal muscle and the central nervous system (CNS) causing local tissue necrosis, in the CNS it is often lethal. Intracellular parasites, including T. gondii, are capable of modulating numerous immune response functions in order to promote the host and themselves survival, consequently achieving successful transmission to a new host.
It is clear that T. gondii has evolved various strategies to take advantage of host cell signaling regulatory mechanism by distorting this balance between positive and negative influences, which allow the parasite to persist through the chronic phase of the disease and prevents host death.4 Here, we discussed how hosts and T. gondii, make use of some immune-regulatory pathways to promote host survival.
Lipoxins
LXA4 or (5S,6S,15S)-5,6,15-trihydroxy-7,9,13-trans-11-cis-eicosatentraenoic acid, is an eicosanoid derived from the lypoxigenase (LO)-metabolism of arachidonic acid (AA). Initial studies found that LXA4 was secreted by neutrophils and inhibited the activating effects of LTB4 on platelets.5 Since then, a growing list of anti-inflammatory effects have been associated with LXA4 and its analogs.6 LO-catalyzed oxygenation of unsaturated fatty acids initiates the formation of a variety of compounds with diverse biological activities. The biosynthetic pathways involved in LXA4 formation are complex, involve the actions of at least two independent LOs (Fig. 1) and can occur through trans-cellular cascades. However, the activity of 5-LO seems to be a common step in LXA4 synthesis.5 5-LO is produced as a propeptide that is activated by cleavage. Low levels of active 5-LO are found in different cell types, including macrophages, platelets, DCs and neutrophils.7 The expression of a 5-LO-activating protein (FLAP) is a key signal for induction of 5-LO activity. Of note, Aspirin acetylation of cyclo-oxygenase (COX),5 leads to a change in functionality that can drive synthesis of the “Aspirin-triggered LXA4” (ATL): 15-epi-LXA4. ATL has the same activity, but is more metabolically stable than LXA4.5

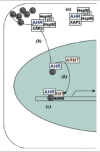
LXA4 is thought to bind to two types of receptors8: (a) a surface membrane seven-transmembrane G-protein-coupled receptor, FPRL-1 or ALX9; and (b) the nuclear ligand-activated transcription factor, the Aryl hydrocarbon receptor (AhR).10 The binding of ligands (i.e., LX) to AhR results in the formation of an active transcription factor that binds to DNA domains—dioxin-responsive elements (DRE)—that activate the expression of a specific panel of genes.11 In the absence of ligand, AhR is located in the cytosol in association with several molecules, Hsp90, the immunophilin-like protein XAP2 and the cochaperone p23. Ligand binding allows for release of the AhR, its translocation to the nucleus and heterodimerization with the AhR nuclear translocator (ARNT; HIF-1β). The AhR/ARNT heterodimer binds to specific DNA sequences (aryl hydrocarbon elements [AHREs], also known as DRE or XRE), where, in concert with the co-activators CBP/p300 (ARNT) and RIPI40 (AhR), they lead to target-gene transcription leading to expression of target genes.11(Fig. 2).
Lipoxins and Toxoplasma Infection
5-LO/LXA4 plays an important counter-regulatory role in mouse models of toxoplasmosis. During infection with T. gondii, serum levels of LXA4 increase steadily over the course of the acute phase and remain at high levels during chronic diseases.12 Such high levels in the serum of chronically infected animals indicated that this mediator might exert relevant biological functions during this stage of the disease. Despite the fact that DCs are one of the main targets for the immune modulatory actions of LXA4 during T. gondii infection, this cell population does not produce detectable levels of the eicosanoid.12 Instead, resident splenic macrophages up-regulate 5-LO expression after in vivo stimulation with parasite extract12 indicating the participation of macrophages in the generation of LXs during experimental toxoplasmosis.
