

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

The recognition of the correct substrate and rejection of the incorrect substrate by RNA-modifying enzymes is of great importance to the process of posttranscriptional base modification. Between 2001 and 2008, structures of nine different rRNA and tRNA base-modifying enzymes in complex with RNA were published and these revealed a plethora of mechanisms for substrate recognition. While some complexes appear to be the result of the RNA being bound primarily as a rigid body, a significant number of enzymes remodel the RNA upon binding, either to increase the affinity of the interaction and/or access an otherwise buried base. Pertinent examples of remodelled RNA are observed in the structures of the ArcTGT-tRNA and RlmD-rRNA complexes. In a number of RNA-modifying enzymes it is clear that there is a degree of modularity, such that the substrate specificity is partly defined by a domain(s) that recognises a particular feature in the RNA while the modification is performed by a suitably positioned catalytic domain. With this chapter we will first review the general principles of protein-RNA interactions and modularity in RNA-modifying enzymes before looking at how they are combined to define substrate specificity in a number of interesting protein-RNA complexes.
Introduction
After transcription by RNA polymerase, many bases of RNAs are modified such that the structure of the RNA is fine-tuned to its final function. Such posttranscriptional modifications are introduced by RNA-modifying enzymes which display the common hallmarks of enzymes, specificity and rate-enhancement, though in many cases there are unanswered questions regarding the mechanisms of modification and substrate recognition employed by the enzymes. Between 2001 and 2008, cocrystal structures of protein-RNA complexes were determined for nine different RNA-modifying enzymes (Table 1) which, in concert with biochemical data, allowed an understanding of the molecular events occurring during substrate recognition and catalysis. While cocrystal structures are the focus of this review a number of interesting articles published in recent years have explored the use of complementary methods in looking at the structures of RNA-modifying enzymes in complex with their substrates, especially when the complex has been recalcitrant to crystallographic analysis.1-3 This review will first look at the principles of protein-RNA recognition and modularity in RNA-modifying enzymes before relating these concepts to a number of the cocrystal structures, looking not just at how the correct substrate is recognised but how the incorrect substrate(s) is rejected.
Table 1A list of RNA-modifying enzyme cocrystal structures
| Enzyme | Function | General Overview | Enzyme Pdb Code(s) | Co-Crystal Pdb Code(s) |
| ArcTGT | Substitution of guanine for Pre-Q0 at position 15 of many archaeal tRNAs | The structure of tRNA is perturbed significantly to form the λ-form, a conformation likely to be important for other enzymes which modify bases in the core of tRNA. G15 is recognised by binding of the tRNA in a specific orientation, counting the positions from position 8 to 13 and insertion of bases 14-16 into pockets. | 1IQ8 1ITZ (guanine) 1IT8 (Pre-Q0)52 | 1J2B12 |
| MnmA | Formation of 2-thiouridine at position 34 of tRNAGln, tRNAGlu and tRNALys | Three cocrystal structures show an initial tRNA-binding form, a prereaction form and an intermediate in which U34 is adenylated. The tRNA is bound as a rigid-body with nonspecific interactions with the D-stem and ASL. U34 is flipped out and recognition is via the bases of the anticodon. | Not available | 2DER, 2DET, 2DEU32 |
| QueTGT | Substitution of guanine for Pre-Q1 at position 34 of tRNAAsn, tRNAAsp, tRNAHis and tRNATyr | Abasic covalent intermediate (1Q2R) and product (1Q2S) structures show that the ASL is recognised by specific interactions with positions 33-35 and remodelling is necessary to prevent steric clashes with QueTGT. Additional stabilising interactions might be made to the D-stem of tRNA through the second monomer of the dimer. | 1PUD57 | 1Q2R, 1Q2S43 |
| RluA | Isomerisation of uridine to pseudouridine at position 32 of various tRNAs and position 746 of 23S rRNA | RluA specifically recognises a limited number of base pairs and specificity comes from base pairs to conserved substrate residues and induced readout. The substrate is dramatically remodelled and contacts are only made to the ASL. | Not available | 2I8225 |
| RlmD (previously RumA) | Methylation of U1939 of 23S rRNA to form 5-methyluridine | RlmD binds the substrate as two sections; the 5′ section adapts a new conformation in which the RNA is remodelled, with the formation of compensating interactions, while the 3′ section interacts with the OB-fold domain through electrostatic and aromatic interactions. | 1UWV55 | 2BH256 |
| TadA | Deamination of adenosine at position 34 of tRNAArg | TadA interacts only with the ASL of tRNA, which is remodelled to prevent steric clashes with helix 5 of TadA. Hydrogen bonds to the anticodon bases allow recognition of the single tRNA substrate. | 1WWR39 2A8N41 | 2B3J40 |
| TrmA | Methylation of position 54 in most tRNAs to form 5-methyluridine | The TΨC-loop is refolded in an induced fit mechanism such that bases stack in a non-sequential manner, similar to RlmD (RumA). The UUC sequence containing the target uridine is recognised in a conserved manner by the catalytic cleft of both enzymes. | 2JJQ, 2VS172 | 3BT758 |
| TruA | Isomerisation of uridine to pseudouridine at positions 38, 39 and 40 in various tRNAs | TruA fixes the D-stem and TΨC-loop, through shape and charge complementarity, with respect to the rest of the enzyme. Substrate bases in the ASL ‘find’ the active site through the inherent flexibility of the tRNA; substrates are distinguished from nonsubstrates by their ability to be modified. | 1DJ049 | 2NQP, 2NR0, 2NRE48 |
| TruB | Isomerisation of uridine to pseudouridine at position 55 in many tRNAs | Structurally, the most studied RNA-modifying enzyme. The cocrystal structures show that TΨC stem and loop of tRNA are bound and subsequent base flipping is vital to substrate recognition and accessing the substrate base. Additional interactions with the RNA occur through the PUA domain of TruB. | 1R3F20 | 1K8W59 1R3E20 1ZE223 1ZL360 2AB424 |
General Principles of Protein-RNA Interactions
Structural biology has been instrumental in revealing key features of protein-protein and protein-DNA interaction surfaces whereas the relative paucity of available protein-RNA complex structures has meant that a detailed understanding of the principles of RNA binding by proteins has lagged behind. Early reviews concentrated on qualitative aspects and were inescapably biased by the small sample of available structures that were not representative of the wide range of functional classes of the protein and RNA partners. Indeed, enzymes involved in RNA metabolism have been particularly underrepresented, with early complex structures being dominated by aminoacyl-tRNA synthetases with their substrates, ribosomal proteins and viral RNA-protein structures. In general these cannot account for some of the features of recognition by RNA-modifying enzymes, such as the ingenious ways in which rather inaccessible targets of modification are delivered to the active sites. The results of previous studies have differed in areas such as the relative importance of different intermolecular bond types in RNA-protein interaction surfaces and the propensities of amino acids to form the protein part of those surfaces. Partly, this is explicable by differences in the datasets used by the authors, but there is an additional complication of differences in definitions, for example with variations in the strictness of what constitutes a hydrogen bond changing the apparent influence of this type of interaction and whether an interaction surface should be defined only by those residues making direct contacts or by the full buried surface area, resulting in disagreements about the apparent composition of that surface.4 Nevertheless, as more structures have become available it has become increasingly possible to investigate the principles of protein-RNA recognition in a more quantitative manner and certain key common features have become apparent. As RNA-modifying enzymes typically make transient, binary complexes, analyses of structures of this class are of particular interest here.5
The negative charge of RNA due to its sugar-phosphodiester backbone ensures that all protein RNA-binding surfaces carry an overall positive charge. Electrostatic interactions contribute to the affinity between RNA and protein and help to stabilise complexes. Direct contacts include nonpolar van der Waals interactions and hydrogen bonds between groups on the protein and RNA molecules. The relative importance of these types of interactions has been disputed, yet the most recent investigations indicate that there are considerably more van der Waals interactions than hydrogen bonds4 and that nonpolar cavities on a protein's surface are often employed to tightly fit exposed bases of RNA.6,7 The specificity of such pockets is enhanced by distinct patterns of hydrogen bonding groups and by steric exclusion; for example by placement of a protein atom in an adenine-binding pocket close to the C2 position of adenine, guanine may be excluded due to a clash with its exocyclic amino group N2.7 The importance of the protein main chain for base-specific bonding has been highlighted. The amide and carbonyl groups of the protein main chain account for about one-third of H-bonds to RNA,5 but dominate the number of bonds made to base donor/acceptor groups, followed by the Arg sidechain.8
Selectivity for RNA over DNA is partly controlled by the heavy involvement of the ribose 2′-OH in hydrogen bonding to protein, comprising one-quarter of all the hydrogen bonds, whereas the sugar plays very little part in hydrogen bonding in protein-DNA complexes. Conversely, hydrogen bonds to DNA phosphates are almost twice as numerous as to RNA phosphates, which account for just over one-third of protein-RNA hydrogen bonds.5 The amount of hydrogen bonding to the bases is similar for RNA-protein and DNA-protein complexes; however the patterns of interaction are different. Exposed regions of RNA allow greater access to the polar groups of the base, but major groove interactions are much less common with RNA as the major groove of the A-form RNA helix is deep and inaccessible compared to that of the B-form DNA helix.
