

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Foxp subfamily genes were recently recognized to be members of the Fox gene family. Foxp subfamily members contain a zinc finger domain and a leucine zipper motif in addition to a forkhead domain and their DNA binding capacities and transcriptional activities are regulated by homo- and heterodimerization via a zinc finger and a leucine zipper motif. Three Foxp subfamily members are abundantly expressed in developing brains. The expression patterns of these genes are overlapping, but they are distinctly expressed in some regions. Thus these genes appear to be involved in the development control of the central nervous system. Recently, FOXP2, a member of the Foxp subfamily, was identified as the first gene to be linked to an inherited form of language and speech disorder. The discovery of a mutation in FOXP2 in a family with a speech and language disorder opened a new window to understanding the genetic cascades and neural circuits that underlie speech and language via molecular approaches. The spatiotemporal FOXP2 mRNA expression pattern suggests that the basic neural network that underlies speech and language may include motor-related circuits, including frontostriatal and/or frontocerebellar circuits. This assumption is supported by brain imaging data obtained by using fMRI and PET on the FOXP2-mutated patients and also by analysis of Foxp2 mutant mice.
Introduction
The Fox gene family encodes a large group of transcription factors that share a common DNA binding domain of sequences called the forkhead or winged helix motif after the founding member of this gene family, forkhead in Drosophila. 1 Many Fox family members are involved in embryonic morphogenesis and mutations in Fox genes have been implicated in a range of human developmental disorders. 2 Foxp subfamily genes were recently recognized to be members of the Fox gene family. Members of the Foxp subfamily contain a zinc finger domain and a leucine zipper motif in addition to a forkhead domain. 3 Recent studies have revealed that three Foxp subfamily members are abundantly expressed in developing brains and that the expression patterns of these genes are overlapping, but distinctly in some regions. Thus these genes appear to be involved in development of the central nervous system. Recently, FOXP2, a member of the Foxp subfamily, was identified. It is the first gene to be linked to an inherited form of language and speech disorder. 4 The discovery of a mutation in FOXP2 in a family with a speech and language disorder opens a new window to understanding of the genetic cascades and neural circuits that underlie speech and language via molecular approaches. In this chapter, we focus on the neural expression of FOXP2 as a ‘Language Gene’ as well as the expression patterns of other Foxp subfamily members and their correlation with anatomical and functional abnormalities in the brains of FOXP2-mutated patients.
The Foxp Subfamily
The Foxp subfamily, which consists of four members, Foxp1, Foxp2, Foxp3 and Foxp4, is characterized on the basis of its members containing a C2H2-type zinc finger domain and a leucine zipper motif in addition to a forkhead domain at the C-terminus. 3 - 6 C-terminal location of the forkhead domain is an atypical feature in the Foxp subfamily, as most other Fox family members have this domain in N-terminal portion. Among the subfamily members, Foxp1, Foxp2 and Foxp4 are highly homologous (showing more than 60% identity at the amino acid level); in particular, their forkhead domains show approximately 80% identity at the amino acid level. Also, Foxp1 and Foxp2, but not Foxp4, have polyglutamine tracts at the N-terminus and these may be involved in protein-protein interactions.
Members of the Fox family of proteins have been demonstrated to bind to target DNA as monomers. By contrast, Foxp1, Foxp2 and Foxp4 proteins require dimerization for DNA binding and their transcriptional activities are regulated by homo- and heterodimerization. 7 The dimerizations are dependent on the conserved stretch of sequence, containing a zinc finger and a leucine zipper motif.
Although one might suspect that the FOXP2 gene, being linked to an inherited language and speech disorder, might be a human-specific gene, because speech and language is unique to humans, orthologs exist in many species. Comparison of the Foxp2 genes of many organisms has revealed that the Foxp2 protein is rather extraordinarily conserved (among the 5% most conserved proteins) among mammals. 8 There are only two amino acids different (out of 715 amino acid residues) between humans and chimpanzees and three different between humans and mice. Surprisingly, the amino acid sequence of the forkhead domain is completely identical among rodents, nonhuman primates and humans. Recently, Krause and colleagues 9 reported that the Neanderthals carried a FOXP2 protein that was identical to that of modern humans in the two positions that differed between humans and chimpanzees.
Discovery of FOXP2 as a ‘Language Gene’
Speech and language disorders are common in childhood. Although twin studies have shown that genetic factors play an important role in the etiology of such disorder, a gene that predisposes individuals to speech and language disorders had not been identified until FOXP2 was discovered.
In 1990, Hurst and colleagues reported a unique case of a large three-generation pedigree (called the KE family), half of whose members have a developmental verbal dyspraxia that is inherited in a pattern consistent with an autosomal dominant penetrance. 10 (Details of the language impairments of the KE family will be addressed later). Using standard positional cloning techniques in combination with bioinformatics, Fisher and colleagues 11 performed a genome-wide search for the candidate gene underlying the speech and language disorders in this family. They mapped the gene locus to the long arm of chromosome 7. In 2001, they finally identified FOXP2 as the gene responsible for this speech and language disorder by further analyzing the breaking point of the genome of a patient, CS, who had similar symptoms to the affected members of the KE family and a translocation between chromosomes 5 and 7. 4
The one point mutation in the FOXP2 gene of the affected members of the KE family is predicted to result in an arginine-to-histidine substitution (R553H) in the forkhead domain of the FOXP2 protein. R553 is invariant among all FOX proteins in species ranging from yeast to humans. This mutation occurred in every affected KE family member, but not in unaffected members, nor in unrelated control subjects. The translocation breakpoint in CS disrupted the gene structure of FOXP2. Furthermore, a nonsense mutation at arginine 328 (R328X) in the FOXP2 gene was found in a family, whose affected members had orofacial dyspraxia. 12 Therefore, it is likely that the amino acid substitution in FOXP2 protein leads to a loss of function of one copy of the FOXP2 gene and that the remaining copy is insufficient for FOXP2 function (haploinsufficiency). There are several examples of human disease states regarded to be s consequence of haploinsufficiency of FOX proteins: mutations in FOXC1, FOXC2, FOXE1 and FOXL2 in humans are associated with congenital hereditary glaucoma, hereditary lymphedema-distichiasis syndrome, thyroid agenesis and ovarian failure with craniofacial anomalies (blephalophimosis/ptosis/epicanthus inversus syndrome) with autosomal dominant inheritance. 13 - 16
FOXP2 and Specific Language Impairment (SLI) and Autism
Although the phenotype in the KE family characterized by verbal dyspraxia does not duplicate the language abnormalities of autism and common forms of specific language impairment, chromosome 7q31, in which FOXP2 is located, has been considered to be a potential susceptibility locus for the language deficits in specific language impairment (SLI) and autism. Therefore, association and mutation screening analyses on FOXP2 gene have been performed in these disease groups.
