

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

The E-Cell System is an advanced platform intended for mathematical modeling and simulation of well-stirred biochemical systems. We have recently implemented the Spatiocyte method as a set of plug in modules to the E-Cell System, allowing simulations of complicated multicompartment dynamical processes with inhomogeneous molecular distributions. With Spatiocyte, the diffusion and reaction of each molecule can be handled individually at the microscopic scale. Here we describe the basic theory of the method and provide the installation and usage guides of the Spatiocyte modules. Where possible, model examples are also given to quickly familiarize the reader with spatiotemporal model building and simulation.
Introduction
The E-Cell System version 3 can model and simulate both deterministic and stochastic biochemical processes.1 Simulated molecules are assumed to be dimensionless and homogeneously distributed in a compartment. Some processes such as cell signaling and cytokinesis, however, depend on cellular geometry and spatially localized molecules to carry out their functions. To reproduce such processes using spatially resolved models in silico, we have developed a lattice-based stochastic reaction-diffuson (RD) simulation method, called Spatiocyte,2 and implemented it as a set of plug in modules to the E-Cell System.3 Spatiocyte allows molecular diffusion and reaction to take place between different compartments: for example, a volume molecule in the cytoplasm can diffuse and react with a surface molecule on the plasma membrane. Since molecules are represented as spheres with dimensions, it can also reproduce anomalous diffusion of molecules in a crowded compartment.4,5 Using Spatiocyte simulated microscopy visualization feature, simulation results of spatiotemporal localization of molecules can be evaluated by directly comparing them with experimentally obtained fluorescent microscopy images.
The theory and algorithm of the Spatiocyte method are provided in Arjunan and Tomita (2010)2 while the implementation details are described in Arjunan and Tomita (2009).3 In this chapter, we provide a guide on how to build spatiotemporal RD models using Spatiocyte modules. We begin with the basic theory of the method and proceed with the installation procedures. The properties of each module are outlined in the subsequent section. Some example models are given to familiarize the reader with the common model structures while describing the modules. We conclude this chapter by outlining the planned future directions of Spatiocyte development.
Spatiocyte Method
In this section, we summarize the underlying features of the Spatiocyte method that are necessary to build an RD model. For a more detailed description of the method we direct the reader to a previous article.2
The Spatiocyte method discretizes the space into a hexagonal close-packed (HCP) lattice of regular sphere voxels with radius rv. Each voxel has 12 adjoining neighbors. To represent a surface compartment such as a cell or a nuclear membrane, all empty voxels of the compartment are occupied with immobile lipid molecules. The method also allows molecules to be simulated at microscopic and compartmental spatial scales simultaneously. In the former, each molecule is discrete and treated individually. For example, each diffusing molecule at the microscopic scale is moved independently by a DiffusionProcess from a source voxel to a target neighbor voxel after a given diffusion step interval. Immobile molecules are also simulated at the microscopic scale. Conversely at the compartmental scale, molecules are assumed to be homogeneously distributed (HD) and thus, the concentration information of each HD species is sufficient without explicit diffusion movements. Depending on the simulated spatial scale and the mobility of the reacting species, molecules can undergo either diffusion-influenced or diffusion-decoupled reactions.
All second-order reactions comprising two diffusing reactants, or a diffusing and an immobile reactant are diffusion-influenced, and are therefore, executed by the DiffusionInfluencedReactionProcess. The remaining reactions, which include all zeroth- and first-order reactions, and second-order reactions that involve two adjoining immobile reactants or at least one HD reactant, can be decoupled from diffusion. These diffusion-decoupled reactions are performed by the SpatiocyteNextReactionProcess.
We proceed with the execution of DiffusionInfluencedReactionProcess for a reaction j. Following our discretized scheme2 of the Collins and Kimball RD approach,6 when a diffusing molecule collides with a reactant pair of j at the target voxel, they react with probability
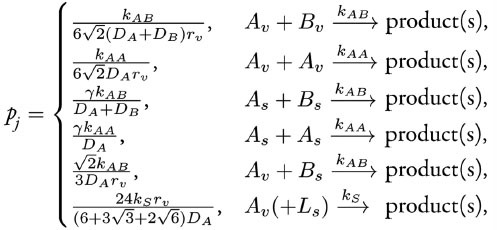
where the constant
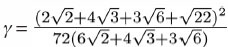
The DiffusionProcess handles the voxel-to-voxel random walk of diffusing molecules and the collisions that take place between each walk. The latter is necessary when a diffusing species participates in a strongly diffusion-limited reaction and the time slice between each walk is too large for an accurate value of pj. Given ts is the current simulation time, the next time a molecule of a diffusing species i with a diffusion coefficient Di can be moved to a randomly selected neighbor voxel is
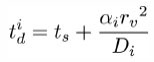
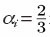
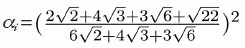
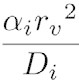
To ensure pj ≤ 1, we reduce the DiffusionProcess interval such that its next execution time becomes
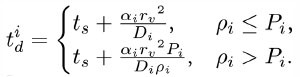
Here Pi is an arbitrarily set reaction probability limit (default value is unity) such that 0 ≤Pi ≤ 1, and ρi = max{p1, ... , pJ} where J is the total number of diffusion-influenced reactions participated by the species i. At each process interval, the molecule can collide as usual with a neighbor reactant pair and react with a scaled probability of pjPi/ρi. In the diffusion-limited case, ρi >Pi and because of the reduced interval, the walk probability becomes less than unity to Pi/ρi.