The requirement for a positively charged protein surface at the interface is largely satisfied by Arg and Lys residues, which according to recent calculations account for between a quarter5 to one-third9 of the amino acid residues contained in an interaction surface. Of the two, Arg is enriched at RNA-binding interfaces compared to the rest of the protein surface whereas Lys is found with about equal frequency on both surfaces.5 The propensities of the amino acids to form part of an RNA-interacting surface are generally similar to the propensities for DNA-binding interfaces10 although the acidic residues Asp and Glu are not so completely excluded as they are from DNA-binding interfaces. Water-mediated hydrogen bonds are important in protein-RNA interfaces and provide partial shielding against the negative charge on these acidic sidechains.10 It has often been noted that the aromatic amino acids (Tyr, Trp and Phe) can interact with unpaired bases in RNA molecules by stacking their side chains next to them. However, although this is seen to be a feature specific to certain RNA-binding domains, such as RRM and the OB-fold, it is not exploited to great extent by other RNA-binding domains.7,8
The overall morphology of protein RNA-binding sites has also been discussed. It was observed that these are often composed of multiple disparate regions.4 The shape of the interacting surface is usually concave on the protein side, as RNA presents a convex surface for recognition whether it is in a single-stranded, helical or loop conformation.5 RNA interacting surfaces of two classes have been distinguished: ‘groove binders’ that use the insertion of a secondary structure element of protein to interact with a groove of an RNA helix through bonds to the phosphodiester backbone and proteins that use a β-sheet surface to create binding pockets for unstacked bases of loops or single stranded RNA.6,11 The average size of a protein-RNA interface, measured by the buried surface area, ranges from about 3500 Å2 for interfaces with tRNA, to 1900 Å2 for interfaces with single-stranded RNA, although within each class there is significant variation about the mean.5 Interfaces with tRNA, although typically large, have relatively fewer direct hydrogen bonding interactions than interfaces with other classes of RNA. The long loops present in tRNA provide features for direct base-contacts and allow discrimination between isotypes of otherwise structurally similar molecules.4
The question of the role of conformational changes in either protein or RNA upon complex formation is still open. Clearly, some systems rely on rather dramatic conformational changes. The tRNA-modifying enzyme ArcTGT, for example, binds to a partially unfolded form of tRNA dubbed the λ-form due to its divergence from the canonical L-shape and it was suggested that this, or a similarly rearranged form of tRNA, could be the substrate for other enzymes whose targets of modification are relatively inaccessible in L-shaped tRNA.12 The protein molecule may also undergo conformational rearrangement, as illustrated by a 10° rigid-body domain movement and ordering of the ‘thumb-loop’ of the tRNA pseudouridine synthase TruB (see details in the chapter by Mueller and Ferre-D'Amare in this volume). Induced fit has been cited as a common feature of RNA-protein complex formation rather than rigid body ‘lock-and-key’ docking.13,14 However, a comparison of RNA-bound and RNA-free forms indicated that drastic changes are unusual and that most RNA-binding proteins are essentially unchanged after complex formation.15
Modularity in RNA-Modifying Enzymes
During studies of the structures of aminoacyl-tRNA synthetases, it was noted how their various substrate specificities were determined by enzyme-specific domains whilst their catalytic cores belong to only one of two highly conserved classes.16 Evolution appears to have taken the efficient path of modularising certain proteins so that, in this case, rather universal enzymatic domains nevertheless perform very specific reactions by their combination with substrate-specific recognition and binding elements. As well as reuse of catalytic domains, proteins that target RNA in similar manner often share certain evolutionarily mobile RNA-binding domains. Domains with the OB-fold are found across multiple classes of RNA-binding proteins, including both aminoacyl-tRNA synthetases and RNA modification enzymes. Another example is the S4-like domain, found in ribosomal S4, tyrosyl-tRNA synthetases and various RNA modification enzymes.17 Particular RNA-binding domains are used to target certain RNAs through sequence or structure specificity and bring these RNAs into the correct orientation and proximity to catalytic domains for modification reactions. It should be remembered that the bases of RNA have the ability to form non Watson-Crick base pairs, by utilising the hydrogen bonding potential of their Hoogsteen and sugar edges in addition to the Watson-Crick edge (as observed in DNA).18,19 These add to the structural diversity of RNA, which ranges from extended single stranded forms to complicated secondary and tertiary structures formed by intra-molecular base pairing and packing of the resulting stem loops, bulges and helices, implying a requirement for a similar diversity in RNA interaction domains.
As RNA-binding domains are discussed in several other chapters in this book, we will here only discuss how they pertain to modularity by looking at pseudouridine synthases. (The enzymes TruB20-24 and RluA25,26 are reviewed in the chapter by Mueller and Ferre-d’Amare in this book and while we will not discuss how they specifically recognise their substrates, it should be noted that the availability of biochemical data and high-resolution cocrystal structures makes them interesting examples of RNA-modifying enzymes.) The different families of pseudouridine synthases have evolved from a common ancestor to produce a group of enzymes which catalyse pseudouridylation in a diverse range of substrates. All families share a catalytic domain with a common fold and the substrate specificity of the enzyme is partly due to the RNA-binding domains which are appended to the catalytic core. Three conserved domains also found in other RNA-modifying enzymes have been identified in pseudouridine synthases: the THUMP domain27 which interacts with the 3′-CCA motif of tRNA in Pus10, the PUA domain28 which interacts with the acceptor stem of tRNA in TruB and the S4-like domain29 which is found in RluB, RluC, RluD, RluE, RluF and RsuA, all of which modify rRNA (Fig. 1).30 The PUA domain is currently the only one of these domains that has been crystallised in the context of a RNA-modifying enzyme with RNA (in the cocrystal structures of ArcTGT, TruB and Cbf5 within the H/ACA box RNP) and these structures reveal differing modes of interaction with RNA.12,20,28 It is likely that the future will lead to an increased focus on the complexes RNA-modifying enzymes form with their substrates and this will allow a greater insight into the role the RNA-binding domains play in substrate binding and recognition.