The action of 5-LO on AA results in the formation of LTA4, which can be rapidly converted to LXA4 through the actions of a second enzyme, 15-LO (Fig. 1A). Although the 5-LO activity after T. gondii infection was known to be associated with splenic macrophages,12 the 15-LO-expressing cell population was not known. In an effort to identify the sources of 15-LO activity after T. gondii infection, Bannenberg and colleagues identified an enzymatic activity in tachyzoite forms exposed to calcium ionophore in the presence of AA in vitro.13 Moreover, proteomics analysis of T. gondii tachyzoite-derived lysates revealed the presence of peptides homologous to plant-derived type 15 Los.13 It therefore seems probable that the induction of LX biosynthesis by T. gondii has been selected through the carrying of a plant-like lipoxygenase gene, which together with the actions of host-derived 5-LO results in the high-level production of LX (Fig. 1B).
The presence of high levels of LX, in turn, dampens ongoing immune responses (i.e., IL-12, IFN-γ and TNF production) so that hosts can control parasite proliferation without succumbing to the damaging consequences of excessive inflammation or tissue destruction. Suggesting that lipoxins-dependent inhibition of pro-inflammatory type 1 responses could provide a favorable environment for transmission and propagation of the pathogen. Furthermore, it is possible to hypothesize that pathogens may take advantage of this regulatory pathway to promote host survival, or even allow a less toxic environment in which replication can occur. It is possible to conjecture that from the perspective of T. gondii, which is a fast-replicating pathogen, the host must be kept alive so that transmission can occur through predation. Thus, the host needs well-balanced immunity against the parasite, the number of which is kept low but not completely eliminated. To accomplish this, lipoxins are induced to keep immunity present, but not intensified.
Experimental Model of Infection with T. gondii and Modulation of Immune Response
Mice that receive extract derived from T. gondii tachyzoites (STAg) triggered endogenous production of LXA4 that was found to inhibit microbe-induced DC migration and IL-12 production in vivo and in vitro.12 Consistent with these findings, 5-LO-deficient animals succumbed to T. gondii infection at the early onset of chronic disease. Upon further investigation, it became clear that immunity against the parasite was actually increased in the absence of 5-LO, with significantly less brain cyst formation than in control animals, indicating that this was not the cause of mortality. By contrast, excessive pro-inflammatory cytokine production and massive cerebral infiltration was found, including atypical meningitis. The conclusion was that the excessive pro-inflammatory response in the brain ultimately caused the death of the 5-LO-deficient hosts.12 T. gondii infection in 5-LO-deficient mice resulted in more extensive tissue pathology, mainly due to lack of LXA4 production, as treatment of 5-LO-deficient mice with LX analogs restored the resistance to tissue pathology with no mortality associated with uncontrolled pro-inflammatory responses, in a similar manner as for wild type animals.12
Among the pro-inflammatory mediators, IL-12 production is critical to the development of innate and adaptive immune responses required for the control of intracellular pathogens by governing the development of IFN-γ-dependent host resistance during T. gondii infection. These initial steps during the interaction of T. gondii with immune system are critical for establishing a balanced host-parasite relationship, such that both host and parasite survive the initial phases of infection, progressing towards chronic disease. In fact, IL-12, IFN-γ and IFN-γ receptor deficient mice are extremely susceptible to T. gondii infection; these mice are unable to survive the early stages of acute infection.14 DCs constitute the major IL-12-producing cell population during T. gondii infection in vivo and consequently, the most relevant cell population for the development of a parasite-specific type 1 immune response.15
