

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Huntington's disease (HD) is an autosomal dominant inherited neurodegenerative disease caused by a CAG repeat expansion in exon 1 of the Huntington gene (HD) also known as IT15. Despite the disease being caused by dysfunction of a single gene, expressed as an expanded polyglutamine in the huntingtin protein, there is a major variability in the symptom profile of patients with Huntington's disease as well as great variability in the neuropathology. The symptoms vary throughout the course of the disease and vary greatly between cases. These symptoms present as varying degrees of involuntary movements, mood, personality changes, cognitive changes and dementia. To determine whether there is a morphological basis for this symptom variability, recent studies have investigated the cellular and neurochemical changes in the striatum and cerebral cortex in the human brain to determine whether there is a link between the pathology in these regions and the symptomatology shown by individual cases. These studies together revealed that cases showing mainly mood symptom profiles correlated with marked degeneration in the striosomal compartment of the striatum, or in the anterior cingulate gyrus of the cerebral cortex. In contrast, in cases with mainly motor symptoms neurodegeneration was especially marked in the primary motor cortex with variable degeneration in both the striosomes and matrix compartments of the striatum. These studies suggest that the variable degeneration of the striatum and cerebral cortex correlates with the variable profiles of Huntington's disease.
INTRODUCTION
Huntington's disease (HD) is an autosomal dominant inherited neurodegenerative disease caused by a mutation in exon 1 of the Huntington gene (HD) also known as the IT15 gene on chromosome 4. In the HD gene a triplet repeat of CAGs which codes for a stretch of glutamines is present in the N-terminal region of the gene with the normal range being up to approximately 35 although this may vary.1 In HD this repeat sequence is expanded to 36 repeats and above resulting in toxic protein causing the disease. The mutant gene causes neurodegeneration in the brain through a variety of proposed mechanisms such as transcriptional dysregulation, synaptic dysfunction, excitotoxic mechanisms, oxidative stress and energy depletion.2 One of the most important features of the disease which has been the subject of recent studies is the highly variable nature of the symptoms and neuropathology in affected subjects. This review will focus on correlations between symptom profile and pattern of degeneration in the basal ganglia and cortex.
SYMPTOM VARIABILITY
Classically, HD expresses a triad of symptoms which include motor, mood and cognitive deficits. However, despite the single-gene etiology of HD there is remarkable variability in the types of behavioural, cognitive, and motor symptoms present in different HD patients both at clinical onset and thereafter during the course of the disease.3,4 Some HD patients exhibit mainly motor dysfunction at clinical onset, and few if any changes in mood for extended periods of time while, at the other extreme, others show mainly mood and/or cognitive changes, with minimal involuntary movements until the late stages of the disease.3,4 Still others experience marked motor, mood and cognitive symptoms simultaneously.4-9 Interestingly, observations in monozygotic twins who inherited identical HD genes with the same repeat length exhibit marked differences in their behavioural symptoms.10 The onset of clinical symptoms in individual HD patients is generally correlated with the number of CAG repeats,11 as does the disease severity, but there is no consistent relationship between CAG repeat length and symptom subtype.4,9,12,13 Thus the source of variability in symptom subtypes is not clear. The clinical diagnosis is usually based on the movement disorder termed Huntington's chorea. These characteristic motor symptoms are expressed as a severe "choreoathetotic" disorder which describes the rapid, irregular, involuntary movements of HD. In addition clumsiness and unsteadiness in walking are also early symptoms. Studies on HD populations have indicated that approximately 50-70% of patients at onset present with chorea14,15 whereas another 30-50% present first with mood, cognitive and behavioural changes. The chorea may develop into rigidity and dystonia later in the disease. Approximately 30-50% of patients may present first with cognitive and emotional problems such as irritability, aggression, anxiety, and obsessive behaviour with the most common being reported as depression.14,15 There is also considerable phenotypic variation in the pattern of symptomatology during the course of the disease. Therefore factors such as different genetic interactions of the HD gene with the individual's genome, the environment effects and epigenetics are all possible influential factors on the course of the disease.