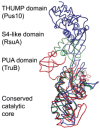
The Various Recognition Modes of RNA Substrates by RNA-Modifying Enzymes
In the following sections we will consider the mechanisms of substrate recognition utilised by a number of RNA-modifying enzymes. As a detailed discussion of each enzyme is not possible, a brief summary of the features of each RNA-modifying enzyme complex is given in Table 1 (including RluA, TrmA and TruB which are not discussed within this chapter) and we would like to refer the interested reader to the original literature for a more detailed overview of each complex.
Predominantly Rigid-Body Docking: Modification of the Anticodon by MnmA
To begin our discussion of substrate recognition by RNA-modifying enzymes, we will look at the 2-thiouridine synthetase (MnmA) which forms its complex with the substrate tRNA (Fig. 2) by binding to it in a predominantly rigid-body manner. MnmA is responsible for the formation of 2-thiouridine (s2U) at position 34 of three tRNAs which lie in mixed codon boxes: tRNAGln(UUG), tRNAGlu(UUC) and tRNALys(UUU). As these boxes of the genetic code are used by more than one amino acid (e.g., GAU and GAC code for Asp while GAA and GAG code for Glu) strict recognition of the third ‘wobble’ position of the codon is necessary for faithful translation of the mRNA. By modifying U34 to s2U34, the C3′-endo conformation is favoured and s2U34 is prevented from forming wobble base pairs, ensuring insertion of the prescribed amino acid.31
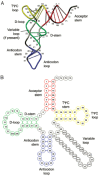
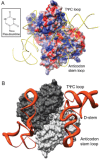
The MnmA-tRNA complex has been crystallised in three different states: the initial tRNA-binding state (form I), the prereaction state (form II) and the adenylated intermediate state (form III).32 In all three forms the domains comprising MnmA (the N-terminal catalytic domain, the central domain and the C-terminal domain with similarity to the C-terminal domain of Ef-Tu)33 possess similar conformations. Similarly, the tRNA is bound by MnmA in a common manner and changes in the conformation of the tRNA between forms I-III are limited to the residues of the anticodon loop (Fig. 3). Interactions between MnmA and the anticodon stem loop (ASL), which sits in the active site of the enzyme and D-stem of tRNA result in a total interface area of ∼1200 Å2 (as calculated by MSDPisa).34 The latter contacts stabilise the complex without conferring specificity on a particular tRNA; a glycine-rich region of MnmA (residues 245-252) interacts with the minor groove formed by the D-stem and anticodon stem through van der Waals interactions and hydrogen bonds to the backbone of the tRNA.
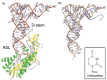
MnmA only modifies tRNAs with the anticodons (positions 34-36) UUC, UUG and UUU, thus while uridine is found at positions 34 and 35, cytidine, guanosine or uridine may be found at position 36.31 How does MnmA use this information to selectively modify only these tRNAs? The form I and II cocrystal structures show that U34 is flipped out of the anticodon base stack and inserted into the active site pocket where it interacts with Gln151 through a hydrogen bond, His128 through an edge-to-face interaction and Asp99 through a hydrogen bond with its sugar hydroxyl.32 The base of other invariant uridine, U35, is involved in hydrogen bond interactions with Gln344 and Tyr312. Thus, the two bases present in all substrates of MnmA are specifically recognised by contacts which would be lost if uridine was not present at these positions. MnmA was cocrystallised with tRNAGlu(UUC) and hydrogen bonds between C36 and the guanidinium group of Arg311 were observed, accounting for the recognition of this base by MnmA. The other substrates have uridine and guanosine at position 36 and the same interaction with Arg311 could be expected with these bases, accounting for the recognition of the base at position 36 by MnmA.
For all RNA-modifying enzymes, it is worth thinking about the effect that pre-existing base modifications might have on the suitability of a particular RNA as a substrate. As the anticodon loop is one of the highly modified regions of tRNA,35,36 Nureki et al considered how modifications at U34 and A37 of tRNA might be accommodated by MnmA. U34 is often modified at position 5 and in all three MnmA structures the region around U34 is either solvent accessible or in an area of the protein exhibiting high B-factors, thus modifications at position 5 of U34 might be accommodated by the inherent flexibility of MnmA in this region. (B-factors are an indicator of the motion displayed by atoms in refined models and they increase with the motion of the atom.)37 With respect to A37, position 2 of the base is exposed to the solvent in all three structures and the formation of N6-threonylcarbamoyladenosine (t6A) at position 37 of the tRNA could potentially lead to the formation of an favourable interaction with Arg313.
Remodelling the Anticodon Stem Loop—TadA and QueTGT
While MnmA binds its tRNA substrate in a predominantly rigid-body manner and only imposes modest conformational changes upon the tRNA, other RNA-modifying enzymes induce more significant changes on the conformation of the substrate. Like MnmA, tRNA adenosine deaminase (TadA) modifies the wobble position of the anticodon (position 34) but it only has one substrate, tRNAArg(ACG). When inosine is formed at this position by TadA, tRNAs with the anticodons GCG and UCG are effectively made redundant as the modified tRNA is able to decode codons with A, C or U at the wobble position.38 With this modification however comes the danger of poor fidelity in translation if nonsubstrate tRNAs are modified at the same position; how does TadA ensure that it only forms inosine at position 34 of tRNAArg(ACG)?
Conformational changes in the protein on binding RNA are minor, with the superposition of A. aeolicus TadA (crystallised without RNA)39 and S. aureus TadA in complex with an analog of the anticodon stem loop (ASL)40 resulting in an rmsd of ∼1.2 Å.41 At the same time, interaction with the protein appears to significantly alter the RNA loop conformation as seen from superposition of the ASL in the TadA structure with yeast tRNAPhe (Fig. 4A).42 The stacking of the anticodon bases in the ASL is distorted to the extent that they no longer stack on top of each other and furthermore, a number of the bases are flipped out. Docking the structure of tRNAPhe into the cocrystal structure of TadA and the ASL shows that this remodelling is a necessity for preventing steric clashes between helix 5 of TadA (residues 133-151) and the ASL (Fig. 4A).

One ASL is bound per TadA dimer and contacts are only made with the five bases of the loop (33-37) and the C32-A38 base pair which caps the loop (Fig. 4A).40 TadA recognises the base pair through a direct minor groove interaction, with Lys106(monomer A) making a hydrogen bonding interaction with C32 whilst Asn138(B) interacts with the base of A38. As this base pair is present in over half of tRNAs, it is unlikely to be important in distinguishing the substrate from nonsubstrates. Likewise, U33 is found in the vast majority of tRNAs but it is hydrogen bonded by both Asp103(A) and Ser138(A). The likely purpose of these interactions is therefore the stabilisation of the complex, rather than the recognition of the substrate.
Similarly to MnmA, TadA recognises its substrate tRNA through the bases of the anticodon (ACG). C35 only forms a single hydrogen bond with TadA, but it is from N4 to the carbonyl oxygen of Gly22(A) and is thus a unique contact that no other base can make.40 G36 is folded back into the ASL where, amongst hydrogen bonds to other regions of the ASL, it hydrogen bonds Arg149(A) through the O6 carbonyl. This interaction is specific to guanine as pyrimidines completely lack this position and the N6 amine of adenine acts as a hydrogen bond donor, not a hydrogen bond acceptor. The substrate base A34 is inserted into the active site pocket where it forms many favourable van der Waals interactions and each of the nitrogen atoms of the base forms a hydrogen bond with TadA; N1 to the backbone amide of Ala54(A), N3 to the side chain amide of Asn42(A) and N7 to a water co-ordinated by a zinc. The positioning of the base is further stabilised by a π-stacking interaction with His53(A) and a hydrophobic interaction with Ile26(A) which lies on the opposite side of the adenine ring to His53(A). Discrimination against other bases at position 34 is thus made by hydrogen bonds and van der Waals interactions; the insertion of a pyrimidine into this pocket would lead to unsatisfied hydrogen bonds and a loss of favourable van der Waals interactions while the insertion of guanine would lead to steric clashes with TadA through the presence of the exocyclic amine at N2 (Fig. 5A).