Chromosome 7q31 has been implicated in SLI. 17 , 18 No mutations were found in exon 14 (where the KE family mutation exists) of the FOXP2 gene, but a strong association to genetic markers adjacent to FOXP2 was found. However, no mutation or association with FOXP2 within SLI patients was found in two studies. 19 , 20 Thus it is still unclear whether the role of FOXP2 in speech and language disorders is generalized to more common and genetically complex forms of language impairment.
Chromosome 7q31 has been repeatedly linked to autism, suggesting that this chromosomal region is likely to harbor a susceptibility gene for autism. Therefore, association studies with FOXP2 and autism were conducted. Although two genetic association studies in Japanese and Chinese subjects showed a positive association, 21 , 22 the results of the majority of association studies of FOXP2 and autism have been negative. 23 - 25 The FOXP2 gene is very large in size (>600 Mb) and novel exons have recently been found. 26 Further genetic studies on the relationship between FOXP2 and SLI or autism will be necessary.
Expression of Foxp Subfamily Members in the Brain
The tissue distributions of Foxp subfamily genes and proteins have been investigated in many species, such as zebrafish, mice, rats, songbirds, nonhuman primates and humans. Foxp1, Foxp2 and Foxp4 are expressed in the brain, whereas Foxp3 is not. Foxp3 is exclusively expressed in the immune system. 27 However, the brain is not the only region where Foxp1, Foxp2 and Foxp4 are expressed: their expression is also seen in other organs, including the lung, heart and gut.
Since this chapter is focused on the relationship between Foxp subfamily members and the nervous system, we will first describe the expression patterns of Foxp1 and Foxp4 in other organs briefly, because their mutant mice have some phenotypes in tissues outside the brain.
Murine Foxp1 is expressed in the developing brain, heart, lung and gut. Foxp1 null embryos have severe defects in the cardiovascular system, including defects in ventricular and outflow tract separation, endocardial cushion development and cardiac myocyte proliferation and maturation. 28 Because Foxp1 null embryos die at E14.5, the role of Foxp1 in the later stages of brain development has not been fully clarified. Foxp1 null embryos showed abnormalities in motor neuronal identity in the spinal cord. 29 , 30 In addition, FOXP1 has received considerable attention in the field of cancer research, as discussed in another chapter. 31
It has been revealed that Foxp4 is essential for cardiac morphogenesis: mouse Foxp4 null mice developed abnormally, with two complete hearts and died in the embryonic stage. 32 Although Foxp4 is expressed in developing rodent brains as described below, the role of Foxp4 in neural development has not yet been fully elucidated, because of early embryonic death.
Several groups have demonstrated the expression patterns of Foxp2 mRNA or protein in rodent, nonhuman primate and human brains. 33 - 38 The expression patterns of Foxp2 in fetal mouse, rat, nonhuman primate and human brains show striking similarities at comparative developmental stages. Therefore, we describe the data on the expression pattern in rodents, unless otherwise commented in this section.
Foxp2 is expressed in several structures of the central nervous system during development, including the cerebral cortex, striatum, thalamus, cerebellum and spinal cord. There are many overlaps between the expression patterns of Foxp2 and those of its paralogs Foxp1 and Foxp4, although detailed analysis revealed a distinct pattern of expression for each member in some neuronal cell types, even though they are expressed in the same anatomical regions. Given that homo- or heterodimerization of Foxp1, Foxp2 and Foxp4 proteins is required for DNA binding and their transcriptional activities, 7 the precise combination of homodimers and heterodimers of different Foxp proteins in the same neurons may regulate the transcription of downstream target genes during brain development and, thus control the patterning of brain structures.
Basal Ganglia
Foxp1, Foxp2 and Foxp4 share partially overlapping and yet differentially regulated expression patterns in the striatum during development. 33 - 37 , 39 , 40 During development in rodents, these three Foxp genes are expressed in the striatal primordia (lateral ganglionic eminence, LGE). Foxp2 and Foxp1 are persistently expressed in adulthood, whereas expression of Foxp4 is developmentally down-regulated in the postnatal stage.
Although these three genes have a common character in that all three Foxp genes are expressed only in the LGE, but not in the MGE (medial ganglionic eminence), there was a subtle difference among the expression patterns of these three genes within the LGE. Recent studies have suggested that the LGE can be divided into a large ventral domain (ventral LGE) giving rise to the striatum and a smaller dorsal domain (dorsal LGE) suggested to give rise to interneurons that migrate in the rostral migratory stream to populate the olfactory bulb. 41 Both Foxp2 and Foxp4 are expressed in the subventricular zone (SVZ) and the mantle zone of the dorsal and ventral LGE, while Foxp1 is only expressed in the SVZ and the mantle zone of the ventral LGE, but not in the dorsal LGE. 36
The ontogeny of Foxp expression is also distinct in the striatal compartments. 35 , 36 The striatum comprises two distinct neurochemical compartments, striosomes (or patch) and the matrix. 42 - 44 Neurons in these two compartments differ in terms of the expression levels of various neurochemical molecules, neurogenesis and neural connectivity.
Expression of Foxp1 starts later than that of Foxp2 and Foxp4 in the early embryonic stage. Foxp1 expression is detected in both striosomal and matrix compartments until adulthood. Although expression of Foxp2 and Foxp4 in the striatum starts at the same early embryonic stage and similar and homogeneous expression patterns continue until the late embryonic stage, Foxp2 expression becomes restricted to the striosomal compartment and continues until adulthood, while Foxp4 expression declines in compartmental order: first in the striosomes and later in the matrix, from the late embryonic to the early postnatal stages. Thus in a certain time window, the striosomes are Foxp2high/Foxp4low, whereas the matrix is Foxp2negative∼low/Foxp4high. The differential expression of Foxp1, Foxp2 and Foxp4 might play an important role in establishing specific types of neuron in each compartment of the striatum.