NEURODEGENERATION IN HUNTINGTON'S DISEASE
Previous studies have shown that neurodegeneration in HD occurs most prominently in the striatum while other regions of the brain are also affected including the "downstream" structures of the striatum, the globus pallidus, substantia nigra and thalamus. Also, various regions of the cerebral cortex, hippocampus and amygdala of the limbic system show variable degeneration.16,17 In the striatum, it is principally the GABAergic medium spiny projection neurons containing the neuropeptides enkephalin and substance P which are affected in HD. The most vulnerable are the GABAergic enkephalin containing medium spiny neurons that project to the external segment of the globus pallidus which degenerate before the GABAergic substance P containing medium spiny neurons which project to the internal segment of the globus pallidus and substantia nigra.18-23 Furthermore, at the regional level the neuronal degeneration appears to be especially marked in the early stages in the tail of the caudate nucleus, and then progresses from the dorsal to the ventral regions and from the rostral to the caudal regions of the striatum.24,25 It is also evident that many neurochemical and neurotransmitter changes occur before any significant cell death can be identified. This has been established in studies on post mortem HD brains which show marked neurotransmitter receptor up and down regulation in the striatum and in the output nuclei of the striatum. In particular, in grade 0 cases which have little or no cell death, the cannabinoid (CB1) receptors are markedly decreased in the striatum, globus pallidus and substantia nigra and are very low in higher grades. With increasing grades GABAA receptor subunits and GABAB receptor subunits are reduced in the striatum but upregulate dramatically in the globus pallidus.20-22,26
STRIOSOME-MATRIX COMPARTMENTS IN THE STRIATUM
The striatum is neurochemically organised into two different compartments, the smaller striosomes and the larger `back-ground' matrix. Many different neurochemical markers including acetylcholinesterase, calcium binding proteins, neurotransmitter receptors and others delineate these two neurochemical compartments by differential intensity of labelling of each compartment. Neurochemicals such as acetylcholinesterase, calbindin and parvalbumin show different intensities of immunoreactivity in the striosomes and matrix compartments.27-29 The striosomes are characterised by intense enkephalin immunoreactivity while the matrix compartment is characterised by dense staining of acetylcholinesterase, calbindin and parvalbumin. Studies of nonhuman primates and humans have revealed differential patterns of connectivity of these compartments with the cerebral cortex, thalamus and substantia nigra. For instance, the frontal and sensory-motor cortex is connected with the matrix whereas the orbitofrontal and limbic cortex is affiliated with the striosomes;30 the striosomes in turn project to the substantia nigra pars compacta.31 These findings regarding connectivity, in combination with preliminary studies investigating functions of the striosomal system in nonhuman primates and rodents suggest a functional role for these compartments (e.g., refs. 32,33). In particular the striosome compartment in the striatum may play a major role in limbic functions such as mood, whereas, the matrix compartment may be more involved with sensory-motor functions.
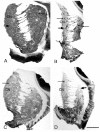
In HD several studies have reported selective degeneration of neurons belonging to either the striosome or matrix compartments.24,27,34-36 Some studies report preferential loss of neurons or neurochemical markers in the matrix compartment in low grade HD20,24,36,37, and others report that neurons or neurochemical markers are lost in striosomes, with clear sparing of the matrix, at least early on.34,35,38 To address the question of the significance of this variable degeneration of neurons in the two major neurochemical compartments of the striatum, the striosomes and the extra striosomal matrix, we recently undertook a study correlating the variable pattern of degeneration of the striatum with the variable pattern of symptomatology in 35 HD cases. In this study we utilised GABAA receptor, calbindin and enkephalin immunohistochemistry, to label specifically the neurodegeneration in the striosomes and matrix compartments in the striatum of the 35 HD cases and 13 normal control cases.39 This study showed a large variability in the loss of GABAA receptors, chemical markers and neurons with respect to their location in the striatal striosome or matrix compartment (Fig. 1). The results of this study on the post-mortem human brain showed that there was a continuum of loss across the different cases, from those which showed predominantly striosomal loss, (Fig. 1C) to those with predominantly matrix loss (Fig. 1B), with a middle group that had a mixed loss in the matrix and striosome compartments (Fig. 1D). To determine whether this variability in compartmental loss was related to the clinical symptomatology, the chemical neuropathology data was compared to the pattern of symptomatology experienced during the life-times of the HD cases, with data collected by researchers blind to the neuroanatomical analyses of the brains. A significant association was found between HD cases with pronounced mood dysfunction and loss of the GABAA receptors and cells in striosomes of the HD striatum. This association held for both clinical onset and end stage assessments of symptoms, suggesting that changes in striosome-related circuits in Huntington's disease brains may lead to mood dysfunction. The cases with accentuated striosome abnormality further exhibited later onset age, lower disease grade, and lower CAG repeat length in the HD gene. However no association was found between CAG repeat length or age of onset and mood dysfunction. No clear association of motor symptoms and matrix pathology was found, although there was a tendency for cases with matrix loss to have higher voluntary motor impairment at end-stage of the disease. Overall we suggest that variation in clinical symptomatology in HD is associated with variation in the relative abnormality of GABAA receptor loss in the striosome or matrix compartments of the striatum.