To complete our discussion of RNA-modifying enzymes which modify the bases of the anticodon of tRNA we will briefly review queuosine tRNA-guanine transglycosylase (QueTGT) which catalyses a transglycosylation reaction in which guanine is replaced with the modified base Pre-Q1 at position 34 of prokaryotic and eukaryotic tRNAs (except those of fungi) with GUN anticodons (tRNAAsn, tRNAAsp, tRNAHis and tRNATyr). Two structures of Z. mobilis QueTGT in complex with an ASL are known.43 The first is a covalent intermediate in which the base of G34 is missing and the ribose at position 34 is covalently linked to the enzyme (the reaction was trapped by withholding Pre-Q1). By soaking these crystals with the base Pre-Q1, the reaction progressed to completion; the bond between QueTGT and the ASL was broken and the ribose of position 34 was instead bonded to Pre-Q1.
Many parallels exist in the mechanisms of substrate recognition utilised by TadA and QueTGT. While the stem of the ASL retains its conformation on binding QueTGT, the conformation of the loop is perturbed from the norm; bases U33, A36 and A38 are flipped out and the conformational changes seen in the ASL occur to prevent otherwise inevitable steric clashes with QueTGT (Fig. 4B). Biochemical data show that the minimal substrate is an ASL with the bases UGU at the positions 33-35, an observation in agreement with the cocrystal structure which revealed specific hydrogen bonds between the bases at these positions and QueTGT. As U33 is highly conserved in tRNAs it is likely that this base is not important in distinguishing substrates from nonsubstrates and that contacts are most likely important for stabilisation of the complex. Huang et al did however make the suggestion that QueTGT might contact U33 and assist in forming a specific ASL conformation suitable for binding,13 a likely possibility given that unmodified ASLs (substrates for QueTGT) are known to be more flexible than the modified ASLs found in mature tRNAs.44
To generalise, the mechanisms of substrate recognition employed by MnmA, TadA and QueTGT rely heavily on the specific recognition of the bases of the anticodon as in these cases they are sufficient to specifically define the substrate; it remains a possibility though that interactions with the RNA backbone which are annotated as nonspecific stabilising interactions might still contribute to substrate specificity through indirect readout.45 For the remainder of this chapter, we will focus on RNA-modifying enzymes which do not bind and recognise their RNA substrates in such a straightforward and intuitive manner.
Exploiting the Dynamics of tRNA—TruA
Pseudouridines are formed at position 38, 39 and 40 of tRNA by the pseudouridine synthase TruA. At these positions in the ASL they act to improve the efficiency and accuracy of translation through improving the stacking between bases and the formation of an additional hydrogen bond with the backbone of tRNA.46,47 What is particularly interesting about TruA from a substrate recognition perspective is the range of tRNAs it modifies; in E. coli 17 tRNAs are substrates and they have a wide-range of sequences in the ASL, with the positional coordinate of the modified uridine(s) varying by up to 15 Å.
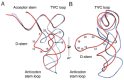
The cocrystal structures of TruA in complex with tRNALeu reveal that the general mode of interaction between TruA and tRNA exploits the dimeric structure of the enzyme.48 The substrate tRNA simultaneously interacts with both subunits, relying on complementarity in both the shape and electrostatic character of the surfaces (Fig. 6A,B). The cleft between the N- and C-terminal domains interacts with the ASL of the tRNA while the other monomer interacts with the D-stem and TΨC-loop of the same tRNA. Comparison of the structures of TruA with and without tRNA shows that there are no significant conformational changes on formation of a complex (rmsd < 1 Å) and while the tRNA is not actively remodelled, its flexibility is essential to the activity of TruA.49,50 By combining the crystal structure data with biochemical data and molecular dynamics simulations, the authors were able to propose a general mechanism for substrate recognition by TruA. The first step is the binding of the D-stem and TΨC-loop of tRNA to TruA through shape and charge complementarity; as these regions of tRNA are conserved with respect to their shape and electrostatic character, this results in a common binding mode for all tRNAs and rationalises why minimal substrates of TruA must contain these loops (Fig. 6A). In this orientation the variable loop projects away from TruA, giving a basis for the observation that TruA does not discriminate against tRNAs with variable loops and the substrate bases are placed in the vicinity of the active site. The low B-factors of the D-stem and TΨC-loops relative to the ASL suggests that these regions are fixed in position with respect to TruA (Fig. 6B).
It was possible to categorise their structures according to the proximity of the ASL to the active site of TruA.48 In the ASL ‘out’ conformation, the ASL of the tRNA is not tightly bound by TruA and molecular dynamics simulations show that bases are able to move such that they may occupy a region of space previously occupied by another base. Given this flexibility, it is possible for each of the substrate bases to access the active site pocket, a finding corroborated by the correlation of ASL flexibility with the efficiency of pseudouridylation. The motions of the ASL with respect to TruA therefore allow the potential points of modification (positions 38, 39 and 40) access to the active site pocket. The binding and recognition of the substrate is therefore a combination of two factors. Firstly, only certain tRNAs have the required flexibility to insert their bases at positions 38, 39 and 40 into the active site and secondly, it is the inability of bases other than uridine to act as substrates which prevents the modification of non-uridine bases.
The λ-form tRNA—ArcTGT
The first structure of a tRNA-modifying enzyme in complex with a complete tRNA was published in 2003.51 Archaeosine tRNA-guanine transglycosylase (ArcTGT) replaces guanine with Pre-Q0, an intermediate in the synthesis of archaeosine which is at position 15 in the core of many archaeal tRNAs where it interacts with the D-stem, TΨC- and variable-loops. G15 is more deeply buried within the core of the tRNA (Fig. 2) than the other bases discussed so far; key questions are therefore how is the base recognised and accessed given its position and the fact that no primary sequence information is required by ArcTGT.
While the conformation of ArcTGT is similar with and without tRNA, apart from a number of residues near the active site (rmsd < 1 Å),52 the conformation of the tRNA on binding ArcTGT is dramatically altered (Fig. 7). In contrast to the canonical L-shaped tRNA (Fig. 2), tRNA bound by ArcTGT has a λ-form conformation in which standard base pairs within the D-stem and the core region (corresponding from U8 to U22) are disrupted. A new region is formed in compensation, the DV helix, which is a double-helical region comprised of the residues which usually form the D-and variable-loops. The presence of nonWatson-Crick base pairs within the DV helix suggests that this is a flexible structure and that ArcTGT binding is vital for its formation and stabilisation. In particular, ArcTGT contains a β-hairpin (residues Lys465 and Thr466) which is essential for ArcTGT activity (Fig. 4C); superposition of L-shaped and λ-form tRNAs shows that this β-hairpin would clash with U8, C25 and C48 in the standard L-shaped tRNA. Much like the examples previously discussed, it appears that the protrusion of the protein into an unperturbed RNA conformation provides (at least in part) a driving force for remodelling. An interesting parallel can be made with the DNA helicase UvrB which also uses β-hairpin to physically separate the DNA duplex.53 The functional implication of the remodelling of tRNA to the λ-form is that the substrate residue G15 can now be accessed by ArcTGT. Yokoyama et al made the appealing suggestion that other enzymes which modify bases of the D-loop might also induce the λ-form on tRNA, perhaps in concert with each other in the form of a weak complex.