Reactions that can be decoupled from diffusion such as zeroth- first-order reactions, and second-order reactions that involve two adjoining immobile reactants or at least one HD reactant, are event-driven by the SpatiocyteNextReactionProcess. The reaction product can be made up of one or two molecules, which can be either HD or nonHD molecules. The SpatiocyteNextReactionProcessis an adapted implementation of the Next Reaction (NR) method,7 which itself is a variation of the Gillespie algorithm.8,9
In the process, the reaction propensity (unit s-1) is calculated from the rate coefficient according to
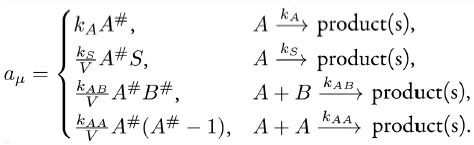
Here, S and V are area and volume of the reaction compartment respectively, while kS (unit ms-1) is the surface-average adsorption rate of an HD volume species A. In the second-order reactions, V is replaced with S if both reactants are in a surface compartment. The next reaction time of a randomly selected molecule in a first order reaction or a pair of molecules in a second-order reaction is given by
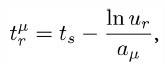
If a reaction has a nonHD product, the new molecule will replace a nonHD reactant in the product compartment. Otherwise if the reaction only involves HD reactants or if the product belongs to a different compartment, the new nonHD molecule will be placed in a random vacant voxel of the product compartment. The placement of a second nonHD product also follows the same procedure. For intercompartmental reactions, a nonHD product will occupy a vacant voxel adjoining both compartments.
Dynamic localization patterns of simulated molecules can be directly compared with experi-mentally obtained fluorescence microscopy images and videos using the MicroscopyTrackingProcess and the SpatiocyteVisualizer. Together, these modules simulate the microphotography process by recording the trajectory of simulated molecules over the camera exposure time and displaying their spatially localized densities. The MicroscopyTrackingProcess records the number of times the molecules of a species occupy each voxel at diffusion step intervals over the exposure time. The SpatiocyteVisualizer then displays the species color at each voxel with intensity and opac-ity levels that are directly proportional the voxel occupancy frequency. Colors from different species occupying the same voxel are blended to mimic colocalization patterns observed in multiple-labeling experiments.
Installing and Running Spatiocyte
The Spatiocyte source code is distributed as open source software under the GNU General Public License and is available at GitHub. At the time of writing, the Spatiocyte modules of the E-Cell System have been tested to run on Linux systems. Spatiocyte does not yet support other operating systems. Here we describe the installation procedures on a Ubuntu Linux system.
On a freshly installed Ubuntu Linux, E-Cell System version 3 and Spatiocyte requires several additional packages:
$ sudo apt-get install automake libtool g++ libgsl0-dev python-numpy python-ply libboost-python-dev libgtkmm-2.4-dev libgtkglextmm-x11-1.2-dev libhdf5-serial-dev git valgrind
The general installation procedure of the E-Cell System version 3 is as follows:
$ cd $ mkdir wrk $ cd wrk $ git clone https://github.com/ecell/ecell3.git $ cd ecell3 $ ./autogen.sh $ ./configure prefix = $HOME/root $ make -j3 (or just make, if there is only one CPU core available) $ make install ( files will be installed in the $HOME/root directory) $ gedit -/.bashrc (other editors such as emacs or vim can also be used here)
The following lines, which specify the environment variables of the E-Cell System should be appended to the .bashrc file:
export PATH = $HOME/root/bin:$PATH export LD_LIBRARY_PATH = $HOME/root/lib:$LD_LIBRARY_PATH:. export PYTHONPATH = $HOME/root/lib/python:$HOME/root/lib/python2.7/site-packages:$PYTHONPATH export ECELL3_DM_PATH = .
In the line 3 above, the Python version number '2.7' should be updated if it is different in the installed system. Next, we load the new environment variables:
$ source -/.bashrc $ ecell3-session-monitor (try opening it, the window shown in Fig. 1 should appear, and then close it)We can now attempt to run a simple model in the E-Cell Model (EM) language, simple.em: $ cd $HOME/wrk/ecell3/doc/samples/simple/ $ ecell3-em2eml simple.em $ ecell3-session-monitor
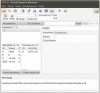
Figure 1.
The E-cell session monitor.
Using ecell3-em2eml, the model file simple.em was converted into simple.eml in Extensible Markup Language (XML) format. The simple.eml file can now be loaded from the File menu of the E-Cell Session Monitor or the File open button (see Fig. 1). Try running the simulation by clicking on the Start button.