To determine whether degeneration in the other major region of the forebrain affected in HD, the cerebral cortex, also varied with symptomatology profile, in a separate study the pattern of degeneration in the primary motor and anterior cingulate cortex was investigated and compared with variable patterns of symptoms in 12 HD cases and 15 control brains. The primary motor cortex is known to be involved in the control of motor functions, whilst there is evidence to show that the anterior cingulate cortex is involved in emotional regulation and mood disturbances.40-44 Detailed stereological studies of post-mortem HD brains were carried out and these revealed a major overall cell loss in these two functionally diverse cortical regions. Interestingly, however, they showed marked variation in the extent of cell loss in the motor and cingulate cortices between individual HD cases. When the pattern of cortical cell loss in the HD cases was compared with the pattern of motor and mood symptoms present during the disease for each case, it was found that motor and mood symptomatology corresponded with the heterogeneity of cell loss in the corresponding functional regions of the cerebral cortex.45 That is, the cell loss in the cingulate gyrus corresponded with the cases with predominant mood symptoms (Figs. 2,4), and the cases with major cell loss in the motor cortex corresponded with those with predominant motor symptoms (Figs. 3,4).
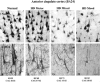
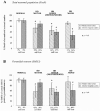

These findings now help to explain the variable results shown in previous quantitative cell studies that have reported variable cell loss across widespread regions of the cerebral cortex.46-51 For example, major losses of pyramidal projection neurons have been documented in HD cases in various regions of the cerebral cortex including the motor cortex,49 superior frontal cortex, cingulate gyrus46 and the angular gyrus of the parietal lobe.50 These findings of variable cortical degeneration add to findings from recent studies using high resolution surface-based analysis of in vivo Magnetic Resonance Imaging (MRI) data to measure cortical thickness.52-55 These in vivo studies in over 30 individuals with HD have shown a heterogeneous pattern of region-specific thinning of the cerebral cortex in Huntington's disease with some of the most marked changes occurring in the sensorimotor cortex and areas of the visual cortex.55,56 In the motor cortex the more dorsal regions associated with the lower limbs showed the most thinning which is reminiscent of the neurodegeneration of the striatum which generally occurs mainly in the dorsal region. Whether the cortical and striatal degeneration is arranged topographically and whether the cortical thinning evident in the imaging data reflect cortical neuronal dysfunction or dysfunction from striatal alterations needs to be investigated further. Interestingly, however, the general pattern of cortical thinning in the MRI studies has been linked with distinct motor phenotypes determined using established clinical tests.56 When the imaging studies are considered along side our findings demonstrating a significant association between patterns of neuronal degeneration in cerebral cortex and the symptom profile, it can be concluded that cortical changes begin early in HD, are regionally heterogeneous and that topologically selective changes in the cerebral cortex might explain much of the clinical heterogeneity found in this disease. The relationship demonstrated between symptom profiles and cortical degeneration provides a novel perspective on understanding the neural basis of clinical heterogeneity found in HD.
MECHANISMS OF DISEASE
The exact mechanisms of neuronal cell death in HD are currently unclear. The expanded CAG repeat of the HD gene is expected to interact with large numbers of other genes as evidenced by the results of gene microarray studies showing large numbers of affected genes in studies on both post mortem HD tissue57 and mouse models of HD.58 These interactions lead to a complex set of parameters that may involve transcriptional dysregulation, excitotoxicity, oxidative stress, changes in neurotransmitters, disruption of cortical BDNF production, and breakdown of cellular and vesicular transport mechanismsin neurons of the striatum, cerebral cortex and other regions throughout the brain.59-64 In the striatum it is the medium spiny neurons which are the most vulnerable, particularly the subset of enkephalin containing striatopallidal neurons. The regional death of these medium spiny neurons however can be quite variable in relation to the striosome-matrix compartments. Therefore other factors such as the connectivity of striatal neurons with other regions of the brain, for instance the topography of the excitatory projection from the cerebral cortex and thalamus may also play a role in this regional cell death. The role of BDNF in the cortico-striatal pathway has been implicated in either causing the death of glutamatergic pyramidal neurons and/or dysfunction of their firing which could be a primary factor in the death of striatal neurons.64-66 It has long been known that the cerebral cortex is not a homogeneous structure as evidenced by the different morphological composition of the Brodmann areas. Furthermore genetic studies show that neurons in the different regions of the cerebral cortex have a variable genetic expression profile which defines their particular subtype.67 Therefore neurons in different regions of the cortex may interact differently with the mutant Htt gene and cause degeneration in variable populations of pyramidal neurons and cortical interneurons.
