

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Metastasis is the leading reason for the resultant mortality of patients with cancer. The past few decades have witnessed remarkable progress in understanding the molecular and cellular basis of this lethal process in cancer. The current article summarizes some of the key progress in this area and discusses the role of cell junctions, cell adhesions, epithelial-mesenchymal transition, angio and lymphangiogenesis and organ specific metastasis.
Introduction
Of primary importance in the prognosis of cancer patients is the sequence of events leading to the development of tumor cell invasion and metastasis. The course of tumor metastasis entails a series of stages that lead to the formation of secondary tumors in distant organs and is, largely, responsible for the mortality and morbidity of cancer.
Once tumor cells acquire the ability to penetrate the surrounding tissues, the process of invasion is instigated as these motile cells pass through the basement membrane and extracellular matrix, progressing to intravasation as they penetrate the lymphatic or vascular circulation. The metastatic cells then journey through the circulatory system invading the vascular basement membrane and extracellular matrix in the process of extravasation. Ultimately, these cells will attach at a new location and proliferate to produce the secondary tumor. Concentrating research efforts on identifying and understanding the mechanisms concerned in tumor cell invasion may lead to limiting tumor progression and, as a result, to a reduction in mortality for many cancer patients. In the following, we have summarized some of the recent progress in the area of cell adhesion, epithelial to mesenchymal transition, angiogenesis, lymphangiogenesis and organ specific metastasis in cancer.
Cancer Invasion and Metastasis: The Role of Cell Adhesion Molecules
Cancer metastasis is the spread of cancer cells to tissues and organs beyond where the tumor originated and the formation of new tumors (secondary and tertiary foci) is the single event that results in the death of most patients with cancer. At the time of cancer diagnosis, at least half of the patients already present clinically detectable metastatic disease.1 A higher number of patients will also have micrometastases that would be beyond conventional detection techniques. Thus, metastasis is the most life threatening event in patients with cancer. The process is composed of a number of sequential events which must be completed in order for the tumor cell to successfully metastasize, the so called metastatic cascade. This process contributes to the complexity of cancer as a multiplex disease. During the metastatic cascade, changes in cell-cell and cell-matrix adhesion are of paramount importance.2
The metastatic cascade can be broadly separated into three main processes: invasion, intravasation and extravasation. The loss of cell-cell adhesion capacity allows malignant tumor cells to dissociate from the primary tumor mass and changes in cell-matrix interaction enable the cells to invade the surrounding stroma; the process of invasion. This involves the secretion of substances to degrade the basement membrane and extracellular matrix and also the expression/ suppression of proteins involved in the control of motility and migration. The tumor must also initialize angiogenesis, without which the tumor would fail to develop, as local diffusion for transport of nutrients to and removal of waste products from the tumor site would suffice for tumors up to 2 mm in diameter.3 The blood vessel within the tumor's vicinity can then provide a route for the detached cells to enter the circulatory system and metastasize to distant sites; the process of intravasation.4,5 Interaction between the tumor cell and the surrounding stroma is extremely important in the development of tumor angiogenesis.6 Once the tumor cell has arrived at a likely point of intravasation, it interacts with the endothelial cells by undergoing biochemical interactions (mediated by carbohydratecarbohydrate locking reactions, which occur weakly but quickly) develops adhesion to the endothelial cells to form stronger bonds, and thus penetrates the endothelium and the basement membrane; the process of extravasation. The new tumor can then proliferate at this secondary focus.
The metastatic cascade is therefore dependent on the loss of adhesion between cells, which results in the dissociation of the cell from the primary tumor, and subsequently the ability of the cell to attain a motile phenotype via changes in cell to matrix interaction.
Cellular Junctions
Epithelial cells are characterized by a remarkable polarization of their plasma membrane, evidenced by the appearance of structurally, compositionally, and functionally distinct surface domains. The cell to cell adhesion complex runs from the apical to the basal membranes and is composed of Tight Junctions (TJ), Adherens Junctions (AJ), Gap Junctions (GJ), Desmosomes and integrins (Fig. 1).
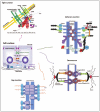
Figure 1.
Schematics showing the arrangement of cell-cell junctions and cell-matrix interactions.
Tight Junctions (TJ)
The permeability of epithelial and endothelial cells is governed by the TJ and they are located at the apical membrane of the cell,7-9 (Fig. 1). The TJ is a region where the plasma membrane of adjacent cells forms a series of contacts that appear to completely occlude the extracellular space thus creating an intercellular barrier and intramembrane diffusion fence.10 In epithelial cells the TJ functions in an adhesive manner and can prevent cell dissociation.11 TJ in endothelial cells function as a barrier through which molecules and inflammatory cells can pass. Interaction with and penetration of the vascular endothelium by dissociated cancer cells is an important step in the formation of cancer metastases. TJ are the first barrier that cancer cells must overcome in order to metastasize. We have previously demonstrated that TJ of vascular endothelium in vivo function as a barrier between blood and tissues against metastatic cancer cells.12 Early studies demonstrated a correlation between the reduction of TJ and tumor differentiation and experimental evidence has emerged to place TJ in the frontline as the structure that cancer cells must overcome in order to metastasize.12-15 Although a considerable body of work exists on TJ and their role in a number of diseases, following the early work of Martinez-Paloma16 and others,17,18 it is only in recent years that there has been an upsurge in studies investigating their possible role in tumorigenesis and metastasis.