How is ArcTGT able to introduce the modification at G15 without any primary structure specificity? The acceptor stem of tRNA is recognised by the PUA domain (domain C3) which fixes the orientation of the tRNA with respect to the remainder of the enzyme by specifically locating the base pair between residues 1 and 72.12 The interaction between the PUA domain28 and tRNA is further stabilised by base stacking interactions between Phe side chains and the unpaired 3′ bases of tRNA. Further nonspecific interactions are made between the positive surface of domain C2 and the backbone of tRNA residues 66 to 69. Together, all base pairs of the acceptor stem up to the 7-66 base pair are recognised (Fig. 8).
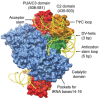
As the position of the tRNA has been fixed, ArcTGT simply has to count the number of residues between the acceptor stem and the substrate base.12 The residues spanning U8 to U13 interact in a nonsequence specific manner with a cleft formed by the two monomers of the ArcTGT dimer; the bases of the tRNA are stacked and face away from ArcTGT. The active site is comprised of three pockets into which A14, the substrate base G15 and U16 are inserted. The pockets containing the bases of positions 14 and 16 do not appear to discriminate between different bases as they are capable of accommodating both purines and pyrimidines and no specific hydrogen bonds are made to the bases. G15 however is specifically contacted and hydrogen bonds exist between O6 and Gln169 and Gly196, the exocyclic N2 and the side chains of Asp95 and Ser98 and N3 and the side chain of Asp95. The positioning of G15 in the active site pocket therefore appears to be a result of three factors in addition to the formation of the λ-form tRNA: the binding of the acceptor stem in a fixed orientation by the C-terminal domains of ArcTGT. the recognition of the six backbone phosphates between positions 8 and 13 and the insertion of the bases at positions 14-16 into active site pockets (Fig. 8).
Identifying Substrates through Probing RNA Flexibility—RlmD
In E. coli, 5-methyluridine (m5U) at position 1939 of the 23S rRNA is formed by the enzyme RlmD (previously known as RumA). While the role of m5U at position 1939 is undetermined, the base lies in the 1942 loop which extends into the major groove of the acceptor stem of tRNA in the A-site,54 suggesting that it may in some way modulate the binding of tRNA to the ribosome. The structure of RlmD revealed the enzyme to have a three domain structure; an N-terminal OB-fold domain, a central Fe4S4 cluster containing domain and a C-terminal catalytic domain with a fold similar to that of class I SAM-dependent methyltransferases.55 In a cocrystal structure with an RNA corresponding to residues 1932-1961 of the 23S rRNA, the RNA was folded into two structural regions bound by a concave surface formed by the three domains of RlmD.56 The 5′ section of the RNA (residues 1932-1943) forms a complex loop while the 3′ section (residues 1945-1961) forms a hairpin. On formation of the complex, an interface of ∼2000 Å2 is formed and comparison of the RlmD structures with and without RNA bound shows that on binding the three domains close around the rRNA substrate (Fig. 9A).
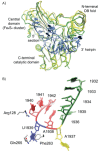
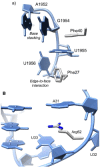
The 5′ loop binds in a groove between the central and C-terminal catalytic domain of RlmD, with the RNA conformation being very different to that observed in structures of ribosome. The bases of this loop form two stacks which are separated by the flipped out bases A1937 and U1939; the first stack interacts with the β-sheet of the central domain while the second stack interacts with a cleft containing the Fe4S4 cluster (Fig. 9A). The bases C1941 and C1942 compensate for the absence of U1939 from the base stack as the RNA chain folds back on itself and bases 1941 and 1942 lie into the void created by the movement of U1939; the importance of this base stacking is indicated by the observation that an abasic residue at position 1942 reduces the rate of catalysis by a factor of at least 1000.56 The new 5′ conformation is stabilised by van der Waals interactions between the base of A1938 and the ribose of A1936 and a specific hydrogen bond, between N2 of G1935 and N1 of A1938, which connects the two base stacks. A complementary surface between RlmD and the RNA exists and this is supplemented by hydrogen bonds from RlmD to the RNA and the extension of the base stack with a cation-π interaction between Arg128 and U1940. These extensive RNA-RNA and RNA-protein interactions are only possible when the RNA is folded into this particular conformation, which is likely to be a function of its sequence.
The substrate base U1939 is stabilised by a number of interactions, principally an edge-to-face interaction with Phe263 and direct hydrogen bonds from N3 and O4 to Gln265 (Fig. 9B). The importance of the latter is demonstrated by the 830-fold reduction in activity on mutating Gln265 to Ala.55 As the conformation of Gln265 is fixed by hydrogen bonds with other residues in RlmD, it requires a hydrogen bond donor at position 3 and a hydrogen bond acceptor at position 4 of the pyrimidine ring. Combined with the small size of the binding pocket, which would not allow the insertion of a purine base, uridine is uniquely specified at this position (Fig. 5B).
The 3′ section of the RNA forms a hairpin consisting of five base pairs and a seven base loop and has a conformation similar to that observed in structures of the 70S ribosome (rmsd ∼2.2 Å).54 The hairpin lies in a cleft formed by the OB-fold and catalytic domains, with most interactions being between the OB-fold domain and the nucleotides of the loop (Fig. 9A). Binding appears to be stabilised by electrostatic and base stacking interactions; A1952, G1954, U1955 and U1956 of the RNA form a network of interactions with Phe27, Phe40 and Tyr61.
The extent to which RlmD remodels the rRNA substrate is surprising, especially when it is considered that other RNA-modifying enzymes with small number of substrates recognise the substrate by interacting with a small number of bases that allow the unique identification of the bound RNA. RlmD differs in that it identifies the RNA as a substrate primarily through indirect readout. As the ability of an RNA to adapt a certain conformation depends upon its sequence, substrate recognition in the case of RlmD can occur without a large number of direct specific contacts to conserved bases.25,45
Conclusions and Future Prospects
Biochemical and structural studies have led to significant advances in our understanding of substrate recognition by RNA-modifying enzymes. In many cases, substrate recognition comes from interactions with specific segments of the RNA which are conserved within substrates, such as the anticodon of tRNA in the cases of MnmA, TadA and QueTGT. More elaborate mechanisms of substrate recognition are used by TruA, ArcTGT and RlmD, which rely upon the dynamics of the substrate and the ability of a substrate to adapt to a new conformation as a method of recognition. The question of how these remodelled complexes form is equally interesting. For the majority of complexes discussed within this chapter, the structure of the enzyme and its substrate are known in the presence and absence of each other and comparisons suggest that the RNA is often remodelled while the enzyme displays only minor conformational changes. Given the flexibility of unmodified RNA, one possibility is that an ensemble of RNA conformers exist and the enzyme binds a nonstandard conformer which is complementary to the active site of the enzyme.14 Though this seems like an attractive proposition, especially when the enzyme does not undergo significant conformational changes on RNA binding, it should be remembered that proteins are also dynamic and that their conformational flexibility is likely to assist RNA binding. Thus protein-RNA complex formation is likely to be a process involving both the dynamics of the enzyme and substrate RNA.
With an ever increasing number of structures of RNA-modifying enzymes, both alone and in complex with their substrate RNA, the future will bring a more detailed understanding of the domains involved in these enzymes (especially those that can only be identified through structure-based searches) and how RNA-modifying enzymes as a whole act to recognise their substrate and catalyse the modification. It should not be forgotten however that biochemical characterisations are equally important as they can both validate the hypotheses made from crystal structures and reveal the importance of each enzyme-substrate interaction.
Acknowledgements
The authors would like to thank the reviewers of this chapter and in particular Henri Grosjean for useful discussions and suggestions on this chapter. Robert Byrne is supported by the BBSRC and Alfred Antson is supported by the Wellcome Trust. David Waterman acknowledges the support of the STFC and the Wellcome Trust to the Diamond Project.