Recent transgenic animal studies have implicated dysfunction of the cortex as one of the major indicators of phenotype; this may occur through cortical synaptic dysfunction even before cell death.65,68 Dysfunction of the cortico-striatal neurons could lead to neurodegeneration of striatal neurons. Also, abnormal glutamate receptor functions in the cerebral cortex have been implicated in behavioural and motor impairments in transgenic mice with physiological and morphological cortical changes predicting the onset and severity of behavioural deficits.69-71 Furthermore, studies in the conditional mouse model where cortical and/or striatal cells selectively express mutant Htt, dysfunction of the cortical neurons was essential to the development of significant behavioural and motor deficits.72 Other transgenic mouse studies have implicated dysfunction of both the cortical projection and interneurons of the cerebral cortex in the development of HD pathology.73,74 All of these animal studies provide accumulating mechanistic evidence that the cortex plays a major role in the initiation and development of the HD phenotype, and that dysfunction in the corticostriate neurons plays a major role in HD forebrain pathology.
In addition to the factors leading to cell death discussed above, the changes in receptor expression in the globus pallidus would also play a role in determining the symptom profile throughout the course of the disease. For example, the upregulation of GABAA and GABAB receptor subunits is thought to be mainly a compensatory mechanism for loss of GABA input to the output nuclei of the basal ganglia and this mechanism may contribute to ameliorating symptoms in HD by maintaining neurochemical balance despite major cell loss.26
CONCLUSION
Our recent studies on the post-mortem HD brain, using stereological counting methods as well as sophisticated MRI imaging techniques on Huntington's disease cases have collectively shown that there is a remarkable variability in the pattern of striatal and cortical degeneration in HD. This variability in neurodegeneration is also correlated with symptomatology. Thus the symptom profile expressed by particular HD cases can be correlated with a pattern of striatal compartmental loss and regional cortical cell loss. The striatum has long been regarded as the primary pathological region of HD causing the movement disorder of HD but the cerebral cortex can now also be regarded as a major contributor to HD symptomatology. Further investigations need to be carried out on the pattern of cell death in the basal ganglia-thalamo-cortical pathways to determine whether the precise pathology that occurs in the cortico-basal ganglia thalamo-cortical loop will correlate with symptomatology in each individual case. This will enable a more complete picture to be produced explaining how the cellular and morphological neurodegeneration of the brain correlates with specific symptom subtypes and these findings may well influence the therapeutic strategies for the treatment of HD in the future.
REFERENCES
- 1.
- Illarioshkin SN, Igarashi S, Onodera O, et al. Trinucleotide repeat length and rate of progression of Huntington's disease. Ann Neurol. 1994;36(4):630–635. [PubMed: 7944295]
- 2.
- Sharp AH, Ross CA. Neurobiology of Huntington's disease. Neurobiol Dis. 1996;3(1):3–15. [PubMed: 9173909]
- 3.
- Andrew SE, Goldberg YP, Kremer B, et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington's disease. Nat Genet. 1993;4(4):398–403. [PubMed: 8401589]
- 4.
- Claes S, Van Zand K, Legius E, et al. Correlations between triplet repeat expansion and clinical features in Huntington's disease. Arch Neurol. 1995;52(8):749–753. [PubMed: 7639626]
- 5.
- Brandt J, Butters N. The neuropsychology of Huntington's disease. TINS. 1986:118–120.
- 6.
- Folstein SE. Huntington's Disease: A Disorder of Families. Baltimore: John's Hopkins University Press; 1989.
- 7.