There have now been numerous studies on colorectal cancer,19-21 pancreatic cancers22-24 and an increasing number of studies performed on breast cancer.25-27 Changes in both tumor and endothelial cells are necessary for successful growth and spread of cancer cells and these changes are somewhat similar. A change in cancer cells by upregulation or downregulation of relevant TJ proteins results in loss of cellcell association, cell contact inhibition, leading to uncontrolled growth, loss of adhesion to and degradation of the basement. These must be a concurrent loss of cellcell association in the endothelium and modulation of TJ proteins involved in facilitating the passage of the cancer cells through this barrier.
HGF/SF (hepatocyte growth factor), a cytokine secreted by stromal cells and key to the development and progression of cancer, particularly during metastasis has been shown to be capable of modulating expression and function of TJ molecules in human breast cancer cell lines.28 HGF decreased trans-epithelial resistance and increased paracellular permeability of human breast cancer cell lines, MDA-MB-231 and MCF-7. Q-PCR showed that HGF modulated the levels of several TJ molecule (occludin, claudin-1 and -5, JAM-1 and -2) mRNA transcripts in MDA-MB-231 and MCF-7 cells. Such data shows that HGF disrupts TJ function in human breast cancer cells by effecting changes in the expression of TJ molecules at both the mRNA and protein levels and that regulation of TJ could be of fundamental importance in the prevention of metastasis of breast cancer cells. Regulation of vascular permeability is one of the most important functions of endothelial cells, and endothelial cells from different organ sites show different degrees of permeability.29 Tumor blood vessels are more permeable on macro-molecular diffusion than normal tissue vessels. However, the cause and mechanism of hyperpermeability of human vessels had not been clear. Tumor cells release a number of factors that can assist their transmigration through the endothelium after treating endothelial cells with conditioned media from a highly invasive and metastatic melanoma cell line,29 with TJ being irreversibly damaged (as assessed using TER-trans-epithelial resistance). In fact, HGF has been shown to decrease TER and increase PCP (paracellular permeability) in human endothelial cells.8
An increasing number of studies have shown that numerous TJ components are directly or indirectly involved in cancer progression including ZO-1, ZO-2, claudin-7, claudin-1 and occludin.25 When human tissues and breast cancer cell lines were amplified for functional regions of occludin, tumor tissues showed truncated and/or variant signals. There was also considerable variation in the expression of occludin in the 10 human breast cancer cell lines investigated. Western blotting demonstrated that variants in the MDA-MB-231 and MCF-7 human breast cancer cell lines did not fit the expected occludin signals for changes in phosphorylation status. Immunostaining showed similarly disparate levels of expression. Ribozyme knockdown resulted in increased invasion, reduced adhesion and significantly reduced TJ functions. Q-RT-PCR analysis of 124 tumor and 33 background human breast tissues showed occludin to be significantly decreased in patients with metastatic disease. Immunohistochemical staining showed a decreased expression of occludin in the tumor sections. This study demonstrated for the first time that occludin is differentially expressed in human breast tumor tissues and cell lines. This loss of or aberrant expression has clear repercussions as to the importance of occludin in maintaining TJ integrity in breast tissues,25 (Fig. 2). Highly differentiated adenocarcinomas with well developed TJ provide an important insight into the usefulness of TJ molecules and are possible prognostic indicators and future targets for therapy. In breast cancer, ZO-1 has been demonstrated to be decreased in poorly differentiated tumors and correlated with increasing Grade and TNM (tumor-nodal) status.30 There are a respectable number of reports describing the dysregulation of transmembrane proteins in human cancers and in cell lines. This dysregulation can be the result of both upregulation and downregulation of expression, epigenetic changes and changes in activation and location of the proteins.