References
- 1.
- Gabant G, Augier J, Armengaud J. Assessment of solvent residues accessibility using three Sulfo-NHS-biotin reagents in parallel: application to footprint changes of a methyltransferase upon binding its substrate. J Mass Spectrom. 2008;43(3):360–370. [PubMed: 17968972]
- 2.
- Gabant G, Auxilien S, Tuszynska I, et al. THUMP from archaeal tRNA : m(2)(2)G10 methyltransferase, a genuine autonomously folding domain. Nucleic Acid Res. 2006;34(9):2483–2494. [PMC free article: PMC1459410] [PubMed: 16687654]
- 3.
- Leulliot N, Chaillet M, Durand D, et al. Structure of the Yeast tRNA m7G Methylation Complex. Structure. 2008;16(1):52–61. [PubMed: 18184583]
- 4.
- Ellis JJ, Broom M, Jones S. Protein-RNA interactions: structural analysis and functional classes. Proteins. 2007;66(4):903–911. [PubMed: 17186525]
- 5.
- Bahadur RP, Zacharias M, Janin J. Dissecting protein-RNA recognition sites. Nucleic Acids Res. 2008;36(8):2705–2716. [PMC free article: PMC2377425] [PubMed: 18353859]
- 6.
- Chen YC, Lim C. Predicting RNA-binding sites from the protein structure based on electrostatics, evolution and geometry. Nucleic Acids Res. 2008;36(5):e29. [PMC free article: PMC2275128] [PubMed: 18276647]
- 7.
- Morozova N, Allers J, Myers J, et al. Protein-RNA interactions: exploring binding patterns with a three-dimensional superposition analysis of high resolution structures. Bioinformatics. 2006;22(22):2746–2752. [PubMed: 16966360]
- 8.
- Allers J, Shamoo Y. Structure-based analysis of protein-RNA interactions using the program ENTANGLE. J Mol Biol. 2001;311(1):75–86. [PubMed: 11469858]
- 9.
- Terribilini M, Lee JH, Yan C, et al. Prediction of RNA binding sites in proteins from amino acid sequence. RNA. 2006;12(8):1450–1462. [PMC free article: PMC1524891] [PubMed: 16790841]
- 10.
- Lejeune D, Delsaux N, Charloteaux B, et al. Protein-nucleic acid recognition: statistical analysis of atomic interactions and influence of DNA structure. Proteins. 2005;61(2):258–271. [PubMed: 16121397]
- 11.
- Draper DE. Themes in RNA-protein recognition. J Mol Biol. 1999;293(2):255–270. [PubMed: 10550207]
- 12.
- Ishitani R, Nureki O, Nameki N, et al. Alternative tertiary structure of tRNA for recognition by a post-transcriptional modification enzyme. Cell. 2003;113(3):383–394. [PubMed: 12732145]
- 13.
- Leulliot N, Varani G. Current topics in RNA-protein recognition: control of specificity and biological function through induced fit and conformational capture. Biochemistry. 2001;40(27):7947–7956. [PubMed: 11434763]
- 14.
- Williamson JR. Induced fit in RNA-protein recognition. Nat Struct Biol. 2000;7(10):834–837. [PubMed: 11017187]
- 15.
- Ellis JJ, Jones S. Evaluating conformational changes in protein structures binding RNA. Proteins. 2008;70(4):1518–1526. [PubMed: 17910059]
- 16.
- Ibba M, Soll D. Aminoacyl-tRNA synthesis. Annu Rev Biochem . 2000;69:617–650 . [PubMed: 10966471]
- 17.
- Aravind L, Koonin EV. Novel predicted RNA-binding domains associated with the translation machinery. J Mol Evol. 1999;48(3):291–302. [PubMed: 10093218]
- 18.
- Leontis NB, Westhof E. Geometric nomenclature and classification of RNA base pairs. RNA. 2001;7(4):499–512. [PMC free article: PMC1370104] [PubMed: 11345429]
- 19.
- Leontis NB, Stombaugh J, Westhof E. The non-Watson-Crick base pairs and their associated isostericity matrices. Nucleic Acid Res. 2002;30(16):3497–3531. [PMC free article: PMC134247] [PubMed: 12177293]
- 20.
- Pan H, Agarwalla S, Moustakas DT, et al. Structure of tRNA pseudouridine synthase TruB and its RNA complex: RNA recognition through a combination of rigid docking and induced fit. Proc Natl Acad Sci USA. 2003;100(22):12648–12653. [PMC free article: PMC240672] [PubMed: 14566049]
- 21.
- Hoang C, Hamilton CS, Mueller EG, et al. Precursor complex structure of pseudouridine synthase TruB suggests coupling of active site perturbations to an RNA-sequestering peripheral protein domain. Protein Sci. 2005;14(8):2201–2206. [PMC free article: PMC2279332] [PubMed: 15987897]
- 22.
- Hamilton CS, Spedaliere CJ, Ginter JM, et al. The roles of the essential Asp-48 and highly conserved His-43 elucidated by the pH dependence of the pseudouridine synthase TruB. Arch Biochem Biophys. 2005;433(1):322–334. [PubMed: 15581587]
- 23.
- Phannachet K, Huang RH. Conformational change of pseudouridine 55 synthase upon its association with RNA substrate. Nucleic Acid Res. 2004;32(4):1422–1429. [PMC free article: PMC390278] [PubMed: 14990747]
- 24.
- Phannachet K, Elias Y, Huang RH. Dissecting the roles of a strictly conserved tyrosine in substrate recognition and catalysis by pseudouridine 55 synthase. Biochemistry. 2005;44(47):15488–15494. [PubMed: 16300397]
- 25.
- Hoang C, Chen JJ, Vizthum CA, et al. Crystal structure of pseudouridine synthase RluA: Indirect sequence readout through protein-induced RNA structure. Molecular Cell. 2006;24(4):535–545. [PubMed: 17188032]
- 26.
- Hamilton CS, Greco TM, Vizthum CA, et al. Mechanistic investigations of the pseudouridine synthase RluA using RNA containing 5-fluorouridine. Biochemistry. 2006;45(39):12029–12038. [PMC free article: PMC2580076] [PubMed: 17002302]
- 27.
- Aravind L, Koonin EV. THUMP -a predicted RNA-binding domain shared by 4-thiouridine, pseudouridine synthases and RNA methylases. TIBS. 2001;26(4):215–217. [PubMed: 11295541]
- 28.
- Perez-Arellano I, Gallego J, Cervera J. The PUA domain -a structural and functional overview. Febs J. 2007;274(19):4972–4984. [PubMed: 17803682]
- 29.
- Davies C, Gerstner RB, Draper DE, et al. The crystal structure of ribosomal protein S4 reveals a two-domain molecule with an extensive RNA-binding surface: one domain shows structural homology to the ETS DNA-binding motif. EMBO J. 1998;17(16):4545–4558. [PMC free article: PMC1170785] [PubMed: 9707415]
- 30.
- Hamma T, Ferre -D'Amare AR. Pseudouridine synthases. Chemistry & Biology. 2006;13(11):1125–1135. [PubMed: 17113994]
- 31.
- Kambampati R, Lauhon CT. MnmA and IscS are required for in vitro 2-thiouridine biosynthesis in Escherichia coli. Biochemistry. 2003;42(4):1109–1117. [PubMed: 12549933]
- 32.
- Numata T, Ikeuchi Y, Fukai S, et al. Snapshots of tRNA sulphuration via an adenylated intermediate. Nature. 2006;442(7101):419–424. [PubMed: 16871210]
- 33.