- Myers RH, Sax DS, Koroshetz WJ, et al. Factors associated with slow progression in Huntington's disease. Arch Neurol. 1991;48(8):800–804. [PubMed: 1832854]
- 8.
- Thompson JC, Snowden JS, Craufurd D, et al. Behavior in Huntington's disease: dissociating cognition-based and mood-based changes. J Neuropsychiatry Clin Neurosci. 2002;14(1):37–43. [PubMed: 11884653]
- 9.
- Zappacosta B, Monza D, Meoni C, et al. Psychiatric symptoms do not correlate with cognitive decline, motor symptoms, or CAG repeat length in Huntington's disease. Arch Neurol. 1996;53(6):493–497. [PubMed: 8660149]
- 10.
- Georgiou N, Bradshaw JL, Chiu E, et al. Differential clinical and motor control function in a pair of monozygotic twins with Huntington's disease. Mov Disord. 1999;14(2):320–325. [PubMed: 10091627]
- 11.
- Wexler NS, Lorimer J, Porter J, et al. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington's disease age of onset. PNAS. 2004;101(10):3498–3503. [PMC free article: PMC373491] [PubMed: 14993615]
- 12.
- MacMillan JC, Snell RG, Tyler A, et al. Molecular analysis and clinical correlations of the Huntington's disease mutation. Lancet. 1993;342(8877):954–958. [PubMed: 8105214]
- 13.
- Telenius H, Kremer B, Goldberg YP, et al. Somatic and gonadal mosaicism of the Huntington disease gene CAG repeat in brain and sperm. Nat Genet. 1994;6(4):409–414. [PubMed: 8054984]
- 14.
- Witjes-Ane MN, Zwinderman AH, Tibben A, et al. Behavioural complaints in participants who underwent predictive testing for Huntington's disease. J Med Genet. 2002;39(11):857–862. [PMC free article: PMC1735005] [PubMed: 12414829]
- 15.
- Di Maio L, Squitieri F, Napolitano G, et al. Onset symptoms in 510 patients with Huntington's disease. J Med Genet. 1993;30(4):289–292. [PMC free article: PMC1016334] [PubMed: 8487272]
- 16.
- Vonsattel JP, Myers RH, Stevens TJ, et al. Neuropathological classification of Huntington's disease. J Neuropath Exp Neurol. 1985;44:559–577. [PubMed: 2932539]
- 17.
- Vonsattel JPG, Difiglia M. Huntington-disease. J Neuropath and Exp Neurol. 1998;57(5):369–384. [PubMed: 9596408]
- 18.
- Albin RL, Makowiec RL, Hollingsworth ZR, et al. Excitatory amino acid binding sites in the basal ganglia of the rat: a quantitative autoradiographic study. Neuroscience. 1992;46:35–48. [PubMed: 1317515]
- 19.
- Deng YP, Albin RL, Penney JB, et al. Differential loss of striatal projection systems in Huntington's disease: a quantitative immunohistochemical study. J Chem Neuroanat. 2004;27(3):143–164. [PubMed: 15183201]
- 20.
- Faull RL, Waldvogel HJ, Nicholson LF, et al. The distribution of GABAA-benzodiazepine receptors in the basal ganglia in Huntington's disease and in the quinolinic acid-lesioned rat. Prog Brain Res. 1993;99:105–123. [PubMed: 8108544]
- 21.
- Glass M, Dragunow M, Faull RLM. The pattern of neurodegeneration in Huntington's disease: a comparative study of cannabinoid, dopamine, adenosine and GABA(A) receptor alterations in the human basal ganglia in Huntington's disease. Neuroscience. 2000;97(3):505–519. [PubMed: 10828533]
- 22.
- Glass M, Faull RL, Dragunow M. Loss of cannabinoid receptors in the substantia nigra in Huntington's disease. Neuroscience. 1993;56(3):523–527. [PubMed: 8255419]
- 23.
- Reiner A, Albin RL, Anderson KD, et al. Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci USA. 1988;85(15):5733–5737. [PMC free article: PMC281835] [PubMed: 2456581]
- 24.
- Ferrante RJ, Kowall NW, Beal MF, et al. Morphologic and histochemical characteristics of a spared subset of striatal neurons in Huntington's disease. J Neuropathol Exp Neurol. 1987;46(1):12–27. [PubMed: 2947977]
- 25.