The steps to install E-Cell3-Spatiocyte are as follows:
$ cd $HOME/wrk $ git clone git://github.com/ecell/ecell3-spatiocyte.git $ cd ecell3-spatiocyte $ make -j3 (or just make, if there is only one CPU core available)
The E-Cell3-Spatiocyte package includes the MinDE model (see Fig. 2) reported in Arjunan and Tomita (2010).2 We can now attempt to run the model with the following steps:
$ cd $HOME/wrk/ecell3-spatiocyte/ $ ecell3-em2eml 2010.arjunan.syst.synth.biol.wt.em $ ecell3-session-monitor

Figure 2.
E-Cell Model (EM) description file for the MinDE model. The file is available in the Spatiocyte source package as 2010.arjunan.syst.synth.biol.wt.em.
Load the model 2010.arjunan.syst.synth.biol.wt.eml and try running the simulation for 90 seconds.
We can also run Spatioctye models using command line interface of the E-Cell System:
$ ecell3-session -f 2010.arjunan.syst.synth.biol.wt.em < 2010.arjunan.syst.synth.biol.wt.eml, t = 0 >>> run(90) < 2010.arjunan.syst.synth.biol.wt.eml, t = 90 >>> exit()
Models that are created using the Python script can be run as,
$ ecell3-session 2012.arjunan.chapter.neuron.py
When running a Spatiocyte model with the VisualizationLogProcess module enabled, the three-dimensional positional information of a logged molecule species will be stored in visual-Log0.dat (default file name). The molecules can be viewed in a separate visualizer window even while the simulation is still running. To view them, we can run SpatiocyteVisualizer by issuing
$ ./spatiocyte
The visualizer will load the visualLog0.dat file by default and display the molecules at every log interval (see Fig. 3). The keyboard shortcuts that are available for the visualizer are listed in the SpatiocyteVisualizer module section.
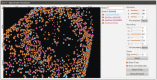
Figure 3.
The SpatiocyteVisualizer displaying simulated membrane-bound proteins of the MinDE model.
If the program fails and crashes when loading or running a model, we can get some debugging information using the Valgrind tool:
$ valgrind tool = memcheck num-callers = 40 leak-check = full python $HOME/ root/bin/ecell3-session -f modelFileName.eml
Spatiocyte Modules
In Spatiocyte modules, the unit of numeric values is given in meters, seconds, radians and mol-ecule numbers. A Spatiocyte model file created using the E-Cell Model (EM) language is shown in Fig. 2. The file contains the wildtype Escherichia coli MinDE cytokinesis regulation model that was reported in Arjunan and Tomita (2010).2 A schematic representation of the model is given in Fig. 4. Python script examples to build models with more complex compartments are provided in Figs. 5 and 6. Figs. 7 and Fig. 8 illustrate 3D visualizations of the resulting models.

Figure 4.
A schematic representation of the MinDE model.

Figure 5.
A Python script to create a neuron-shaped model. The file is available in the Spatiocyte source package as 2012.arjunan.chapter.neuron.py.

Figure 6.
A Python script to create a compartment with randomly distributed microtubules. The file is available in the Spatiocyte source package as 2012.arjunan.chapter.microtubules.py.

Figure 7.
A neuron-shaped compartment created from a combination of rod and ellipsoid compartment geometries. The model is created from the Python script shown in Fig. 5.

Compartment
Compartments are defined hierarchically and follow the format used by the E-Cell System version 3 (see the E-Cell Simulation Environment Version 3 Users Manual for details). Each sub-compartment within a parent compartment is created according to the alphabetical order of the compartment names. Predefined Variables that specify the Compartment properties include DIMENSION, GEOMETRY, LENGTHX, LENGTHY, LENGTHZ, ORIGINX, ORIGINY, ORIGINZ, ROTATEX, ROTATEY, ROTATEZ, XYPLANE, XZPLANE, YZPLANE, VACANT, DIFFUSIVE and REACTIVE. Examples of these variable definitions can be seen in Fig. 2 (lines 4-7 and 33-34), 5 (lines 5-8, 17-24, 31-32, 41-49 and 52-54) and 6 (lines 46-49, 59-60, 63-69 and 71-72).
Molecule species within a Compartment are also defined as a Variable. The Value property of each species stipulates the molecule number during initialization. All species by default are nonHD.
Examples of nonHD species definitions can be seen in Fig. 2 (lines 8-10 and 35-38), 5 (line 25) and 6 (line 50). To define a HD species, the Name property of the Variable should be set to "HD" as shown in the EM and Python examples below:
Variable Variable(A) {
Value 100;
Name "HD"; }
A = theSimulator.createEntity('Variable', 'Variable:.:A')
A.Value = 100
A.Name = "HD"
DIMENSION
The DIMENSION variable defines the spatial dimension of the compartment, whether it is a line ('1'), surface ('2') or a volume ('3') type. At the time of writing, the line compartment type is still in development. A surface compartment encloses its parent volume compartment, and as a result, it cannot be defined independently without a volume compartment to enclose with. A surface compartment does not have any child volume or surface compartment. The root compartment should always be defined as a volume compartment. Since the default DIMENSION value is '3', a volume compartment can be defined without the DIMENSION variable. A volume compartment can also use the predefined variables GEOMETRY, LENGTHX, LENGTHY, LENGTHZ, ORIGINX, ORIGINY, ORIGINZ, ROTATEX, ROTATEY, ROTATEZ, XYPLANE, XZPLANE, YZPLANE, DIFFUSIVE and VACANT, whereas a surface compartment only requires the DIMENSION and VACANT variables and inherits the remaining relevant properties from its parent compartment. In addition, surface compartments can also define the DIFFUSIVE and REACTIVE variables. See Fig. 2 (line 33), 5 (lines 31 and 52) and 6 (lines 59 and 71) for examples of the DIMENSION variable definition.