In the fetal human brain, FOXP2 and FOXP1 mRNAs are also expressed in the striatal primordia. 34 , 38 The expression of FOXP2 is developmentally regulated: the expression is quite low in the adult according to northern blot analysis. 4
In developing nonhuman primates, FoxP2 is selectively expressed in the striosomal compartment of the basal ganglia in the perinatal period. 37 Thus the Foxp2/FOXP2 expression pattern in the basal ganglia seems to be conserved in rodents and primates. Nevertheless, there are several aspects in which Foxp2/FoxP2 striatal expression differs in monkey and rodent brains. First, the striosomal FoxP2 expression pattern in the monkey striatum is only detected during the perinatal and early postnatal periods and expression declines during postnatal development. By contrast, Foxp2 striosomal expression in the rodent striatum persists into adulthood. Second, regional differences in FoxP2 mRNA expression exist within the striatum. The monkey striatum comprises the caudate nucleus and the putamen. In the monkey brain, the FoxP2 mRNA expression level in the striosomal compartment is differentially regulated in the caudate nucleus and putamen; specifically, FoxP2 expression in the caudate nucleus is higher than that in the putamen in the perinatal stage and expression is barely detectable in the putamen in the postnatal period.
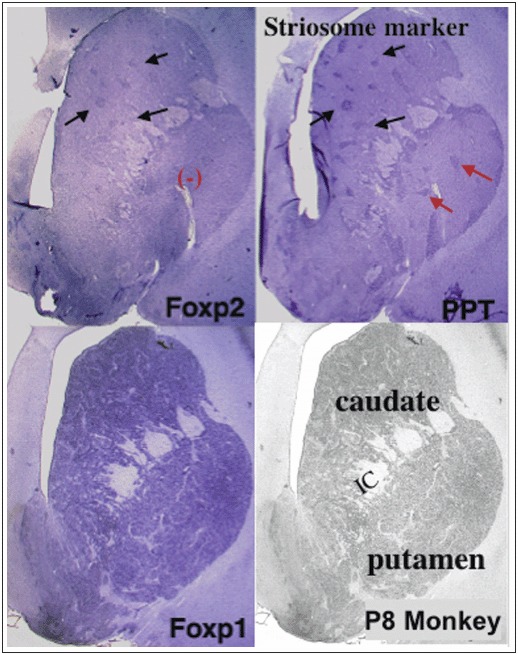
Figure 1
Expression of FoxP2 and FoxP1 in postnatal monkey striatum. FoxP2-positive patches were aligned with PPT (preprotachykinin)-positive striosomes in the monkey caudate nucleus. In contrast, FoxP1 was expressed homogeneously in the caudate nucleus and the putamen.
In contrast to the striosomal FoxP2 expression pattern, FoxP1 mRNA expression is homogeneous in the caudate nucleus and putamen in the prenatal and postnatal periods in monkeys. 37 Then, FoxP1 expression decreases both in the caudate nucleus and the putamen.
Cerebral Cortex and Hippocampus
Foxp family genes are also differentially expressed in the developing cerebral cortex. The expression patterns of Foxp1, Foxp2 and Foxp4 genes show characteristic medio-lateral differences and layer specificity. 33 - 36
In the early embryonic stage, Foxp4 has a mediolateral graded expression in the cerebral cortex: high expression in the medial cortex, low expression in the lateral cortex. By contrast, the cortical expression pattern of Foxp2 mRNA is very different from that of Foxp4. Foxp2 is only expressed in the lateral telencephalon without any gradient. Foxp1 expression starts a little later than expression of Foxp2 and Foxp4 and becomes apparent in the medial telencephalon.
In the embryonic and postnatal periods, Foxp4, Foxp2 and Foxp1 genes are expressed in specific layers of the cerebral cortex. During the early developmental stages in the cerebral cortex, Foxp4 is expressed in proliferating cells in the ventricular zone/subventricular zone (VZ/SVZ) and migrating neurons in the intermediate zone (IZ). Expression of Foxp1 mRNA is observed in the upper half of the cortical plate (CP), while that of Foxp2 mRNA is observed in the lower part of the cortical plate (CP). Thus the expression pattern of Foxp2 appears complementary to that of Foxp1.
During the late developmental stages of corticogenesis, Foxp2 is expressed in the lower cortical layers, where early-born neurons reside and Foxp1 is expressed in the upper cortical layers, where late-born neurons reside. On the other hand, Foxp4 is expressed in the entire cortical layers, including the subplate (SP). This complex pattern of expression may suggest the possibility that a layer-specific identity of cortical neurons may be defined, at least in part, by combinatorial codes of Foxp genes.
In the postnatal cerebral cortex, expression of each Foxp gene shows regional specificity. Foxp1 is expressed in the dorsal and medial cortex, while Foxp2 is not expressed in the dorsal or medial cortex, but is expressed in the lateral cortex, such as in the insular cortex. In addition, both Foxp2 and Foxp1 are expressed in the olfactory tubercle, but not in the piriform cortex, although the expression patterns of these two genes are different within the olfactory tubercle. By contrast, the regional expression of Foxp4 is different from that of Foxp1 or Foxp2. Foxp4 is expressed in the piriform cortex, but not in the olfactory tubercle. These expression patterns of Foxp1 and Foxp2 in the cerebral cortex persist until adulthood, while Foxp4 expression declines by adulthood.
In addition to the developing cerebral cortex, Foxp1, Foxp2 and Foxp4 are differentially expressed in the developing hippocampus. Foxp4 expression is first observed in the medial telencephalon, including the hippocampal anlage, but is absent from the most medial part called the cortical hem. During this developmental stage, there is no expression of Foxp2 or Foxp1 in the hippocampal anlage.
In the postnatal hippocampus, Foxp4 is expressed in the hilar region and from CA3 to CA1, while Foxp1 is expressed mainly in CA1. There is no Foxp2 expression in the hippocampus throughout development. Foxp4 expression declines in the mature hippocampus, while Foxp1 expression persists until adulthood. In human fetal cortex, FOXP1 is expressed in more superficial layers than FOXP2, as in rodents. The expression of FOXP1 and FOXP2 in the cerebral cortex is not asymmetrical.
In the developing monkey cerebral cortex, differential expression of FoxP1 and FoxP2 is evident. FoxP2 is expressed in the deeper cortical layers, whereas FoxP1 is highly expressed in the more superficial layers. The layer-specific expression of these two FoxP genes is similar to that in mouse, rat and human brain. 33 , 35 , 38 FoxP2 is widely expressed throughout the cortical areas, including frontal, parietal, temporal, insular and occipital cortices, although expression is faint in the cingulate cortex. By contrast, moderate FoxP1 expression is detected in all areas examined, including the cingulate cortex. Foxp2/FoxP2 expression in the developing cerebral cortex appears to be conserved in rodents and primates.
Hippocampal FoxP2 and FoxP1 expression patterns in the nonhuman primate are also similar to those in the rat: 35 , 37 that is, low FoxP2 expression is detected in the hippocampus, whereas FoxP1 is expressed in the CA1-CA3 region of the hippocampus. On the whole, regional Foxp2/FoxP2 expression is very similar in rats and nonhuman primates, although the temporal expression pattern of Foxp2/FOXP2 is different.