- Vonsattel JP, Ge P, Kelly L. The Neuropathology of Dementia. Cambridge: Cambridge University Press UK; 1997. Huntington's disease. In: Esiri M, Morris JH, editors; pp. 219–240.
- 26.
- Allen KL, Waldvogel HJ, Glass M, et al. Cannabinoid (CB(1)), GABA(A) and GABA(B) receptor subunit changes in the globus pallidus in Huntington's disease. J Chem Neuroanat. 2009;37(4):266–281. [PubMed: 19481011]
- 27.
- Graybiel AM, Ragsdale CW Jr. Histochemically distinct compartments in the striatum of human, monkeys and cat demonstrated by acetylthiocholinesterase staining. Proc Nat Acad Sci USA. 1978;75(11):5723–5726. [PMC free article: PMC393041] [PubMed: 103101]
- 28.
- Holt DJ, Graybiel AM, Saper CB. Neurochemical architecture of the human striatum. J Comp Neurol. 1997;384:1–25. [PubMed: 9214537]
- 29.
- Waldvogel HJ, Faull RLM. Compartmentalization of parvalbumin immunoreactivity in the human striatum. Brain Res. 1993;610:311–316. [PubMed: 8319092]
- 30.
- Eblen F, Graybiel AM. Highly restricted origin of prefrontal cortical inputs to striosomes in the macaque monkey. J Neurosci. 1995;15(9):5999–6013. [PMC free article: PMC6577677] [PubMed: 7666184]
- 31.
- Gerfen CR. The neostriatal mosaic: multiple levels of compartmental organization. TINS. 1992;15:133–138. [PubMed: 1374971]
- 32.
- Saka E, Goodrich C, Harlan P, et al. Repetitive behaviors in monkeys are linked to specific striatal activation patterns. J Neurosci. 2004;24(34):7557–7565. [PMC free article: PMC6729641] [PubMed: 15329403]
- 33.
- White NM, Hiroi N. Preferential localization of self-stimulation sites in striosomes/patches in the rat striatum. Proc Natl Acad Sci USA. 1998;95(11):6486–6491. [PMC free article: PMC27819] [PubMed: 9600993]
- 34.
- Hedreen JC, Folstein SE. Early loss of neostriatal striosome neurons in Huntington's disease. J Neuropathol Exp Neurol. 1995;54(1):105–120. [PubMed: 7815073]
- 35.
- Morton AJ, Nicholson LF, Faull RL. Compartmental loss of NADPH diaphorase in the neuropil of the human striatum in Huntington's disease. Neuroscience. 1993;53(1):159–168. [PubMed: 7682296]
- 36.
- Seto-Ohshima A, Emson PC, Lawson E, et al. Loss of matrix calcium-binding protein-containing neurons in Huntington's disease. Lancet. 1988;1234:1252–1254. [PubMed: 2897519]
- 37.
- Olsen JM, Penney JB, Shoulson I, et al. Inhomogeneities of striatal receptor binding in Huntington's disease. Neurology. 1986;36:342.
- 38.
- Augood SJ, Faull RL, Love DR, et al. Reduction in enkephalin and substance P messenger RNA in the striatum of early grade Huntington's disease: a detailed cellular in situ hybridization study. Neuroscience. 1996;72(4):1023–1036. [PubMed: 8735227]
- 39.
- Tippett LJ, Waldvogel HJ, Thomas SJ, et al. Striosomes and mood dysfunction in Huntington's disease. Brain. 2007;130(Pt 1):206–221. [PubMed: 17040921]
- 40.
- Alexopoulos GS, Gunning-Dixon FM, Latoussakis V, et al. Anterior cingulate dysfunction in geriatric depression. Int J Geriatr Psychiatry. 2008;23(4):347–355. [PubMed: 17979214]
- 41.
- Davidson RJ, Pizzagalli D, Nitschke JB, et al. Depression: perspectives from affective neuroscience. Annu Rev Psychol. 2002;53:545–574. [PubMed: 11752496]
- 42.
- Ebert D, Ebmeier KP. The role of the cingulate gyrus in depression: from functional anatomy to neurochemistry. Biol Psychiatry. 1996;39(12):1044–1050. [PubMed: 8780840]
- 43.
- Harrison PJ. The neuropathology of primary mood disorder. Brain. 2002;125(Pt 7):1428–1449. [PubMed: 12076995]
- 44.