GEOMETRY
The GEOMETRY variable of a volume compartment specifies one of the six supported geometric primitives: cuboid ('0'), ellipsoid ('1'), cylinder ('2'), rod ('3'), torus ('4') and pyramid ('5'). More complex forms can be constructed using a combination of these primitives. Figs. 4 and 6 illustrate the construction of a neuron-shaped model using a combination of ellipsoid and rod compartments. Compartments without the GEOMETRY definition is set to the cuboid form since the default value is '0'. For examples of GEOMETRY definition see Fig. 2 (line 4), 5 (lines 17 and 41) and 6 (lines 46 and 63).
LENGTH[X, Y, Z]
The three variables LENGTH[X, Y, Z] can specify the compartment lengths in the direc-tions of [x, y, z]-axes, respectively. The cuboid, ellipsoid and pyramid compartments use all three variables. If all three lengths are equal, a cube or a sphere compartment can be created with a cuboid or an ellipsoid geometry, respectively. For the pyramid compartment, LENGTH[X, Y, Z] stipulate its base length, height and base width, respectively. For a cylinder compartment, LENGTHX defines the cylinder length, while its diameter is given by LENGTHY. In the case of a rod compartment, LENGTHX indicates the length from the tip of one pole to the other while LENGTHY defines its diameter. For a torus, its larger diameter (from the torus center to the edge) is given by LENGTHX, whereas LENGTHY determines the tube diameter. LENGTH[X, Y, Z] definitions examples are given in Fig. 2 (lines 5-6), 5 (lines 5-7, 18-20, and 42-43) and 6 (lines 47-48 and 64-65).
[XY, XZ, YZ]PLANE
When a volume compartment has the cuboid geometry, the boundary type or the presence of the [xy, xz, yz]-plane surfaces enclosing the compartment can be specified using [XY, XZ, YZ] PLANE variables. The boundary type can be reflective ('0'), periodic ('1') or semi-periodic ('2'). A semi-periodic boundary allows nonHD molecules to move unidirectionally from one boundary to the other. When a surface compartment is defined to enclose the cuboid compartment, we can remove one or both faces of the cuboid in a given [XY, XZ, YZ]PLANE. To remove the surface on the upper or the lower face of the cuboid in a plane, we can set the variable to '3' or '4', respectively, whereas to remove both faces we can set it to '5'. If the variable is not defined, the boundary type is set to the default reflective ('0') type. Examples in EM and Python to remove both of the cuboid XYPLANE faces are given below:
Variable Variable(XYPLANE) { Value 5; }
theSimulator.createEntity('Variable', 'Variable:.:XYPLANE').Value = 5
ORIGIN[X, Y, Z]
A child volume compartment can be placed at any location within a parent compartment using the variables ORIGIN[X, Y, Z]. The variables define the origin (center) coordinates of the child compartment relative to its parent center point. The variable values '-1' and '1' correspond to the normalized lowest and the highest points of the parent compartment in a given axis, respectively. Since the default value of these variables is '0', the child compartment will be placed at the center of its parent if they are not defined. Fig. 5 (lines 21-24 and 44-46) and 6 (lines 66-68) give some examples of the ORIGIN[X, Y, Z] variables definition.
ROTATE[X, Y, Z]
A compartment can be rotated along the [x, y, z]-axis with the origin at the compartment center using the ROTATE[X, Y, Z] variables respectively. The unit of the variables is in radians. If there are multiple rotation definitions, they follow the [x, y, z]-axis rotation order. Compartments are not rotated if the variables are not defined since their default value is '0'. An example of compartment rotation definition is given in Fig. 5 (line 47).