Mouse CD8α+ DCs produce IL-12 in response to T. gondii in the absence of costimulatory signals,16 indicating that these cells can activate the immune system by recognition of parasite-derived molecules. In fact, splenic DCs stimulated with a soluble extract derived from T. gondii tachyzoites (STAg) produce high levels of IL-12 in vitro and in vivo in a MyD88-independent and -dependent pathways. The MyD88-independent induction of IL-12 production by DCs has been attributed to the CC-chemokine receptor (CCR5), interacting with the secreted, parasite-derived peptide, cyclophilin-18 (C-18), which mimic that signals through CCR5 inducing IL-12 production by DCs in vitro and in vivo.17-19 CCR5 deficient mice are susceptible to T. gondii, with short survival times, high parasite proliferation and low detectable IL-12 and IFN-γ responses, supporting the biological significance of the unusual requirement for a chemokine receptor to participate in microbial recognition by DCs in vivo. However, the IL-12 levels observed after stimulation of DCs with C-18 are much lower than those seen after stimulation with whole parasite lysate or with a pool of tachyzoite-secreted proteins, indicating that pathways other than those initiated by CCR5/C-18 might also be important for IL-12 production by these cells.18 In fact, studies showed that Toll Like Receptor (TLR) adaptor protein MyD88-deficient cells display severely impaired STAg-induced IL-12 responses in vivo and in vitro. Moreover, upon T. gondii infection, MyD88-deficient hosts had high mortality due to a lack of protective IFN-γ-mediated immunity.19
Recently, it was reported that TLR-11 interaction with T. gondii-derived prophillin is also an important signal for IL-12 induction by DCs.20 Although the molecular basis of IL-12 gene expression is unclear, no doubt remains regarding the critical function of IL-12 in response to T. gondii infection. IL-12 produced in vivo triggers NK-cells to release IFN-γ. IFN-γ production is also amplified by tumor necrosis factor and IL-1β in a positive feedback loop. Together these cytokines drive the generation of type 1 CD4+ and CD8+ T-cells,21,22 which produce further IFN-γ, a key requirement for inducing and maintaining control of acute and chronic infection.23
IFN-γ is a potent microbicidal/microbiostatic factor for upregulating the expression of inducible nitric oxide synthase (iNOS) expression and the production of nitric oxide (NO) by T. gondii-infected macrophages. During the chronic phase, some parasites escape immune control and survive within the host for long periods, even though the immune system is continuing to survey the tissues for parasite proliferation. Some T. gondii virulent strains escape immune responses and eventually kill the host, despite stimulating vigorous IL-12 production and type 1 T-cell induction. It is possible that such parasite strains represent the result of random abnormal virulence not based on evolutionary selection pressure but based on the naturally occurring host-parasite relationships. It is known that a growing list of pathogens escape immune responses through the actions of membrane receptors or cytoplasmic enzymes that inactivate or neutralize effector molecules, such a complement factors,24 superoxide or nitric oxide.25 Alternatively, the induction of modulatory or anti-inflammatory factors by the pathogen could inhibit the accumulation of effector cells or their activation in situ, favoring the escape of the pathogen from immune responses and the development of chronic disease.
Intracellular Mechanisms of Anti-Inflammatory Actions of LXs: SOCS Proteins
It was recently shown in an experimental ischemia model that LXA4-analog administration prevented renal failure. Notably, such protection was associated with increased in vivo expression of several suppressors of cytokine signaling (SOCS) genes, including SOCS-1 and SOCS-2,26 providing clues to the intracellular mechanics of LXA4-dependent anti-inflammatory responses. SOCS are a family of intracellular proteins, typically characterized by the presence of an SH2 domain and a so-called, “SOCS Box”.27 This family of proteins was initially identified by several groups after precipitation of signaling elements of diverse cytokine receptor intracellular cascades, i.e., STATS or JAK2, to which SOCS members were associated via the SH2 domain.28 Since then, extensive research has been reported on the first three members of this family: SOCS-1, -2 and -3.28
SOCS-1-deficient mice are runted and die at 3 weeks of age. At that point, the animals exhibit severe lymphopenia, fatty degeneration of the liver and macrophage infiltration of major organs.29 Apparently, this complex phenotype is due to uncontrolled secretion and signaling of IFN-γ.29 These results indicate that a major role of SOCS-1 is to contain deleterious IFN-γ responses. When investigating the physiological function of SOCS-3, Marine et al, found that this protein is highly expressed in fetal liver, suggesting its participation in the regulation of fetal erythropoiesis. This role was then confirmed in SOCS-3-deficient mice, which have embryonic lethality due to massive expansion of erythroid progenitors.30 These results indicate that the primary physiological regulatory target for SOCS-3 is the erythropoietin receptor-induced signaling cascade.