Thalamus
In the developing diencephalon, Foxp1, Foxp2 and Foxp4 are expressed in the epithalamus, the dorsal thalamus (DT), the ventral thalamus and the hypothalamus. However, the distributions and expression levels of these family members in each region are distinct; for example, Foxp4 is highly expressed in the proliferating cells in the DT, whereas Foxp2 expression is high in differentiated cells located in the lateral part of the DT, but low in proliferating cells. Thus expression pattern of Foxp2 is complementary to that of Foxp4. The expression of Foxp1 in differentiated cells is similar to that of Foxp2, although the level of Foxp1 expression is much lower than that of Foxp2.
Human FOXP1 and FOXP2 expression overlaps in the developing thalamus. 38 FOXP2 is highly expressed in the centromedian nucleus and mediodorsal nucleus of the thalamus. More moderate expression of FOXP2 is observed in the anterior nucleus and parafascicular nucleus. In the neonatal nonhuman primate brains, expression of FoxP1 and FoxP2 is quite similar to that in human counterparts.
Cerebellum
Foxp1, Foxp2 and Foxp4 are all expressed in the cerebellar primordia. Expression of these members is detected in Purkinje cells, cerebellar nuclear neurons, but not in granular neurons or cerebellar interneurons. They are also expressed in the inferior olive. The expression of these members in cerebellum and inferior olive declines postnatally and diminished in the adulthood.
Spinal Cord
Foxp1 is expressed in most of the motor neurons in the brachial spinal cord, while Foxp4 is expressed in a subset of motor neurons and a subset of spinal interneurons. 29 Foxp2 is not expressed in motor neurons, but is expressed in a large number of interneurons.
Language Impairments in the Affected Members of the KE Family
Despite the extensive behavioral analyses of the KE family, there have been inconsistencies in the analyses and it is still unclear how many core deficits there are. However, there is at least one core deficit, verbal and orofacial dyspraxia underlying the speech and language disorders, in the affected members of the KE family. The verbal and orofacial apraxia in the affected members strikingly resembles ‘Broca's aphasia', which is usually seen in patients suffering from brain damage in the ‘Broca's language center' located in the left frontal lobe, often caused by cerebrovascular diseases. There KE patients have no hearing loss or neurological deficits that affect swallowing or limb movements. Nor do they have abnormality in other organs. In that sense, the disease observed in the affected family members in the KE family is really regarded as distinctive speech and language disorders.
Imaging Studies on the KE Family
Although there is still much to be learned about neuropathology, there is no reported autopsy of an affected member of the KE family. The structural and functional abnormalities in the brains of the affected members of the KE family have been investigated using MRI. 45 , 46 The structural brain abnormalities in the affected members of the KE family were investigated using voxel-based morphometric (VBM) methods of MRI analysis. The VBM analyses showed bilateral abnormalities in the caudate nucleus, the inferior frontal gyrus (Broca's area), the precentral gyrus, temporal pole and the cerebellum (lobules VIIB and VIIIB) in the affected members, compared with unaffected members and age-matched controls, who did not differ from each other. The abnormality in the caudate nucleus was of particular interest, because functional abnormality was also found in a related positron emission tomography (PET) study. Moreover, the reduction in volume was significantly correlated with the performance of the affected members on several language tasks.
The affected members of the KE family showed highly atypical fMRI brain activation when performing both covert (silent) and overt (spoken) verb generation tasks, as well as word repetition tasks. 47 The unaffected family members showed a typical left-dominant activation in the inferior frontal gyrus, including Broca's area, in both generation tasks and a more bilateral activation in the repetition tasks, whereas the affected members showed significant underactivation of Broca's area and its right homolog, as well as language-related cortical regions and the putamen. Also, in affected cases, paradoxical activation was observed in cortical regions that are not usually involved in language tasks.
The underactivation of Broca's area in affected members reminds us of patients suffering from motor aphasia caused by cerebrovascular brain damage. The language deficits in both cases are very similar. The functional abnormality in the putamen suggests dysfunction of the striatum.
The FOXP2 Expression Pattern in the Brain and Its Relation to the Cognitive Functions of Speech and Language
FOXP2 is extensively expressed in the developing brain and its expression is down-regulated in the adult. This fact suggests the possibilities that the speech and language impairments found in the affected members of the KE family are due to the developmental defects of the neural network critically involved in speech and language function and that formation of this network is dependent upon a gene network via FOXP2. The spatiotemporal FOXP2 mRNA expression pattern suggests that the basic neural network that underlies speech and language may include motor-related circuits, including corticostriatal and/or corticocerebellar circuits.
This assumption is supported by brain imaging studies on FOXP2-mutated KE family members. Morphometrical analysis using MRI and a functional anatomical study, using PET and fMRI, revealed a bilateral abnormality in the inferior frontal cortex, caudate-putamen and cerebellum. 46 - 48 In particular, FOXP2 expression in the striosomes of the caudate nucleus might have important implications for brain abnormalities induced by FOXP2 mutations in KE family patients. Graybiel and colleagues have shown that striosomes with patches containing low levels of acetylcholinesterase activity are more prominent in the caudate nucleus than in the putamen, 43 , 49 suggesting that striosomes may fully engage in the neural circuits running through the caudate nucleus. If FOXP2 expression levels in the caudate nucleus of the human brain are truly higher than those in the putamen, the FOXP2 mutation may result in a stronger phenotype in the caudate nucleus, which is observed in the brains of affected KE family members.
The affected members of the KE family show impaired movement of mouth, lips and tongue during speech. A study using monkeys has shown that a striosome-dominant activation, as marked by immediate-early gene expression, could occur under conditions in which repetitive movements are induced by dopamine agonists. 50 Moreover, dopamine agonist-induced dyskinesia of repetitive movements is present in experimental parkinsonism. 51 , 52 Therefore, we hypothesize that the symptoms of orofacial dyspraxia in KE family patients may be related to dysfunction of the striosomal system in the striatum.
As described above, language impairments in the affected members of the KE family resemble those in Broca's aphasia, which usually involves the inferior frontal lobe. Although the basal ganglia are not generally considered to be necessary for language acquisition, several recent reports suggest that the caudate nucleus, in particular, is involved in language processing. 53 - 55 Damasio and Damasio 56 hypothesize that the basal ganglia circuitry contributes to grammatical rule processing in conjunction with the frontal lobe. Ullman and colleagues 54 , 55 found that grammatical mistakes occurred in patients suffering from Parkinson's disease or Huntington's disease and developed a declarative/procedural model of language. According to the model, the mental grammar involves procedural memory-like skills and habits and is rooted in the frontal lobe-basal ganglia, whereas the mental lexicon depends on declarative memory and is rooted in the temporal lobe.