VACANT
Every compartment must have a VACANT variable that represents the 'species' of empty voxels within the compartment. The VACANT voxels of a surface compartment are analogous to the lipid molecules mentioned in the Spatiocyte Method section and in Arjunan and Tomita (2010).2 Examples of the VACANT variable definition are shown in Fig. 2 (lines 7 and 34), 5 (lines 8, 24, 32, 48 and 53) and 6 (lines 49, 60, 69 and 72). The variable can be used to define sink (e.g., A - > VACANT) and membrane binding reactions (e.g., BV + VACANTS -> BS) of non-HD species, as shown in the EM and Python examples below:
First-Order Sink Reaction, A → Ø
Process SpatiocyteNextReactionProcess(sink) {
VariableReferenceList [_ Variable:/:A -1]
[_ Variable:/:VACANT 1];
k 0.3; }
Second-Order Surface-Adsorption Reaction, Bv + Surface.VACANT → Bs
Process DiffusionInfluencedReactionProcess(bind) {
VariableReferenceList [_ Variable:/:B -1]
[_ Variable:/Surface:VACANT -1]
[_ Variable:/Surface:B 1];
k 2e-8; }
First-Order Sink Reaction, A → Ø
sinker = theSimulator.createEntity('SpatiocyteNextReactionProcess', 'Process:/:sink')
sinker.VariableReferenceList = [['_', 'Variable:/:A', '-1']]
sinker.VariableReferenceList = [['_', 'Variable:/:VACANT', '1']]
sinker.k = 0.3
Second-Order Surface-Adsorption Reaction, Bv + Surface.VACANT → Bs
binder = theSimulator.createEntity('DiffusionInfluencedReactionProcess', 'Process:/:bind')
binder.VariableReferenceList = [['_', 'Variable:/:B', '-1']]
binder.VariableReferenceList = [['_', 'Variable:/Surface:VACANT', '-1']]
binder.VariableReferenceList = [['_', 'Variable:/Surface:B', '1']]
binder.k = 2e-8
For a volume compartment, the Value of the VACANT variable determines if the compartment has a higher occupancy priority when it intersects with a peer compartment. Fig. 9 displays cross-sections of various intersection forms of two spherical peer compartments with different volume and surface VACANT values (listed in Table 1). In the case of a surface compartment, the VACANT variable determines if it fully encloses a parent compartment that has an intersection. A nonzero value indicates that the parent will be fully enclosed even at the location of intersection. Otherwise if the value is '0', the surface will be open at the intersecting region. Fig. 10 shows four possible enclosure forms when a compartment intersects with a root compartment. Fig. 7 illustrates the intersection of various compartments to create a unified neuron-shaped compartment.

Table 1.
Combinations of volume and surface VACANT values and their corresponding intersected peer compartment forms. In all cases X is an integer and the DIFFUSIVE variable is not set .

Figure 10.
Cross-sections of intersected root and child compartment. The VACANT surface voxels of the cuboid root compartment are shown in green while those of the ellipsoid child compartment are in white. The blue molecules belong to the child volume compartment. (more...)
DIFFUSIVE
To unify intersecting compartments, the DIFFUSIVE variable can be specified. It enables nonHD molecules to diffuse into and from an intersecting compartment. The Name property of the DIFFUSIVE variable defines the path and name of the diffusible intersecting compartment. With the DIFFUSIVE variable defined, the VACANT species of the unified compartments become identical. Fig. 5 (lines 49 and 54) gives some examples of the DIFFUSIVE variable definition and usage.
REACTIVE
The REACTIVE variable enables nonHD molecules in a surface compartment to collide and react with the VACANT voxels (i.e., lipids) and nonHD molecules in an adjacent surface compartment. The Name property of the REACTIVE variable specifies the path and name of the reactive adjacent surface compartment. Examples of the REACTIVE variable definition in EM and Python are given below:
Variable Variable(REACTIVE) { Name "/Cell:Surface"; }
theSimulator.createEntity('Variable', 'Variable:/Surface:REACTIVE').Name = "/ Cell:Surface"
SpatiocyteStepper
The SpatiocyteStepper is the only stepper used by Spatiocyte in the E-Cell System and must be defined to run all simulations. It advances the simulation in an event-driven manner. Initialization examples of the SpatiocyteStepper are shown in Fig. 2 (line 1), 5 (line 2) and 6 (line 44). In each compartment, the StepperID must be set to the SpatiocyteStepper ID. Examples of SpatiocyteStepperID definition in compartments are given in Fig. 2 (lines 3 and 32), 5 (lines 4, 16, 30, 40 and 51) and 6 (lines 45, 58, 62 and 70).
VoxelRadius
The radius of the HCP lattice voxels can be set in the SpatiocyteStepper using the VoxelRadius property. The default radius is 10e-9 m. Fig. 2 (line 1), 5 (line 2) and 6 (line 44) show some examples of the VoxelRadius initialization.
SearchVacant
The SearchVacant property of the SpatiocyteStepper provides an option to direct the simulator to search for all adjacent voxels for vacancy during dissociation reactions that result in nonHD product molecules. The reaction can only take place if there is an available target vacant voxel. This option is useful when evaluating the effects of a crowded compartment. The value of SearchVacant by default is true ('1'). To disable it, we can set it to '0'. When disabled, an adjacent target voxel is selected randomly and the reaction is only executed if the voxel is vacant. EM and Python examples of SearchVacant initialization are as follows:
Stepper SpatiocyteStepper(SS) { SearchVacant 0; }
theSimulator.createStepper('SpatiocyteStepper', 'SS').SearchVacant = 0
MoleculePopulateProcess
The initial positions of all nonHD species with nonzero initial molecule numbers must be speci-fied with the MoleculePopulateProcess. The molecules can be either uniformly or normally distributed within the compartment. By default, without any MoleculePopulateProcess parameter definition, molecules are uniformly distributed over the entire compartment. Otherwise if the GaussianSigma is set to a nonzero value, the compartment will be populated according to the Gaussian distribution. MoleculePopulateProcess definitions can be seen in Fig. 2 (lines 26-28), 5 (lines 13-14) and 6 (line 57). A Python example showing two different species populated at the poles of a rod surface compartment is also listed in Fig. 11 with the corresponding output in Fig. 12.