SOCS-2 has previously mainly been linked to the growth hormone (GH)/insulin-like growth factor-1 (IGF-1) signaling due the phenotype observed in SOCS-2 deficient mice. SOCS-2-deficient mice are characterized by a 40% increase in body weight,31 a phenotype resembling that observed in GH transgenic mice,32 acromegalic patients33 and high-growth (hg) mice.34
Machado et al found that SOCS-2 is an essential intracellular mediator of the anti-inflammatory actions of LXs in vivo during toxoplasmosis.35 Unlike WT mice, 5-LO- and AhR-deficient mice fail to up-regulate SOCS-2 expression in vivo after STAg injection, indicating that induction of SOCS-2 expression depends on LX-generated enzymes and receptors. Confirming the involvement of SOCS-2 in LXA4-triggered immune regulatory pathways, SOCS-2-deficient and AhR-deficient mice do not show repression of IL-12 production after in vivo restimulation with STAg (DC paralysis model).35 During infection with T. gondii, SOCS-2 deficient mice have significantly higher IL-12, IFN-γ and TNF production, reduced number of brain cysts, higher frequency of TNF+ and iNOS+ cells infiltrating the central nervous system and higher mortality rates—consistent with the results found in T. gondii-infected, 5-LO deficient mice. Additionally, T. gondii-infected SOCS-2 deficient mice produce significantly higher levels of chemokines and show increased monocyte and neutrophil migration into the peritoneal cavity, compared with WT counterparts.35 Taken together, these studies demonstrate that, during T. gondii infection pro-inflammatory cytokine responses, leukocyte infiltration and T-cell activation are all controlled by a LX-regulatory pathway dependent on SOCS-2 in vivo.
Furthermore was also shown that SOCS-2 has a role in the intracellular pathways triggered by ATLs.35 ATLs share the same receptors and mediate the same biological effects, as LXA4.36 Indeed, we have shown that WT mice, but not BOC2 (a peptide antagonist for ALX receptor)-treated or AhR-deficient mice, upregulate SOCS-2 expression after treatment with Aspirin. Moreover, Aspirin-treated SOCS-2-deficient mice fail to inhibit neutrophil infiltration into the peritoneal cavity after thioglycollate challenge and SOCS-2 is also essential for in vivo Aspirin-dependent inhibition of lipopolysaccharide (LPS)-induced secretion of TNF, a hallmark of the anti-inflammatory actions of this drug.35 Thus, we have shown that endogenous LXA4 and ATL, signaling through AhR and LXAR, modulate innate and acquired immune responses through inducing SOCS-2 expression.35
LX-Induced SOCS2 Mediated TRAF2 and TRAF6 Proteosomal Degradation: A Major Pathway for the Anti-Inflammatory Actions of LXA4 and ATL
SOCS family proteins are thought to mediate their actions by docking to the intracellular domains of cytokine or hormone receptors, thereby preventing binding and activation of downstream signaling elements.28 Some SOCS proteins also facilitate proteasome-dependent degradation of transcription factors through the induction of ubiquitination.27,28 Regardless of the target, the basic mode of action of all SOCS proteins studied to date is similar. In general, SOCS proteins associate with their targets via the SH2 domain, however this does not seem to be sufficient for inhibition. In the case of the association of JAK proteins with SOCS-1 and -3, a second domain in the amino terminus of the molecule is responsible for blocking the kinase activity.27 In other cases, i.e., association with STATs, the inhibition occurs merely by steric hindrance of the docking site. Ultimately, the SOCS/target protein complex is subject to proteasome-mediated degradation. This occurs via poly-ubiquitination catalyzed by the complex ElonginBC/Cullin-2 bound to the SOCS-box domain in the carboxy-terminus of SOCS proteins (Fig. 3).