- Nissen P, Kjeldgaard M, Thirup S, et al. Crystal Structure of the Ternary Complex of Phe-tRNAPhe, EF-Tu, and a GTP Analog. Science. 1995;270(5241):1464–1472. [PubMed: 7491491]
- 34.
- Krissinel E, Henrick K. Inference of Macromolecular Assemblies from Crystalline State. J Mol Bio. 2007;372(3):774–797. [PubMed: 17681537]
- 35.
- Sprinzl M, Vassilenko KS. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res. 2005;33:D139–140. [PMC free article: PMC539966] [PubMed: 15608164]
- 36.
- Sprinzl M, Horn C, Brown M, et al. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acid Res. 1998;26(1):148–153. [PMC free article: PMC147216] [PubMed: 9399820]
- 37.
- Wlodawer A, Minor W, Dauter Z, et al. Protein crystallography for non-crystallographers, or how to get the best (but not more) from published macromolecular structures. Febs J. 2008;275(1):1–21. [PMC free article: PMC4465431] [PubMed: 18034855]
- 38.
- Wolf J, Gerber AP, Keller W. tadA, an essential tRNA-specific adenosine deaminase from Escherichia coli. EMBO J. 2002;21(14):3841–3851. [PMC free article: PMC126108] [PubMed: 12110595]
- 39.
- Kuratani M, Ishii R, Bessho Y, et al. Crystal Structure of tRNA Adenosine Deaminase (TadA) from Aquifex aeolicus. J Biol Chem 22. 2005;280(16):16002–16008. [PubMed: 15677468]
- 40.
- Losey HC, Ruthenburg AJ, Verdine GL. Crystal structure of Staphylococcus aureus tRNA adenosine deaminase TadA in complex with RNA. Nat Struct Mol Biol. 2006;13(2):153–159. [PubMed: 16415880]
- 41.
- Elias Y, Huang RH. Biochemical and Structural Studies of A-to-I Editing by tRNA:A34 Deaminases at the Wobble Position of Transfer RNA. Biochemistry. 2005;44(36):12057–12065. [PubMed: 16142903]
- 42.
- Shi HJ, Moore PB. The crystal structure of yeast phenylalanine tRNA at 1.93 angstrom resolution: A classic structure revisited. RNA. 2000;6(8):1091–1105. [PMC free article: PMC1369984] [PubMed: 10943889]
- 43.
- Xie W, Liu X, Huang RH. Chemical trapping and crystal structure of a catalytic tRNA guanine transglycosylase covalent intermediate. Nat Struct Mol Biol. 2003;10(10):781–788. [PubMed: 12949492]
- 44.
- Stuart JW, Koshlap KM, Guenther R, et al. Naturally-occurring Modification Restricts the Anticodon Domain Conformational Space of tRNAPhe. J Mol Bio. 2003;334(5):901–918. [PubMed: 14643656]
- 45.
- Perona JJ, Hou YM. Indirect Readout of tRNA for Aminoacylation. Biochemistry. 2007;46(37):10419–10432. [PubMed: 17718520]
- 46.
- Davis DR. Biophysical and Conformational Properties of Modified Nucleosides in RNA (Nuclear Magnetic Resonance Studies). In: Grosjean H, Benne R, editors. Modification and Editing of RNA. 1st ed. American Society for Microbiology; 1998. pp. 85–102.
- 47.
- Helm M. Post-transcriptional nucleotide modification and alternative folding of RNA. Nucleic Acid Res. 2006;34(2):721–733. [PMC free article: PMC1360285] [PubMed: 16452298]
- 48.
- Hur S, Stroud RM. How U38, 39, and 40 of many tRNAs become the targets for pseudouridylation by TruA. Molecular Cell. 2007;26(2):189–203. [PMC free article: PMC3562137] [PubMed: 17466622]
- 49.
- Foster PG, Huang LX, Santi DV, et al. The structural basis for tRNA recognition and pseudouridine formation by pseudouridine synthase I. Nat Struct Biol. 2000;7(1):23–27. [PubMed: 10625422]
- 50.
- Foster PG, Huang LX, Santi DV, et al. The crystal structure of E-coli tRNA pseudouridine synthase I. FASEB J. 1997;11(9):A862–A862.
- 51.
- Ishitani R, Nureki O, Nameki N, et al. Alternative tertiary structure of tRNA for recognition by a posttranscriptional modification enzyme. Cell. 2003;113(3):383–394. [PubMed: 12732145]
- 52.
- Ishitani R, Nureki O, Fukai S, et al. Crystal Structure of Archaeosine tRNA-guanine Transglycosylase. J Mol Bio. 2002;318(3):665–677. [PubMed: 12054814]
- 53.
- Theis K, Chen PJ, Skorvaga M, et al. Crystal structure of UvrB, a DNA helicase adapted for nucleotide excision repair. EMBO J. 1999;18(24):6899–6907. [PMC free article: PMC1171753] [PubMed: 10601012]
- 54.
- Selmer M, Dunham CM, Murphy FVIV, et al. Structure of the 70S Ribosome Complexed with mRNA and tRNA. Science. 2006;313(5795):1935–1942. [PubMed: 16959973]
- 55.
- Lee TT, Agarwalla S, Stroud RM. Crystal Structure of RumA, an Iron-Sulfur Cluster Containing E. coli Ribosomal RNA 5-Methyluridine Methyltransferase. Structure. 2004;12(3):397–407. [PubMed: 15016356]
- 56.
- Lee TT, Agarwalla S, Stroud RM. A Unique RNA Fold in the RumA-RNA-Cofactor Ternary Complex Contributes to Substrate Selectivity and Enzymatic Function. Cell. 2005;120(5):599–611. [PubMed: 15766524]
- 57.
- Romier C, Reuter K, Suck D, et al. Crystal structure of tRNA-guanine transglycosylase: RNA modification by base exchange. EMBO J. 1996;15(11):2850–2857. [PMC free article: PMC450223] [PubMed: 8654383]
- 58.
- Alian A, Lee TT, Griner SL, Stroud RM, Finer-Moore J. Structure of a TrmA-RNA complex: A consensus RNA fold contributes to substrate selectivity and catalysis in m5U methyltransferases. Proc Natl Acad Sci USA. 2008;105(19):6876–6881. [PMC free article: PMC2383949] [PubMed: 18451029]
- 59.
- Hoang C, Ferre-D'Amare AR. Cocrystal structure of a tRNA Psi 55 pseudouridine synthase: Nucleotide flipping by an RNA-modifying enzyme. Cell. 2001;107(7):929–939. [PubMed: 11779468]
- 60.
- Hoang C, Hamilton CS, Mueller EG, et al. Precursor complex structure of pseudouridine synthase TruB suggests coupling of active site perturbations to an RNA-sequestering peripheral protein domain. Protein Sci. 2005;14(8):2201–2206. [PMC free article: PMC2279332] [PubMed: 15987897]
- 61.
- Pettersen EF, Goddard TD, Huang CC, et al. UCSF chimera - A visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. [PubMed: 15264254]
- 62.
- Sekine S, Nureki O, Dubois DY, et al. ATP binding by glutamyl-tRNA synthetase is switched to the productive mode by tRNA binding. EMBO J. 2003;22(3):676–688. [PMC free article: PMC140737] [PubMed: 12554668]
- 63.
- Dolinsky TJ, Nielsen JE, McCammon JA, et al. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acid Res. 2004;32:W665–W667. [PMC free article: PMC441519] [PubMed: 15215472]
- 64.
- Baker NA, Sept D, Joseph S, et al. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98(18):10037–10041. [PMC free article: PMC56910] [PubMed: 11517324]
- 65.
- Fukai S, Nureki O, Sekine S, et al. Structural basis for double-sieve discrimination of L-valine from L-isoleucine and L-threonine by the complex of tRNA(Val) and valyl-tRNA synthetase. Cell. 2000;103(5):793–803. [PubMed: 11114335]
- 66.