Figure 11.
A python script to populate molecules at the poles of a rod surface compartment. The file is available in the Spatiocyte source package as 2012.arjunan.chapter.populate.py.

Figure 12.
Visualization of molecules populated at the poles of a rod surface compartment. The model is created from the Python script shown in Fig. 11.
Origin[X, Y, Z]
Origin[X, Y, Z] is the origin point relative to the compartment center point for a species population. The molecules may have a uniform or a Gaussian distribution from this point. The range of the point along each axis covering the entire compartment is [-1, 1]. Therefore, the origin is at the center of the compartment if Origin[X, Y, Z] is fixed to [0, 0, 0], the default set of values.
GaussianSigma[X, Y, Z]
GaussianSigma[X, Y, Z] stipulates the sigma value for a Gaussian distributed population from the origin in [x, y, z]-axis, respectively.
UniformRadius[X, Y, Z]
The uniformly distributed normalized population radius from the origin point in [x, y, z]-axis is given by the UniformRadius[X, Y, Z] parameter. Since the default values of UniformRadius[X, Y, Z] and Origin[X, Y, Z] are [1, 1, 1] and [0, 0, 0], respectively, the molecules are spread uniformly within the entire compartment when the parameters are not defined.
ResetTime
To place the molecules at a certain interval after the simulation has started, we can use the ResetTime parameter. This parameter is useful when the positions of a molecule species need to be actively altered after a simulation interval.
DiffusionProcess
The DiffusionProcess handles the voxel-to-voxel random walk of diffusing molecules and the collisions that take place between each walk. Examples of the DiffusionProcess usage are shown in Fig. 2 (lines 11-16 and 39-50), 5 (lines 26-28) and 6 (lines 51-53).
D
In the DiffusionProcess, the diffusion coefficient of the molecule species is set with D, which has the unit m2s-1. The default value is 0 m2s-1.
P
P is an arbitrarily set reaction probability limit of the diffusing species, within the range [0, 1]. The default value is '1', which is sufficient to produce accurate simulations. We can set it to a smaller value to perform reaction-diffusion processes at smaller intervals.
PeriodicBoundaryDiffusionProcess
We can use the PeriodicBoundaryDiffusionProcess in place of the DiffusionProcess when a molecule species needs to be diffused across periodic two-dimensional surface edges. The surface compartment must be enclosing a cuboid parent compartment. The process overcomes the limitation of setting [XY, XZ, YZ]PLANE of the Compartment variable to periodic, which only supports periodic volume edges. It inherits the diffusion coefficient, D and the reaction probability limit, P from the DiffusionProcess. Examples of PeriodicBoundaryDiffusionProcess in EM and Python are as follows:
Process PeriodicBoundaryDiffusionProcess(diffuse) {
VariableReferenceList [_ Variable:/Surface:A];
D 0.2e-12; }
diffuser = theSimulator.createEntity('PeriodicBoundaryDiffusionProcess', 'Process:/:diffuse')
diffuser.VariableReferenceList = [['_', 'Variable:/Surface:A']]
diffuser.D = 0.2e-12
DiffusionInfluencedReactionProcess
The DiffusionInfluencedReactionProcess is used to execute all second-order reactions comprising two diffusing reactants, or a diffusing and an immobile reactant that are diffusion-influenced. Fig. 2 (lines 51-60 and lines 67-69) shows several usage examples of DiffusionInfluencedReactionProcess. A python example of the process definition is provided below:
Second-Order Reaction A + B → C
binder = theSimulator.createEntity('DiffusionInfluencedReactionProcess', 'Process:/:associate')
binder.VariableReferenceList = [['_', 'Variable:/:A', '-1']]
binder.VariableReferenceList = [['_', 'Variable:/:B', '-1']]
binder.VariableReferenceList = [['_', 'Variable:/:C', '1']]
binder.p = 0.5
k
The intrinsic rate constant of the diffusion-influenced reaction is set to k. The relationship between the intrinsic rate constant with the macroscopic rate constant kon is given by 1/kon =1/k + 1/kd, where kd = 4πDR is the maximally diffusion-limited reaction rate, D is the diffusion coefficient and R is the contact radius. The units of k for various reaction types are given in Table 2.
Table 2.
Units of the rate constant, k in DiffusionInfluencedReactionProcess and SpatiocyteNextReactionProcess .
p
The absolute reactive collision probability of the reaction is given by p. This process requires either the value of k or p.