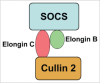
Figure 3
SOCS Ubiquitin Ligase complex. SOCS1 forms an E3 ubiquitin ligase complex with elongins B and C, Cullin-2. The ubiquitinated substrate is subsequently recognized and degraded by the proteasome.
Recently it was shown that LXAs-induced SOCS-2 associate with TNF receptor-associated factors (TRAFs) 2, 6.37 TRAFs constitute a family of highly conserved adapter proteins among several species, including protozoa, nematodes, insects and mammalians. TRAFs are important in diverse biological functions, including innate and adaptive immune response, embryonic development, stress responses and bone metabolism. As adapter proteins, TRAFs elaborate receptor signal transduction as both a convergent and a divergent platform. TRAF proteins are characterized by the presence of a unique TRAF domain at the C-terminus, the TRAF-C domain. This domain plays an important role in defining biological function by mediating self-association as well as upstream interactions with receptors and other signaling molecules. The N-terminus usually contains a RING finger motif as well as several zinc finger motifs, which are important for downstream signaling events.38
TRAF6 is the only TRAF family member that participates in signal transduction from both TNF receptor superfamily members and IL-1/Toll-like receptor superfamily members. It is a critical element in signal transduction during innate microbial recognition (via TLRs), adaptive immune responses (via CD40) as well as TNF-mediated responses (via TNFR). TRAF6 undergoes ubiquitination when activated and can act as a junction, linking the signaling cascades initiated upon triggering of the above receptors to their downstream elements, such as NF-κB and AP-1 activation pathways.39 It is important to point out that the ubiquitin ligation that mediates TRAF6 activation does not signal for proteasome degradation, which only occurs when ubiquitin chains are bound to its targets. Genetic deficiency of TRAF6 causes a lack of responsiveness to TLR-ligands, IL-1, IL-18 and TNF, thus leading to increased susceptibility to infection with several pathogens.39
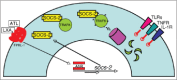
Interesting that LX-induced SOCS2 association with TRAF2, 6 was described as a new molecular target and mechanism of action of LX/ATL-induced SOCS. In fact, LX-induced SOCS2 targets TRAF2 and TRAF6 for proteasomal degradation; degradation of TRAF6 renders cells exposed to LX unable to respond to stimulation through TLRs, TNF-R, IL-1R and CD40, resulting in the inhibition of pro-inflammatory cytokine production by DCs; proteasome-mediated degradation of TRAF6 plays a major role in the mechanism of action of ASA in restraining DC production of pro-inflammatory cytokines; and this pathway of targeted proteasomal degradation of signaling intermediates is critical for successful modulation of inflammatory responses during infection with T. gondii.37 The biological role of LX and ASA-driven TRAF2 degradation remains to be defined.
Concluding Remarks
Researches have traditionally focused on the mechanisms that underlie and control the activation of inflammatory responses. The last decade has seen growing awareness and experimental attention paid to the fact that restraint and resolution of inflammatory responses is also, perforce, under tight, active regulation. In turn, this has led to growing interest in facilitation of resolution as a therapeutic strategy for the dysregulated inflammation seen as central to the pathogenesis and expression of an increasingly wide spectrum of diseases. The mechanism(s) of action of the lipoxins, at the level of cellular signaling, are of interest. Along these lines, LXA4 and ATL restrain innate and acquired immune responses via stimulating SOCS-2 expression in an AhR-dependent manner. LX-induced SOCS-2 targets TRAF2 and TRAF6 for proteasomal degradation that results in degradation of TRAF2, 6 inhibiting pro-inflammatory cytokine production during T. gondii infection and Aspirin treatment (Fig. 4). The biological role of LX and ASA-driven TRAF2 degradation remains to be defined. In addition, the emerging role for lipoxins as immune-modulatory mediators and the potential use of their inhibitory effects for pathogen survival and replication, is still a new and unclear field. The nature of the pathogen-derived signal that contributes to lipoxin generation, or whether the anti-inflammatory effects of LXA4 have a critical role in the balance between type 1, type 2 and regulatory T-cell responses await to be clarified.