The role of the cerebellum in language function is partially understood. 57 It is not certain whether the speech and language disturbances in the KE family are caused by defects of cortico-striatal or cortico-cerebellar circuits, or both. 58
Phenotype in Foxp2 Mutant Mice
Foxp2 mutant mice have been generated and analyzed by three groups. 59 - 61 Homozygotes deficient for both Foxp2 alleles (null mutant) showed severe motor impairment (delayed rightening.rlex maturation), premature death and an absence of ultrasonic vocalization when pups were isolated from their mothers. Shu and colleagues 61 reported abnormalities in the cerebella of homozygotes. Specifically, alignment of Purkinje cells was irregular and the external granular layer (EGL), which should not be retained at the comparative age, was retained. Heterozygotes also showed a modest developmental delay, cerebellar abnormalities and a significant change in ultrasonic vocalization in response to isolation.
Other groups 59 , 60 generated knock-in mice with a point mutation in the Foxp2 gene to give rise to a R552H mutation (corresponding to the human FOXP2 R553H mutation). Homozygous and heterozygous R552H mice reported by Fujita and colleagues 59 showed largely similar phenotypes to the KO mice reported by Shu. 61 Of particular interest, in the homozygous R552H mutants, some neurons had nuclear aggregates of Foxp2 protein. In addition to the immature cerebellar development, the nuclear aggregates might further compromise the function of Purkinje cells and cerebral neurons, resulting in their death.
There were some different phenotypes and interpretations in another line of mutant mice with the same R552H mutation. 60 Homozygous R552H mice showed severe reductions in cerebellar growth and postnatal weight gain, but were able to produce complex ultrasonic vocalization in response to isolation. Heterozygous R552H mice were overtly normal in brain structure and development. The most interesting findings in their study were that heterozygous R552H mice show significant deficits in motor-skill learning and abnormal synaptic plasticity is observed in striatal and cerebellar brain slices by electrophysiological analysis.
Thus findings in these mutant mice seem to support a role for Foxp2 in the development and function of the striatum and cerebellum and a possible involvement of corticostriatal and/or corticocerebellar circuits in the brains of the FOXP2-mutated KE family members.
Transcriptional Activity of the FOXP2 Protein
Protein expression, subcellular localization, DNA-binding and transactivation properties of disease-causing mutations in FOXP2 have been studied using cultured cell models. 62 , 63
Wild-type FOXP2 protein expressed in human cell lines is localized mainly in the nucleus. This intracellular localization is disrupted in the mutants: FOXP2 with a R553H mutation is localized in both nucleus and cytoplasm, whereas FOXP2 with R328X localized predominantly in the cytoplasm. In addition, R328X yields an unstable protein product possibly by nonsense mediated RNA decay.
The DNA-binding properties of wild-type and mutant forms of FOXP2 were investigated via electrophoretic mobility shift assays (EMSAs) using an oligonucleotide probe bound to mouse Foxp1. Wild-type FOXP2 protein possesses DNA-binding capacity, while neither R553H nor R328X mutants bound to the target DNA.
It was reported that mouse Foxp1 and Foxp2 proteins strongly repress transcription from the SV40 promoter, via binding to a naturally occurring target site in the promoter sequence. 64 The transcription capacities of FOXP2 variants were determined by luciferase reporter gene assays. Wild-type FOXP2, FOXP1 and FOXP4 function as transcriptional repressor for SV40 promoter, whereas R528H and R328X mutants lose the repressor activity. 62
In sum, FOXP2 disease-causing mutations disrupt normal subcellular localization, DNA-binding or transactivation capacities in mammalian cell model systems. Thus similar functional changes caused by the mutations are expected to occur in vivo in affected humans.
Foxp2 Upstream and Downstream Genes
FOXP2 gene mutation is so far the only known cause of developmental speech and language disorders in humans. Identifying the molecular network of this gene and its encoded protein will provide a unique window into neural processes involved in speech and language. The upstream regulatory mechanisms that control FOXP2 expression and the downstream molecular events that are regulated by the FOXP2 gene are being investigated by several approaches as follows.
Since FOXP2 is a transcription factor, its potential transcriptional targets can be identified by using the technique of chromatin immunoprecipitation followed by the microarray analysis of promoter regions (ChIP-chip assay) and the functional regulation of targets by FOXP2 can be validated in vitro and in vivo. Two groups have identified targets of FOXP2 in vivo in two brain regions (basal ganglia and inferior frontal cortex) of the human fetal brain and also in a human neuronal cell models. 65 , 66 Interestingly, half of the target genes identified by the these studies are overlapped. FOXP2 protein bound to the promoters of genes involved in diverse biological functions, including cell signaling, synaptic transmission, neural development, ion transport and axon guidance in fetal human brain and living neuron-like cell lines. The expression of a majority of the target genes (such as ANK1, KCNJ15 and LBR) was repressed by FOXP2 in cell culture models, while expression of a minority of the targets (such asTAGLN and CALCRL) was activated by FOXP2. Thus FOXP2 can act as both a repressor and an activator under certain circumstances, possibly dependent upon FOXP2 cofactors (such as Foxp1, Foxp4 and CtBP1) or its posttranslational modifications.

Figure 2
Perspectives. Identification of Foxp2-downstream genes may provide insights into the molecular mechanisms underlying the neural development such as striatal compartmentalization and the neural network formation of frontostriatal and frontocerebellar circuits, potentially related to language acquisition. The identification of the Foxp2-downstream genes might also lead to the discovery of the susceptible genes for SLI (specific language Impairment) and autism. Further understanding the gene network may be valuable for development of the therapeutics for SLI and autism.
It has been reported that Foxp1 works as a cofactor for Hox protein in establishing spinal motor neuronal identity. 29 , 30 A number of transcription factors have been shown to be preferentially and highly expressed in the developing striatum. 67 A gene expression study using in situ hybridization revealed that two transcription factor genes, Pbx3 and Meis2, belonging to the TALE (three amino acid loop extension) superclass of the homeobox gene family, 68 as well as Foxp2, to be preferentially expressed in the striosomes of the developing rat and nonhuman primate striatum. 35 , 37 Because Pbx and Meis proteins act as cofactors for various transcription factors, such as Hox proteins and bHLH proteins, 69 , 70 and Foxp2, Pbx3 and Meis2 are co-expressed in the developing striatum, a direct or indirect interaction between Foxp2 and Pbx/Meis proteins is expected.