SpatiocyteNextReactionProcess
The SpatiocyteNextReactionProcess is used to execute all reactions that can be decoupled from diffusion such as zeroth- and first-order reactions, and second-order reactions that involve two adjoining immobile reactants or at least one HD reactant. Each reaction is performed according to the Next Reaction method.7 Unlike in the DiffusionInfluencedReactionProcess, the membrane-adsorption reaction where a HD species binds to the membrane is represented as a first-order reac-tion (see example below). EM examples of the SpatiocyteNextReactionProcess are given in Fig. 2 (lines 61-66 and 70-75), while Python examples of zeroth- and first-order (surface-adsorption) reactions are given below:
Zeroth-Order Reaction, Ø → A
zero = theSimulator.createEntity('SpatiocyteNextReactionProcess', 'Process:/:create')
zero.VariableReferenceList = [['_', 'Variable:/:A', '-1']]
zero.k = 0.01
First-Order Surface-Adsorption Reaction, Av → As
uni = theSimulator.createEntity('SpatiocyteNextReactionProcess', 'Process:/:dissociate')
uni.VariableReferenceList = [['_', 'Variable:/:A', '-1']]
uni.VariableReferenceList = [['_', 'Variable:/Surfact:A', '1']]
uni.k = 0.01
k
The rate constant of the event-driven reaction. For second-order reactions, the units are listed in Table 2. For all first-order reactions, the unit is in s-1.
VisualizationLogProcess
We can use the VisualizationLogProcess to log the coordinates of nonHD species at a speci-fied periodic interval. The SpatiocyteVisualizer can load the log file to display the molecules in 3D. Fig. 2 (lines 17-20), 5 (lines 9-12) and 6 (lines 54-56 and 73) show some examples of VisualizationLogProcess usage.
FileName
FileName is the name of the binary log file. The default name is 'visualLog0.dat', which is also the default file name loaded by SpatiocyteVisualizer.
LogInterval
The interval for logging the coordinates is determined by LogInterval. The default value is '0', which means that the interval would be set to the smallest diffusion or collision interval of the logged nonHD species. If LogInterval > 0, the log interval will be set to the specified value. The unit of LogInterval is in seconds.
MicroscopyTrackingProcess
The MicroscopyTrackingProcess mimics the fluorescent microphotography process by logging the trajectory of nonHD molecules averaged over a specified camera exposure time. It inherits the FileName and LogInterval properties from the VisualizationLogProcess. After each LogInterval, the number of times a voxel is occupied by a molecule species is counted. At the end of a given ExposureTime, the frequency is averaged over the total number of intervals and logged. Fig. 2 (lines 21-25) shows an example of the MicroscopyTrackingProcess definition. A Python example is given below:
tracker = theSimulator.createEntity('MicroscopyTrackingProcess', 'Process:/:track)
tracker.VariableReferenceList = [['_', 'Variable:/Surface:MinEE', '2']]
tracker.VariableReferenceList = [['_', 'Variable:/Surface:MinDEE', '3']]
tracker.VariableReferenceList = [['_', 'Variable:/Surface:MinE', '-2']]
tracker.VariableReferenceList = [['_', 'Variable:/Surface:MinDE', '-2']]
tracker.VariableReferenceList = [['_', 'Variable:/Surface:MinE', '-1']]
tracker.FileName = "microscopyLog0.dat"
MicroscopyTrackingProcess enables representation of different fluorescent colored subunits within a complex according to the coefficient assigned to each variable. In the Python example above, the coefficient of the first variable MinEE is 2, representing two subunits of MinE within the complex MinEE. Similarly for MinDEE, the three subunits (one MinD and two MinEs) are represented by the coefficient 3. Each unique variable with a negative coefficient is assigned a different color during visualization. The first negative variable, MinE, has a coefficient of -2, which means that two subunits from the first positive variable, MinEE, are assigned a unique color of MinE. The second negative variable MinDE also has a coefficient of -2, specifying that two subunits of the second positive variable, MinDEE, is assigned the color of MinDE. The third negative variable MinE has a coefficient of -1, corresponding to the color of the remaining one MinE subunit of the second positive variable MinDEE.
ExposureTime
The simulated camera exposure time is specified by ExposureTime. The default value is 0.5 s.
MeanCount
MeanCount is the maximum number of voxel occupancy frequency before it is averaged. The default value is '0', which indicates that the specified LogInterval or the smallest collision or dif-fusion interval should be used. In this case, the MeanCount will be ExposureTime/LogInterval. Otherwise if MeanCount > 0, the LogInterval is set to ExposureTime/MeanCount.
IteratingLogProcess
The IteratingLogProcess executes multiple simulation runs with different random seeds and logs the averaged physical values of molecules, such as their displacement or survival probability, over the total runs. The values are logged in a file using the comma-separated values (csv) format. By default the process logs the number of available molecules of recorded species at the specified interval periodically.
LogDuration
LogDuration is the total duration of a simulation run (i.e., an iteration).
LogInterval
LogInterval is the interval for logging physical values of molecules within an iteration.
Iterations
The number of simulation runs before the logged values are averaged and saved in the log file is specified by the Iterations parameter.
FileName
The file name of the log file is given by FileName. The default file name is "Log.csv".
SaveInterval
When running many iterations, it is useful to save the logged data in a backup file for quick analysis, or to avoid restarting the runs because of some unexpected failures (e.g., power failure). To this end, a backup file of the logged values can be saved at the iteration intervals given by Iterations/SaveInterval. The default value of SaveInterval is '0', which indicates that a backup file will not be saved.
Survival
The Survival parameter can be set to '1' to log the survival probability of a molecule species. The default value of the parameter is '0'.