- Perrett S, Zahn R, Stenberg G, et al. Importance of electrostatic interactions in the rapid binding of polypeptides to GroEL. J Mol Bio. 1997;269(5):892–901. [PubMed: 9223649]
- 67.
- van Holde KEJW, Ho PS. Principles of physical biochemistry. Prentice-Hall; 1998.
- 68.
- Baker EN, Hubbard RE. Hydrogen-bonding in globular-proteins. Prog Biophys Mol Biol. 1984;44(2):97–179. [PubMed: 6385134]
- 69.
- Waters ML. Aromatic interactions in model systems. Curr Opin Chem Biol. 2002;6(6):736–741. [PubMed: 12470725]
- 70.
- Justin PG, Dennis AD. Cation-pi interactions in structural biology. Proc Natl Acad Sci USA. 1999;96(17):9459–9464. [PMC free article: PMC22230] [PubMed: 10449714]
- 71.
- Fersht A. Structure and mechanism in protein science: a guide to enzyme catalysis and protein folding. WH Freeman and company; 1999.
- 72.
- Walbott H, Leulliot N, Grosjean H, et al. The crystal structure of Pyrococcus abyssi tRNA (uracil-54, C5)-methyltransferase provides insights into its tRNA specificity. Nucleic Acids Res. 2008;36(15):4929–4940. [PMC free article: PMC2528175] [PubMed: 18653523]
Supplementary Information — The Physical Forces Involved in Protein-RNA Interactions
In this section we will briefly describe the forces involved in protein-RNA interactions and how they are involved in the formation of a complex and contribute to substrate specificity. While values for interaction distances and free energies are given, it must be emphasised that these are typical values; a review of the given references will reveal that the interconnection of enthalpy and entropy for a particular interaction is very much case dependent and this affects free energy of interaction formation. For this reason, the interested reader is referred to the provided references which describe the structural and thermodynamic factors of intermolecular forces in greater detail.
Electrostatic interactions in their simplest form occur between a pair of ions which are separated by a distance r. As the electrostatic potential of these interactions has a 1/r dependence, they are effective over longer distances than other intermolecular interactions and are thus involved in initial attraction of two oppositely-charged molecules towards each other in solution.66 With macromolecular complexes, the total electrostatic interaction is a sum of these many individual ion-ion interactions, though the situation is complicated by the number of charges (both full and partial) and the effect of the buffer and ionic strength on the electrostatic interaction.67 The role of electrostatics in complex formation can be visualised qualitatively by mapping the electrostatic potential on to the surface of the molecule under investigation or, more elaborately, by contouring the surface so that it reveals how the potential varies at different distances from the macromolecule. In both cases, the colour of the surface corresponds to the strength and sign of the electrostatic potential at that point; negatively charged regions are coloured red while positively charged regions are coloured blue. Such visualisations immediately reveal the charge complementarity of many protein-RNA interactions; the RNA-interacting surface of the protein has a positive electrostatic potential, a consequence of high proportion of positively-charged residues on the surface, while the RNA has a negative electrostatic potential because of its anionic polyphosphate backbone (Fig. S1).

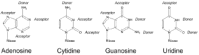
Hydrogen bonds play a crucial role in protein-RNA interactions. Briefly, these consist of a donor (a nitrogen or oxygen atom with a covalently bound hydrogen atom) and an acceptor (an electronegative element).68 In proteins, hydrogen bonding groups are found in the backbone (in the NH and carbonyl oxygen) and most of the amino acid side chains (all but Ala, Ile, Leu, Phe, Pro and Val). In RNA, the sugar-phosphate backbone is a source hydrogen bonding groups which can be utilised to stabilise an interaction without conferring specificity on a particular RNA. Specific recognition occurs through hydrogen bonds to bases which are rich in hydrogen bond donors and acceptors (Fig. S2). Optimal hydrogen bonding occurs when the three atoms are close to colinear and the two nonhydrogen atoms are separated by ∼3 Å and, in this case, the bond has a typical free energy ∼10 kJ mol-1. It should be noted that the strength of the interaction increases to ∼ 20 kJ mol-1 if either the donor or the acceptor is charged and to ∼30 kJ mol-1 for salt bridges, when both the donor and the acceptor are charged.
Aromatic systems, such as the bases of RNA and the side chains phenylalanine, tryptophan and tyrosine have delocalised electron systems. While the planes above and below the rings have increased electron density, the edges of the ring have decreased electron density. These properties lead to three forms of aromatic interactions; ring stacking, edge-to-face and cation-π interactions.69 Favourable π-π interactions result in the stacking of aromatic residues such as in the helices of DNA and RNA, or when an aromatic amino acid side chain is inserted into stack of RNA bases. Edge-to-face interactions occur when two aromatic residues interact and the planes of the rings are perpendicular to each other, such that the increased electron density around the face of one ring interacts with the decreased electron density around the edge of the perpendicular ring (Fig. S3A). Cation-π interactions take the form of a positively-charged group (such as the guanidinium cation of Arg) interacting with the electron rich face of an aromatic residue (Fig. S3B).70
Van der Waals (or London) interactions exist between all atoms in nonpolar bonds and consist of an attractive and repulsive components. The attractive component has a distance dependence of 1/r6 (where r is the distance between the atoms) and is a result of the favourable electrostatic interaction that exists between temporarily induced dipoles on a pair of atoms. The repulsive component has a distance dependence of 1/r12 and results from the interpenetration of the electron orbitals of two different atoms.71 The combination of these terms results in an potential energy minimum at the optimal separation for a pair of atoms (the van der Waals contact distance); this is typically ∼3.5 Å in macromolecular structures and at this distance, the free energy ∼4 kJ mol-1. While increasing the separation beyond this distance leads to a modest increase in potential energy, decreasing the separation leads to a massively unfavourable increase in potential energy; for this reason contacts which lead to steric clashes are forbidden. Despite the fact that the energy provided by a single optimal van der Waals interaction is relatively small, the stabilisation provided by the many instances of these interactions over an entire protein-RNA interface is much greater.
- Introduction
- General Principles of Protein-RNA Interactions
- Modularity in RNA-Modifying Enzymes
- The Various Recognition Modes of RNA Substrates by RNA-Modifying Enzymes
- Predominantly Rigid-Body Docking: Modification of the Anticodon by MnmA
- Remodelling the Anticodon Stem Loop—TadA and QueTGT
- Exploiting the Dynamics of tRNA—TruA
- The λ-form tRNA—ArcTGT
- Identifying Substrates through Probing RNA Flexibility—RlmD
- Conclusions and Future Prospects
- Acknowledgements
- References
- Supplementary Information — The Physical Forces Involved in Protein-RNA Interactions
- Enzyme-RNA Substrate Recognition in RNA-Modifying Enzymes - Madame Curie Bioscie...Enzyme-RNA Substrate Recognition in RNA-Modifying Enzymes - Madame Curie Bioscience Database
- DNA Demethylation - Madame Curie Bioscience DatabaseDNA Demethylation - Madame Curie Bioscience Database
- Role of Mammalian Coronin 7 in the Biosynthetic Pathway - Madame Curie Bioscienc...Role of Mammalian Coronin 7 in the Biosynthetic Pathway - Madame Curie Bioscience Database
- Lysosomal Storage Disorders - Madame Curie Bioscience DatabaseLysosomal Storage Disorders - Madame Curie Bioscience Database
- Neural Crest Delamination and Migration: Integrating Regulations of Cell Interac...Neural Crest Delamination and Migration: Integrating Regulations of Cell Interactions, Locomotion, Survival and Fate - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...