There is so far only one report on the upstream regulation of the Foxp2 gene. 71 There are six Lef1 binding sites common between zebrafish, mouse and humans in the Foxp2 genomic region and expressions of Lef1 and Foxp2 is overlapped in the zebrafish brain during development. Lef1 is a transcription factor activated by the canonical Wnt/β-catenin signaling pathway involved in body patterning, neuronal cell specification and axon pathfinding. Knockdown of Lef1 using siRNA caused loss of Foxp2 expression in a restricted part of the brain in zebrafish experiments. Also, a ChIP experiment confirmed that Lef1 binds to sites in the Foxp2 enhancer region. Thus Lef1 may also regulate the expression of Foxp2 in humans.
Perspectives
Molecular network analysis regarding FOXP2 will provide at least two important opportunities in the field of cognitive and behavioral neurology. First, uncovering the gene and protein networks related to FOXP2/FOXP2 will aid elucidation of the molecular mechanisms underlying neural development potentially involved in language acquisition (especially in the striatal compartments, cerebral and cerebellar structures). Second, understanding the genes and pathways that are regulated by FOXP2 might lead to discovery of candidate genes for SLI (specific language Impairment) and autism. Finally, the progresses that we make in the ‘neurobiology of language' will give us hints in developing pharmacological tools not only for treating speech and language disorders, but also for potentiating or improving speech and language skills.
References
- 1.
- Kaufmann E, Knochel W. Five years on the wings of fork head. Mech Dev. 1996;57:3–20. [PubMed: 8817449]
- 2.
- Carlsson P, Mahlapuu M. Forkhead transcription factors: key players in development and metabolism. Dev Biol. 2002;250:1–23. [PubMed: 12297093]
- 3.
- Shu W, Yang H, Zhang L, et al. Characterization of a new subfamily of winged-helix/forkhead (Fox) genes that are expressed in the lung and act as transcriptional repressors. J Biol Chem. 2001;276:27488–27497. [PubMed: 11358962]
- 4.
- Lai CSL, Fisher SE, Hurst JA, et al. A forkhead-domain gene is mutated in a severe speech and language disorder. Nature. 2001;413:519–523. [PubMed: 11586359]
- 5.
- Lu MM, Li S, Yang H, et al. Foxp4: a novel member of Foxp subfamily of winged-helix genes co-expressed with Foxp1 and Foxp2 in pulmonary and gut tissues. Mech Dev. 2002;119S:S197–S202. [PubMed: 14516685]
- 6.
- Teufel A, Wong EA, Mukhopadhyay N, et al. FoxP4, novel forkhead transcriptional factor. Biochim Biophys Acta. 2003;1627:147–152. [PubMed: 12818433]
- 7.
- Li S, Weidenfeld J, Morrisey EE. Transcriptional and DNA binding activity of the Foxp1/2/4 family is modulated by heterotypic and homotypic protein interactions. Mol Cell Biol. 2004;24:809–822 . [PMC free article: PMC343786] [PubMed: 14701752]
- 8.
- Enard W, Przeworski M, Fisher SE, et al. Molecular evolution of FOXP2, a gene involved in speech and language. Nature. 2002;418:869–872. [PubMed: 12192408]
- 9.
- Krause J, Lalueza-Fox C, Orlando L, et al. The derived FOXP2 variant of modern humans was shared with Neandertals. Curr Biol. 2007;17:1908–1912. [PubMed: 17949978]
- 10.
- Hurst J, Baraitser M, Auger E, et al. An extended family with a dominantly inherited speech disorder. Dev Med Child Neurol. 1990;32:347–355. [PubMed: 2332125]
- 11.
- Fisher SE, Vargha-Khadem F, Watkins KE, et al. Localisation of a gene implicated in a severe speech and language disorder. Nature Genet. 1998;18:168–170. [PubMed: 9462748]
- 12.
- MacDermot KD, Bonora E, Sykes N, et al. Identification of FOXP2 truncation as a novel cause of developmental speech and language deficits. Am J Hum Genet. 2005;76:1074–1080. [PMC free article: PMC1196445] [PubMed: 15877281]
- 13.
- Clifton-Bligh RJ, Wentworth JM, Heinz P, et al. Mutation of the gene encoding human TTF-2 associated with thyroid agenesis, cleft palate and choanal atresia. Nat Genet. 1998;19:399–401. [PubMed: 9697705]
- 14.
- Crisponi L, Deiana M, Loi A, et al. The putative forkhead transcription factor FOXL2 is mutated in blepharophimosis/ptosis/epicanthus inversus syndrome. Nat Genet. 2001;27:159–166. [PubMed: 11175783]
- 15.
- Fang J, Dagenais SL, Erickson RP, et al. Mutations in FOXC2(MFH-1), a forkhead family transcription factor, are responsible for the hereditary lymphedema-distichiasis syndrome. Am J Hum Genet. 2000;67:1382–1388. [PMC free article: PMC1287915] [PubMed: 11078474]
- 16.
- Nishimura DY, Swiderski RE, Alward WL, et al. The forkhead transcription factor gene FKHL7 is responsible for glaucoma phenotypes which map to 6p25. Nat Genet. 1998;19(2):140–147. [PubMed: 9620769]
- 17.
- Kaminen N, Hannula-Jouppi K, Kestilä M, et al. A genome scan for developmental dyslexia confirms linkage to chromosome 2p11 and suggests a new locus on 7q32. J Med Genet. 2003;40:340–345. [PMC free article: PMC1735449] [PubMed: 12746395]
- 18.
- O'Brien EK, Zhang X, Nishimura C, et al. Association of specific language impairment (SLI) to the region of 7q31. Am J Hum Genet. 2003;72:1536–1543. [PMC free article: PMC1180313] [PubMed: 12721956]
- 19.
- Meaburn E, Dale PS, Craig IW, et al. Language-impaired children: no sign of the FOXP2 mutation. NeuroReport. 2002;13:1075–1077. [PubMed: 12060812]
- 20.
- Newbury DF, Bonora E, Lamb JA, et al. FOXP2 is not a major susceptibility gene for autism or specific language impairment. Am J Genet. 2002;70:1318–1327. [PMC free article: PMC447606] [PubMed: 11894222]
- 21.
- Gong X, Jia M, Ruan Y, et al. Association between the FOXP2 gene and autistic disorder in Chinese population. Am J Med Genet B Neuropsychiatr Genet. 2004;127B:113–116. [PubMed: 15108192]
- 22.
- Li H, Yamagata T, Mori M, et al. Absence of causative mutations and presence of autism-related allele in FOXP2 in Japanese autistic patients. Brain Dev. 2005;27:207–210. [PubMed: 15737702]
- 23.