- Konarski JZ, McIntyre RS, Kennedy SH, et al. Volumetric neuroimaging investigations in mood disorders: bipolar disorder versus major depressive disorder. Bipolar Disord. 2008;10(1):1–37. [PubMed: 18199239]
- 45.
- Thu DC, Oorschot DE, Tippett LJ, et al. Cell loss in the motor and cingulate cortex correlates with symptomatology in Huntington's disease. Brain. 2010;133(Pt 4):1094–1110. [PubMed: 20375136]
- 46.
- Cudkowicz M, Kowall NW. Degeneration of pyramidal projection neurons in Huntington's disease cortex. Ann Neurol. 1990;27:200–204. [PubMed: 2138444]
- 47.
- Hedreen JC, Peyser CE, Folstein SE, et al. Neuronal loss in layers V and VI of cerebral cortex in Huntington's disease. Neurosci Lett. 1991;133(2):257–261. [PubMed: 1840078]
- 48.
- Heinsen H, Strik M, Bauer M, et al. Cortical and striatal neurone number in Huntington's disease. Acta Neuropathol. 1994;88(4):320–333. [PubMed: 7839825]
- 49.
- Macdonald V, Halliday G. Pyramidal cell loss in motor cortices in Huntington's disease. Neurobiol Dis. 2002;10(3):378–386. [PubMed: 12270698]
- 50.
- Macdonald V, Halliday GM, Trent RJ, et al. Significant loss of pyramidal neurons in the angular gyrus of patients with Huntington's disease. Neuropathol Appl Neurobiol. 1997;23(6):492–495. [PubMed: 9460715]
- 51.
- Selemon LD, Rajkowska G, Goldman-Rakic PS. Evidence for progression in frontal cortical pathology in late-stage Huntington's disease. J Comp Neurol. 2004;468(2):190–204. [PubMed: 14648679]
- 52.
- Rosas HD, Feigin AS, Hersch SM. Using advances in neuroimaging to detect, understand and monitor disease progression in Huntington's disease. NeuroRx. 2004;1(2):263–272. [PMC free article: PMC534942] [PubMed: 15717027]
- 53.
- Rosas HD, Hevelone ND, Zaleta AK, et al. Regional cortical thinning in preclinical Huntington disease and its relationship to cognition. Neurology. 2005;65(5):745–747. [PubMed: 16157910]
- 54.
- Rosas HD, Koroshetz WJ, Chen YI, et al. Evidence for more widespread cerebral pathology in early HD: an MRI-based morphometric analysis. Neurology. 2003;60(10):1615–1620. [PubMed: 12771251]
- 55.
- Rosas HD, Liu AK, Hersch S, et al. Regional and progressive thinning of the cortical ribbon in Huntington's disease. Neurology. 2002;58(5):695–701. [PubMed: 11889230]
- 56.
- Rosas HD, Salat DH, Lee SY, et al. Cerebral cortex and the clinical expression of Huntington's disease: complexity and heterogeneity. Brain. 2008;131(Pt 4):1057–1068. [PMC free article: PMC2657201] [PubMed: 18337273]
- 57.
- Hodges A, Strand AD, Aragaki AK, et al. Regional and cellular gene expression changes in human Huntington's disease brain. Hum Mol Genet. 2006;15(6):965–977. [PubMed: 16467349]
- 58.
- Luthi-Carter R, Strand A, Peters NL, et al. Decreased expression of striatal signaling genes in a mouse model of Huntington's disease. Hum Mol Genet. 2000;9(9):1259–1271. [PubMed: 10814708]
- 59.
- Cattaneo E, Rigamonti D, Goffredo D, et al. Loss of normal huntingtin function: new developments in Huntington's disease research. Trends Neurosci. 2001;24(3):182–188. [PubMed: 11182459]
- 60.
- Cha JH. Transcriptional dysregulation in Huntington's disease. Trends Neurosci. 2000;23(9):387–392. [PubMed: 10941183]
- 61.
- Morton AJ, Faull RL, Edwardson JM. Abnormalities in the synaptic vesicle fusion machinery in Huntington's disease. Brain Res Bull. 2001;56(2):111–117. [PubMed: 11704347]
- 62.
- Petersen A, Mani K, Brundin P. Recent advances on the pathogenesis of Huntington's disease. Exp Neurol. 1999;157(1):1–18. [PubMed: 10222105]
- 63.