References
- 1.
- Martinez AJ, Sell M, Mitrovics T, et al. The neuropathology and epidemiology of AIDS. A Berlin experience. A review of 200 cases. Pathol Res Pract. 1995;191(5):427–443. [PubMed: 7479362]
- 2.
- Morisaki JH, Heuser JE, Sibley LD. Invasion of Toxoplasma gondii occurs by active penetration of the host cell. J Cell Sci. 1995;108(Pt 6):2457–2464. [PubMed: 7673360]
- 3.
- Black MW, Boothroyd JC. Lytic cycle of Toxoplasma gondii. Microbiol Mol Biol Rev. 2000;64(3):607–623. [PMC free article: PMC99006] [PubMed: 10974128]
- 4.
- Hay J, Hutchison WM. Toxoplasma gondii—An environmental contaminant. Ecol Dis. 1983;2(1):33–43. [PubMed: 6678762]
- 5.
- Serhan CN, Hamberg M, Samuelsson B. Lipoxins: Novel series of biologically active compounds formed from arachidonic acid in human leukocytes. Proc Natl Acad Sci USA. 1984;81(17):5335–5339. [PMC free article: PMC391698] [PubMed: 6089195]
- 6.
- Aliberti J. Host persistence: exploitation of anti-inflammatory pathways by Toxoplasma gondii. Nat Rev Immunol. 2005;5(2):162–170. [PubMed: 15662369]
- 7.
- Funk CD, Chen XS, Johnson EN, et al. Lipoxygenase genes and their targeted disruption. Prostaglandins Other Lipid Mediat. 2002;68-69:303–312. [PubMed: 12432925]
- 8.
- Fiore S, Romano M, Reardon EM, et al. Induction of functional lipoxin A4 receptors in HL-60 cells. Blood. 1993;81(12):3395–3403. [PubMed: 8389617]
- 9.
- Fiore S, Maddox JF, Perez HD, et al. Identification of a human cDNA encoding a functional high affinity lipoxin A4 receptor. J Exp Med. 1994;180(1):253–260. [PMC free article: PMC2191537] [PubMed: 8006586]
- 10.
- Schaldach CM, Riby J, Bjeldanes LF. Lipoxin A4: A new class of ligand for the Ah receptor. Biochemistry. 1999;38(23):7594–7600. [PubMed: 10360957]
- 11.
- Mandal PK. Dioxin: a review of its environmental effects and its aryl hydrocarbon receptor biology. J Comp Physiol [B] 2005;175(4):221–230. [PubMed: 15900503]
- 12.
- Aliberti J, Hieny S, Reis e Sousa C, et al. Lipoxin-mediated inhibition of IL-12 production by DCs: A mechanism for regulation of microbial immunity. Nat Immunol. 2002;3(1):76–82. [PubMed: 11743584]
- 13.
- Bannenberg GL, Aliberti J, Hong S, et al. Exogenous pathogen and plant 15-lipoxygenase initiate endogenous lipoxin A4 biosynthesis. J Exp Med. 2004;199(4):515–523. [PMC free article: PMC2211821] [PubMed: 14970178]
- 14.
- Scharton-Kersten TM, Wynn TA, Denkers EY, et al. In the absence of endogenous IFN-gamma, mice develop unimpaired IL-12 responses to Toxoplasma gondii while failing to control acute infection. J Immunol. 1996;157(9):4045–4054. [PubMed: 8892638]
- 15.