- Gauthier J, Joober R, Mottron L, et al. Mutation screening of FOXP2 in individuals diagnosed with autistic disorder. Am J Med Genet A. 2003 Apr 15;118A:172–175. [PubMed: 12655497]
- 24.
- Marui T, Koishi S, Funatogawa I, et al. No association of FOXP2 and PTPRZ1 on 7q31 with autism from the Japanese population. Neuroscience Res. 2005;53:91–94. [PubMed: 15998549]
- 25.
- Wassink TH, Piven J, Vieland VJ, et al. Evaluation of FOXP2 as an autism susceptibility gene. Am J Med Genet. 2002;114:566–569. [PubMed: 12116195]
- 26.
- Bruce HA, Margolis RL. FOXP2: novel exons, splice variants and CAG repeat length stability. Hum Genet. 2002;111:136–144. [PubMed: 12189486]
- 27.
- Brunkow ME, Jeffery EW, Hjerrild KA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001;27:68–73. [PubMed: 11138001]
- 28.
- Wang B, Weidenfeld J, Lu MM, et al. Foxp1 regulates cardiac outflow tract, endocardial cushion morphogenesis and myocyte proliferation and maturation. Development. 2004;131:4477–4487. [PubMed: 15342473]
- 29.
- Dasen JS, De Camilli A, Wang B, et al. Hox repertoires for motor neuron diversity and connectivity gated by a single accessory factor, FoxP1. Cell. 2008;134:304–316. [PubMed: 18662545]
- 30.
- Rousso DL, Gaber ZB, Wellik D, et al. Coordinated actions of the forkhead protein Foxp1 and Hox proteins in the columnar organization of spinal motor neurons. Neuron. 2008;59:226–240. [PMC free article: PMC2547125] [PubMed: 18667151]
- 31.
- Banham AH, Beasley N, Campo E, et al. The FOXP1 winged helix transcription factor is a novel candidate tumor suppressor gene on chromosome 3p. Cancer Res. 2001;61:8820–8829. [PubMed: 11751404]
- 32.
- Li S, Zhou D, Lu MM, et al. Advanced cardiac morphogenesis does not require heart tube fusion. Science. 2004;305:1619–1622. [PubMed: 15361625]
- 33.
- Ferland RJ, Cherry TJ, Preware PO, et al. Characterization of Foxp2 and Foxp1 mRNA and protein in the developing and mature brain. J Comp Neurol. 2003;460:266–279. [PubMed: 12687690]
- 34.
- Lai CS, Gerrelli D, Monaco AP, et al. FOXP2 expression during brain development coincides with adult sites of pathology in a severe speech and language disorder. Brain. 2003;126:2455–2462. [PubMed: 12876151]
- 35.
- Takahashi K, Liu F-C, Hirokawa K, et al. Expression of Foxp2, a gene involved in speech and language, in the developing and adult striatum. J Neurosci Res. 2003;73:61–72. [PubMed: 12815709]
- 36.
- Takahashi K, Liu FC, Hirokawa K, et al. Expression of Foxp4 in the developing and adult rat forebrain. J Neurosci Res. 2008;86:3106–3116. [PubMed: 18561326]
- 37.
- Takahashi K, Liu FC, Oishi T, et al. Expression of FOXP2 in the developing monkey forebrain: comparison with the expression of the genes FOXP1, PBX3 and MEIS2. J Comp Neurol. 2008;509:180–189. [PubMed: 18461604]
- 38.
- Teramitsu I, Kudo LC, London SE, et al. ParallelFoxP1 and FoxP2 expression in songbird and human brain predicts functional interaction. J Neurosci. 2004;24:3152–3163. [PMC free article: PMC6730014] [PubMed: 15056695]
- 39.
- Tamura S, Morikawa Y, Iwanishi H, et al. Expression pattern of the winged-helix/forkhead transcription factor Foxp1 in the developing central nervous system. Gene Expression Patterns. 2003;3:193–197. [PubMed: 12711548]
- 40.
- Tamura S, Morikawa Y, Iwanishi H, et al. Foxp1 gene expression in projection neurons of the mouse striatum. Neuroscience. 2004;124:261–267. [PubMed: 14980377]
- 41.
- Campbell K. Dorsal-ventral patterning in the mammalian telencephalon. Curr Opin Neurobiol. 2003;13:50–56. [PubMed: 12593982]
- 42.
- Graybiel AM. Neurotransmitters and neuromodulators in the basal ganglia. Trends In Neuroscience. 1990;15:244–254. [PubMed: 1695398]
- 43.
- Graybiel AM, Penney JB. Bloom FE, Björklund A, Hökfelt T, editors. Chemical architecture of the basal ganglia. Amsterdam: Elsevier Science; Handbook of Chemical Neuroanatomy. The Primate Nervous System. Part III. 1999;15:227–284.
- 44.
- Gerfen CR, Wilson CJ. Swanson LW, Björklund A, Hökfelt T, editors. The basal ganglia. Amsterdam: Elsevier Science; Handbook of Chemical Neuroanatomy. Integrated Systems of the CNS. Part III. 1996;12:371–468.
- 45.
- Belton E, Salmond CH, Watkins KE, et al. Bilateral brain abnormalities associated with dominantly inherited verbal and orofacial dyspraxia. Hum Brain Mapp. 2003;18:194–200. [PMC free article: PMC6872113] [PubMed: 12599277]
- 46.
- Watkins KE, Vargha-Khadem F, Ashburner J, et al. MRI analysis of an inherited speech and language disorder: structural brain abnormalities. Brain. 2002;125:465–478. [PubMed: 11872605]
- 47.
- Liégeois F, Baldeweg T, Connelly A, et al. Language fMRI abnormalities associated with FOXP2 gene mutation. Nat Neurosci. 2003;6:1230–1237. [PubMed: 14555953]
- 48.
- Vargha-Khadem F, Watkins KE, Price CJ, et al. Neural basis of an inherited speech and language disorder. Proc Natl Acad Sci USA. 1998;95:12695–12700. [PMC free article: PMC22893] [PubMed: 9770548]
- 49.
- Graybiel AM, Ragsdale CW Jr. Histochemically distinct compartments in the striatum of human, monkey and cat demonstrated by acetylthiocholinesterase staining. Proc Natl Acad Sci USA. 1978;75:5723–5726. [PMC free article: PMC393041] [PubMed: 103101]
- 50.
- Canales JJ, Graybiel AM. A measure of striatal function predicts motor stereotypy. Nat Neurosci. 2000;3:377–383. [PubMed: 10725928]
- 51.