- Rosas HD, Salat DH, Lee SY, et al. Complexity and heterogeneity: what drives the ever-changing brain in Huntington's disease? Ann N Y Acad Sci. 2008;1147:196–205. [PMC free article: PMC2813569] [PubMed: 19076442]
- 64.
- Zuccato C, Cattaneo E. Role of brain-derived neurotrophic factor in Huntington's disease. Prog Neurobiol. 2007;81(5-6):294–330. [PubMed: 17379385]
- 65.
- Cepeda C, Wu N, Andre VM, et al. The corticostriatal pathway in Huntington's disease. Prog Neurobiol. 2007;81(5-6):253–271. [PMC free article: PMC1913635] [PubMed: 17169479]
- 66.
- Strand AD, Baquet ZC, Aragaki AK, et al. Expression profiling of Huntington's disease models suggests that brain-derived neurotrophic factor depletion plays a major role in striatal degeneration. J Neurosci. 2007;27(43):11758–11768. [PMC free article: PMC6673215] [PubMed: 17959817]
- 67.
- Molyneaux BJ, Arlotta P, Menezes JR, et al. Neuronal subtype specification in the cerebral cortex. Nat Rev Neurosci. 2007;8(6):427–437. [PubMed: 17514196]
- 68.
- Cummings DM, Andre VM, Uzgil BO, et al. Alterations in cortical excitation and inhibition in genetic mouse models of Huntington's disease. J Neurosci. 2009;29(33):10371–10386. [PMC free article: PMC2754238] [PubMed: 19692612]
- 69.
- Andre VM, Cepeda C, Venegas A, et al. Altered cortical glutamate receptor function in the R6/2 model of Huntington's disease. J Neurophysiol. 2006;95(4):2108–2119. [PubMed: 16381805]
- 70.
- Laforet GA, Sapp E, Chase K, et al. Changes in cortical and striatal neurons predict behavioral and electrophysiological abnormalities in a transgenic murine model of Huntington's disease. J Neurosci. 2001;21(23):9112–9123. [PMC free article: PMC6763893] [PubMed: 11717344]
- 71.
- Sapp E, Schwarz C, Chase K, et al. Huntingtin localization in brains of normal and Huntington's disease patients. Ann Neurol. 1997;42(4):604–612. [PubMed: 9382472]
- 72.
- Gu X, Andre VM, Cepeda C, et al. Pathological cell-cell interactions are necessary for striatal pathogenesis in a conditional mouse model of Huntington's disease. Mol Neurodegener. 2007;2:8. [PMC free article: PMC1885431] [PubMed: 17470275]
- 73.
- Gu X, Li C, Wei W, et al. Pathological cell-cell interactions elicited by a neuropathogenic form of mutant Huntingtin contribute to cortical pathogenesis in HD mice. Neuron. 2005;46(3):433–444. [PubMed: 15882643]
- 74.
- Spampanato J, Gu X, Yang XW, et al. Progressive synaptic pathology of motor cortical neurons in a BAC transgenic mouse model of Huntington's disease. Neuroscience. 2008;157(3):606–620. [PMC free article: PMC2802129] [PubMed: 18854207]
- 75.
- Van Roon-Mom WM, Hogg VM, Tippett LJ, et al. Aggregate distribution in frontal and motor cortex in Huntington's disease brain. Neuroreport. 2006;17(6):667–670. [PubMed: 16603932]
- SELECTIVE NEURODEGENERATION, NEUROPATHOLOGY AND SYMPTOM PROFILES IN HUNTINGTON'S...SELECTIVE NEURODEGENERATION, NEUROPATHOLOGY AND SYMPTOM PROFILES IN HUNTINGTON'S DISEASE - Madame Curie Bioscience Database
- The Restriction Point of the Cell Cycle - Madame Curie Bioscience DatabaseThe Restriction Point of the Cell Cycle - Madame Curie Bioscience Database
- RNA Modification Subsystems in the SEED Database - Madame Curie Bioscience Datab...RNA Modification Subsystems in the SEED Database - Madame Curie Bioscience Database
- Origin, Recognition, Signaling and Repair of DNA Double-Strand Breaks in Mammali...Origin, Recognition, Signaling and Repair of DNA Double-Strand Breaks in Mammalian Cells - Madame Curie Bioscience Database
- Protein Misassembly: Macromolecular Crowding and Molecular Chaperones - Madame C...Protein Misassembly: Macromolecular Crowding and Molecular Chaperones - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...