Displacement
Set the Displacement to '1' to log the displacement of a molecule species. The default value of Displacement is '0'.
Diffusion
If the Diffusion parameter is set to '1', the apparent diffusion coefficient of a molecule species will be logged. The default Diffusion value is '0'.
SpatiocyteVisualizer
The SpatiocyteVisualizer can be started by executing ./spatiocyte in the Spatiocyte directory. Fig. 3 illustrates the SpatiocyteVisualizer interface, whereas its features and keyboard shortcuts are listed in Table 3. To change the color of a species, right mouse click on the species and select a desired color. The visualizer can display each species within a specified range in each axis using the bounding feature. Fig. 8 displays the output after specifying a set of ranges for the cell membrane. Each displayed frame can be saved into the Portable Network Graphics (PNG) image format. A quick way to create a movie from the saved images is to use the ffmpeg program:
$ ffmpeg -i image%07d.png -sameq out.mp4
Table 3.
SpatiocyteVisualizer features and keyboard shortcuts.
Conclusion
Building computational models of biochemical processes is usually a demanding task, especially for experimental biologists without modeling experience. This chapter aims to provide a guide on how one can quickly build and simulate spatially resolved biochemical models with the available Spatiocyte modules. We started with the basic theory of the Spatiocyte method and continued with the installation and simulation procedures. The various modules available to Spatiocyte users were also explained with accompanying model examples.
We plan to continuously develop and improve the Spatiocyte software and user experience. The contents of this guide will also therefore, evolve with the addition of new features and enhance-ments. The latest version of this guide will be available along with the Spatiocyte source code, which at the time of writing, is hosted at GitHub. The Spatiocyte website, http://spatiocyte.org also contains the latest information about the Spatiocyte method and software.
In future, we would like to introduce the ability of subunits to polymerize on the membrane and in the cytoplasm. A polymerization strategy using the HCP lattice was proposed recently.10 Diffusion of compartments, and molecules with different shapes and sizes are also in the future development plan. Parallel implementation of the Spatiocyte method to run on multi-core ar-chitectures and graphics processing units is also being considered. We are also currently working on introducing compartments with complex surface geometries. Spatiocyte users are encouraged to submit feature requests and bug reports, while independent developers can submit their own algorithm modules, code improvements and bug fixes.
References
- 1.
- Takahashi K, Kaizu K, Hu B, et al. A multi-algorithm, multi-timescale method for cell simulation. Bioinformatics. 2004;20(4):538–546. [PubMed: 14990450]
- 2.
- Arjunan SNV, Tomita M. A new multicompartmental reaction-diffusion modeling method links transient membrane attachment of E. coli MinE to E-ring formation. Syst Synth Biol. 2010;4(1):35–53. [PMC free article: PMC2816228] [PubMed: 20012222]
- 3.
- Arjunan SNV, Tomita M. Modeling reaction-diffusion of molecules on surface and in volume spaces with the E-Cell System. IJCSIS. 2009;3(1):211–216.
- 4.
- Dix JA, Verkman AS. Crowding effects on diffusion in solutions and cells. Annu Rev Biophys. 2008;37:247–263. [PubMed: 18573081]
- 5.
- Hall D, Hoshino M. Effects of macromolecular crowding on intracellular diffusion from a single particle perspective. Biophys Rev. 2010;2(1):39–53. [PMC free article: PMC2957576] [PubMed: 21088688]
- 6.
- Collins FC, Kimball GE. Diffusion-controlled reaction rates. J Colloid Sci. 1949;4(4):425–437.
- 7.
- Gibson M, Bruck J. Efficient exact stochastic simulation of chemical systems with many species and many channels. J Phys Chem A. 2000;104(9):1876–1889.
- 8.
- Gillespie DT. A general method for numerically simulating the stochastic time evolution of coupled chemical reactions. J Comput Phys. 1976;22(4):403–434.
- 9.
- Gillespie DT. Exact stochastic simulation of coupled chemical reactions. J Phys Chem. 1977;81(25):2340–2361.
- 10.
- Arjunan SNV. Modeling three-dimensional spatial regulation of bacterial cell division. PhD Thesis, Keio University. 2009
- A Guide to Modeling Reaction-Diffusion of Molecules with the E-Cell System - Mad...A Guide to Modeling Reaction-Diffusion of Molecules with the E-Cell System - Madame Curie Bioscience Database
- Therapeutic Utilities of SOD Mimetics: Cancer, Radiotherapy and SOD Mimetics - M...Therapeutic Utilities of SOD Mimetics: Cancer, Radiotherapy and SOD Mimetics - Madame Curie Bioscience Database
- TRAFs in RANK Signaling - Madame Curie Bioscience DatabaseTRAFs in RANK Signaling - Madame Curie Bioscience Database
- Keratins As Targets in and Modulators of Liver Diseases - Madame Curie Bioscienc...Keratins As Targets in and Modulators of Liver Diseases - Madame Curie Bioscience Database
- Embryonic Salivary Gland Branching Morphogenesis - Madame Curie Bioscience Datab...Embryonic Salivary Gland Branching Morphogenesis - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...