- Cenci MA, Tranberg A, Andersson M, et al. Changes in the regional and compartmental distribution of FosB-and JunB-like immunoreactivity induced in the dopamine-denervated rat striatum by acute or chronic L-dopa treatment. Neuroscience. 1999;94:515–527. [PubMed: 10579213]
- 52.
- Saka E, Elibol B, Erdem S, et al. Compartmental changes in expression of c-Fos and FosB proteins in intact and dopamine-depleted striatum after chronic apomorphine treatment. Brain Res. 1999;825:104–114. [PubMed: 10216178]
- 53.
- Crinion J, Turner R, Grogan A, et al. Language control in the bilingual brain. Science. 2006;312:1537–1540. [PubMed: 16763154]
- 54.
- Ullman MT, Corkin S, Coppola M, et al. A neural dissociation within language: evidence that the mental dictionary is part of declarative memory and that grammatical rules are processed by the procedural system. J Cogn Neurosci. 1997;9:266–276. [PubMed: 23962016]
- 55.
- Ullman MT. A neurocognitive perspective on language: the declarative/procedural model. Nature Rev Neurosci. 2001;2:717–726. [PubMed: 11584309]
- 56.
- Damasio AR, Damasio H. Brain and language. Sci Am. 1992;267:88–95. [PubMed: 1502527]
- 57.
- Ackermann H. Cerebellar contributions to speech production and speech perception: psycholinguistic and neurobiological perspectives. Trends Neurosci. 2008;31:265–272. [PubMed: 18471906]
- 58.
- Vargha-Khadem F, Gadian DG, Copp A, et al. FOXP2 and the neuroanatomy of speech and language. Nat Rev Neurosci. 2005;6:131–138. [PubMed: 15685218]
- 59.
- Fujita E, Tanabe Y, Shiota A, et al. Ultrasonic vocalization impairment of Foxp2 (R552H) knockin mice related to speech-language disorder and abnormality of Purkinje cells. Proc Natl Acad Sci USA. 2008;105:3117–3122. [PMC free article: PMC2268594] [PubMed: 18287060]
- 60.
- Groszer M, Keays DA, Deacon RM, et al. Impaired synaptic plasticity and motor learning in mice with a point mutation implicated in human speech deficits. Curr Biol. 2008;18:354–362. [PMC free article: PMC2917768] [PubMed: 18328704]
- 61.
- Shu W, Cho JY, Jiang Y, et al. Altered ultrasonic vocalization in mice with a disruption in the Foxp2 gene. Proc Natl Acad Sci USA. 2005;102:9643–9648. [PMC free article: PMC1160518] [PubMed: 15983371]
- 62.
- Vernes SC, Nicod J, Elahi FM, et al. Functional genetic analysis of mutations implicated in a human speech and language disorder. Hum Mol Genet. 2006;15:3154–3167. [PubMed: 16984964]
- 63.
- Mizutani A, Matsuzaki A, Momoi MY, et al. Intracellular distribution of a speech/language disorder associated FOXP2 mutant. Biochem Biophys Res Commun. 2007;353:869–874. [PubMed: 17196932]
- 64.
- Wang B, Lin D, Li C, et al. Multiple domains define the expression and regulatory properties of Foxp1 forkhead transcriptional repressors. J Biol Chem. 2003;278:24259–24268. [PubMed: 12692134]
- 65.
- Spiteri E, Konopka G, Coppola G, et al. Identification of the transcriptional targets of FOXP2, a gene linked to speech and language, in developing human brain. Am J Hum Genet. 2007;81:1144–1157. [PMC free article: PMC2276350] [PubMed: 17999357]
- 66.
- Vernes SC, Spiteri E, Nicod J, et al. High-throughput analysis of promoter occupancy reveals direct neural targets of FOXP2, a gene mutated in speech and language disorders. Am J Hum Genet. 2007;81:1232–1250. [PMC free article: PMC2276341] [PubMed: 17999362]
- 67.
- Takahashi H, Liu F-C. Genetic patterning of the mammalian telencephalon by morphogenetic molecules and transcription factors. Birth Defects Res C Embryo Today. 2006;78:256–266. [PubMed: 17061260]
- 68.
- Burglin TR. Analysis of TALE superclass homeobox genes (MEIS, PBC, KNOX, Iroquois, TGIF) reveals a novel domain conserved between plants and animals. Nucleic Acids Res. 1997;25:4173–4180. [PMC free article: PMC147054] [PubMed: 9336443]
- 69.
- Berkes CA, Bergstrom DA, Penn BH, et al. Pbx marks genes for activation by MyoD indicating a role for a homeodomain protein in establishing myogenic potential. Mol Cell. 2004;14:465–477. [PubMed: 15149596]
- 70.
- Mann RS, Affolter M. Hox proteins meet more partners. Curr Opin Genet Dev. 1998;8:423–429. [PubMed: 9729718]
- 71.
- Bonkowsky JL, Wang X, Fujimoto E, et al. Domain-specific regulation of foxP2 CNS expression by lef1. BMC Dev Biol. 2008;8:103. [PMC free article: PMC2579431] [PubMed: 18950487]
- Introduction
- The Foxp Subfamily
- Discovery of FOXP2 as a ‘Language Gene’
- FOXP2 and Specific Language Impairment (SLI) and Autism
- Expression of Foxp Subfamily Members in the Brain
- Language Impairments in the Affected Members of the KE Family
- Imaging Studies on the KE Family
- The FOXP2 Expression Pattern in the Brain and Its Relation to the Cognitive Functions of Speech and Language
- Phenotype in Foxp2 Mutant Mice
- Transcriptional Activity of the FOXP2 Protein
- Foxp2 Upstream and Downstream Genes
- Perspectives
- References
- FOXP Genes, Neural Development, Speech and Language Disorders - Madame Curie Bio...FOXP Genes, Neural Development, Speech and Language Disorders - Madame Curie Bioscience Database
- Regulation of Paracellular Transport across Tight Junctions by the Actin Cytoske...Regulation of Paracellular Transport across Tight Junctions by the Actin Cytoskeleton - Madame Curie Bioscience Database
- Development of Manganic Porphyrin Mimetics of Superoxide Dismutase Activity - Ma...Development of Manganic Porphyrin Mimetics of Superoxide Dismutase Activity - Madame Curie Bioscience Database
- The Role of Maspin in Tumor Progression and Normal Development - Madame Curie Bi...The Role of Maspin in Tumor Progression and Normal Development - Madame Curie Bioscience Database
- Rapid Evolution of Sex-related Genes: Sexual Conflict or Sex-specific Adaptation...Rapid Evolution of Sex-related Genes: Sexual Conflict or Sex-specific Adaptations? - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...