- Liu CH, Fan YT, Dias A, et al. Cutting Edge: Dendritic cells are essential for in vivo IL-12 production and development of resistance against Toxoplasma gondii infection in mice. J Immunol. 2006;177(1):31–35. [PubMed: 16785494]
- 16.
- Reis e Sousa C, Hieny S, Scharton-Kersten T, et al. In vivo microbial stimulation induces rapid CD40 ligand-independent production of interleukin 12 by dendritic cells and their redistribution to T-cell areas. J Exp Med. 1997;186(11):1819–1829. [PMC free article: PMC2199158] [PubMed: 9382881]
- 17.
- Aliberti J, Reis e Sousa C, Schito M, et al. CCR5 provides a signal for microbial induced production of IL-12 by CD8 alpha+ dendritic cells. Nat Immunol. 2000;1(1):83–87. [PubMed: 10881180]
- 18.
- Aliberti J, Valenzuela JG, Carruthers VB, et al. Molecular mimicry of a CCR5 binding-domain in the microbial activation of dendritic cells. Nat Immunol. 2003;4(5):485–490. [PubMed: 12665855]
- 19.
- Scanga CA, Aliberti J, Jankovic D, et al. Cutting edge: MyD88 is required for resistance to Toxoplasma gondii infection and regulates parasite-induced IL-12 production by dendritic cells. J Immunol. 2002;168(12):5997–6001. [PubMed: 12055206]
- 20.
- Yarovinsky F, Zhang D, Andersen JF, et al. TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science. 2005;308(5728):1626–1629. [PubMed: 15860593]
- 21.
- Hunter CA, Chizzonite R, Remington JS. IL-1 beta is required for IL-12 to induce production of IFN-gamma by NK cells. A role for IL-1 beta in the T-cell-independent mechanism of resistance against intracellular pathogens. J Immunol. 1995;155(9):4347–4354. [PubMed: 7594594]
- 22.
- Sher A, Oswald IP, Hieny S, et al. Toxoplasma gondii induces a T-independent IFN-gamma response in natural killer cells that requires both adherent accessory cells and tumor necrosis factor-alpha. J Immunol. 1993;150(9):3982–3989. [PubMed: 8473745]
- 23.
- Yap GS, Sher A. Cell-mediated immunity to Toxoplasma gondii: Initiation, regulation and effector function. Immunobiology. 1999;201(2):240–247. [PubMed: 10631573]
- 24.
- Karp CL, Wills-Karp M. Complement and IL-12: yin and yang. Microbes Infect. 2001;3(2):109–119. [PubMed: 11251297]
- 25.
- Son ES, Song KJ, Shin JC, et al. Molecular cloning and characterization of peroxiredoxin from Toxoplasma gondii. Korean J Parasitol. 2001;39(2):133–141. [PMC free article: PMC2721090] [PubMed: 11441500]
- 26.
- Leonard MO, Hannan K, Burne MJ, et al. 15-Epi-16-(para-fluorophenoxy)-lipoxin A(4)-methyl ester, a synthetic analogue of 15-epi-lipoxin A(4), is protective in experimental ischemic acute renal failure. J Am Soc Nephrol. 2002;13(6):1657–1662. [PubMed: 12039996]
- 27.
- Kile BT, Schulman BA, Alexander WS, et al. The SOCS box: A tale of destruction and degradation. Trends Biochem Sci. 2002;27(5):235–241. [PubMed: 12076535]
- 28.
- Alexander WS, Hilton DJ. The role of suppressors of cytokine signaling (SOCS) proteins in regulation of the immune response. Annu Rev Immunol. 2004;22:503–529. [PubMed: 15032587]
- 29.
- Starr R, Metcalf D, Elefanty AG, et al. Liver degeneration and lymphoid deficiencies in mice lacking suppressor of cytokine signaling-1. Proc Natl Acad Sci USA. 1998;95(24):14395–14399. [PMC free article: PMC24384] [PubMed: 9826711]
- 30.
- Marine JC, McKay C, Wang D, et al. SOCS3 is essential in the regulation of fetal liver erythropoiesis. Cell. 1999;98(5):617–627. [PubMed: 10490101]
- 31.
- Metcalf D, Greenhalgh CJ, Viney E, et al. Gigantism in mice lacking suppressor of cytokine signalling-2. Nature. 2000;405(6790):1069–1073. [PubMed: 10890450]
- 32.
- Kopchick JJ, Bellush LL, Coschigano KT. Transgenic models of growth hormone action. Annu Rev Nutr. 1999;19:437–461. [PubMed: 10448532]
- 33.
- Colao A, Merola B, Ferone D, et al. Acromegaly. J Clin Endocrinol Metab. 1997;82(9):2777–2781. [PubMed: 9284694]
- 34.
- Bradford GE, Famula TR. Evidence for a major gene for rapid postweaning growth in mice. Genet Res. 1984;44(3):293–308. [PubMed: 6530139]
- 35.
- Machado FS, Johndrow JE, Esper L, et al. Anti-inflammatory actions of lipoxin A4 and aspirin-triggered lipoxin are SOCS-2 dependent. Nat Med. 2006;12(3):330–334. [PubMed: 16415877]
- 36.
- Hachicha M, Pouliot M, Petasis NA, et al. Lipoxin (LX)A4 and aspirin-triggered 15-epi-LXA4 inhibit tumor necrosis factor 1alpha-initiated neutrophil responses and trafficking: Regulators of a cytokine-chemokine axis. J Exp Med. 1999;189(12):1923–1930. [PMC free article: PMC2192964] [PubMed: 10377187]
- 37.
- Machado FS, Esper L, Dias A, et al. Native and aspirin-triggered lipoxins control innate immunity by inducing proteasomal degradation of TRAF6. J Exp Med. 2008;205(5):1077–1086. [PMC free article: PMC2373840] [PubMed: 18411340]
- 38.
- Choi Y. Role of TRAF6 in the immune system. Adv Exp Med Biol. 2005;560:77–82. [PubMed: 15932023]
- 39.
- Lomaga MA, Yeh WC, Sarosi I, et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40 and LPS signaling. Genes Dev. 1999;13(8):1015–1024. [PMC free article: PMC316636] [PubMed: 10215628]
- Introduction
- Lipoxins
- Lipoxins and Toxoplasma Infection
- Experimental Model of Infection with T. gondii and Modulation of Immune Response
- Intracellular Mechanisms of Anti-Inflammatory Actions of LXs: SOCS Proteins
- LX-Induced SOCS2 Mediated TRAF2 and TRAF6 Proteosomal Degradation: A Major Pathway for the Anti-Inflammatory Actions of LXA4 and ATL
- Concluding Remarks
- References
- Lipoxins as an Immune-Escape Mechanism - Madame Curie Bioscience DatabaseLipoxins as an Immune-Escape Mechanism - Madame Curie Bioscience Database
- Slits and Their Receptors - Madame Curie Bioscience DatabaseSlits and Their Receptors - Madame Curie Bioscience Database
- FOXP Genes, Neural Development, Speech and Language Disorders - Madame Curie Bio...FOXP Genes, Neural Development, Speech and Language Disorders - Madame Curie Bioscience Database
- Regulation of Paracellular Transport across Tight Junctions by the Actin Cytoske...Regulation of Paracellular Transport across Tight Junctions by the Actin Cytoskeleton - Madame Curie Bioscience Database
- Development of Manganic Porphyrin Mimetics of Superoxide Dismutase Activity - Ma...Development of Manganic Porphyrin Mimetics of Superoxide Dismutase Activity - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...