Adherens Junctions (AJ)
AJ are cellcell microdomains that provide adherent strength and localize to the basal side of the TJ31 (Fig. 1). The integral membrane proteins of the AJ are of the cadherin family, with E-cadherin being most abundant in epithelia and VE-cadherin in endothelia (Fig. 1). Nectins are also found in AJ of epithelia. In polarized epithelia of vertebrates, the AJ is part of the tripartite junctional complex localized at the juxtaluminal region, which comprises the TJ, AJ, and desmosome aligned in this order from the apical end of the junction.32 In this type of epithelia, the AJ is specifically termed the zonula adherens or adhesion belt, as it completely encloses the cells along with the F-actin lining, called the circumferential actin belt.33 The AJs in other cell types assume different morphologies with the AJ in fibroblastic cells being spotty and discontinuous34 while those in neurons are organized into tiny puncta as a constituent of the synaptic junctions.35 A major function of AJs is to maintain the physical association between cells, as disruption of them causes loosening of cellcell contacts, leading to disorganization of tissue architecture.33
Classical or type I cadherins mediate adhesion at the adherens, cellcell or cellmatrix adhesive junctions that are linked to microfilaments. Type I classical cadherins are composed of five tandem extracellular cadherin domains (EC1-EC5), a single segment transmembrane domain and a distinct, highly conserved cytoplasmic tail that specifically binds catenins.36 In addition to cadherin homophilic binding, it has been reported that cadherin is also capable of heterophilic interactions with numerous extracellular and intracellular proteins. The key to their adhesive activity is the interaction between the catenin-binding sequence and submembrane plaque proteins β-catenin or plakoglobin (γ-catenin), which form the link to the actin cytoskeleton. α-catenin binds to a short region close to the N terminus of β-catenin forming a stable bond between the complex and the actin cytoskeleton.36 In addition to α-, β-, and γ-catenin, a fourth catenin-like protein capable of binding cadherin, p120ctn, has emerged as a key regulator of cadherin function.37 p120ctn was originally identified as a substrate for receptor tyrosine kinases and like the other catenin molecules, binds directly to the cytoplasmic domain of cadherin.37
Nectins are transmembrane proteins that are found in both TJ and AJ. In AJ, during the process of early cellcell contacts, nectins first accumulate at the contacts, and then cadherins follow them, suggesting that the former may guide the latter in their junctional localization. Nectin interaction serves for recruiting cadherins to heterotypic cellcell borders, which are otherwise distributed throughout cellcell borders.33 Thus, nectins recruit cadherins to the synaptic contacts formed between two distinct domains of hippocampal neurons, i.e., axons and dendrites, which express nectin-1 and nectin-3, respectively.38 Thus, nectins show important cooperation with classic cadherins in generating heterotypic cellcell contacts.33
Evidence has long accumulated to point toward a pivitol role for E-cadherin and the catenin complex in the control of cancer cell dissociation and spread. Tumor invasion and metastasis, both hallmarks of tumor malignancy, frequently coincide with the loss of E-cadherin-mediated cell-cell adhesion. Expression of E-cadherin, the most abundant adhesion molecule in adherens junctions of epithelia, is downregulated in most, if not all, epithelial cancers.39 Several studies have shown that reconstitution of a functional E-cadherin adhesion complex suppresses the invasive phenotype of many different tumor cell types.40-42 In the context of cancer, E-cadherin has been categorized as a tumor suppressor, given its essential role in the formation of proper intercellular junctions, and its downregulation in the process of epithelial-mesenchymal transition (EMT) in epithelial tumor progression.
Recent studies in triple-negative breast cancer (TNBC), which is characterized by negativity for estrogen receptor, progesterone receptor and human epidermal growth factor receptor 2 (HER2), have shown there is a high risk breast cancer that lacks specific targets for treatment selection. Chemotherapy is, therefore, the primary systemic modality used in the treatment of this disease, but reliable parameters to predict the chemosensitivity of TNBC have not been clinically available.43 Patients with E-cadherin-negative and Ki67-positive expression showed significantly worse overall survival time than those with either E-cadherin-positive or Ki67-negative expression. Multivariate analysis showed that the combination of E-cadherin-negative and Ki67-positive expression was strongly predictive of poor overall survival in TNBC patients receiving adjuvant chemotherapy. The authors demonstrated that adjuvant therapy is beneficial for Stage II TNBC patients and that the combination of E-cadherin and Ki67 status might be a useful prognostic marker indicating the need for adjuvant chemotherapy in Stage II TNBC patients.43
E-cadherin inactivation with loss of cell adhesion is the hallmark of lesions of the lobular phenotype and E-cadherin is typically absent, as seen by immunohistochemistry in both lobular carcinoma in situ and invasive lobular lesions, suggesting it occurs early in the neoplastic process. In invasive lobular lesions, the cadherin-catenin complex was examined; complete complex dissociation was defined as negative membranous E-cadherin, α- and β-catenin expression.44 E-cadherin was found to be absent in all lesions and positive in all normal tissues. Membranous a and β-catenin expressions decreased with the transition from lobular lesions to invasive lesions, while TWIST expression increased. Gene expression paralleled IHC-staining patterns with a stepwise downregulation of E-cadherin, α and β-catenins from normal to lobular to invasive lesions, and increasing expression of TWIST from normal to lobular to invasive lesions. The decreasing membranous catenin expression in tandem with increasing levels of TWIST across the spectrum of lobular lesions suggests that cadherin-catenin complex dissociation is a progressive process in human breast cancer.44
Desmosomes
In cell-cell junctions, desmosomes form adherent points in the form of a continuum of cells within tissues by linkage of their integral membrane proteins (desmocollin and desmoglein) via desmoplakins (plakophilin and plakoglobin) to intermediate filaments31,45 (Fig. 1). Desmosomes are crucial for tissue integrity by their very strong adherence that resists calcium-depletion in developed tissue, but can be regulated by protein kinase C when dynamic remodelling of cellcell adhesion is required.45 Desmosomes not only provide mechanical stability but also facilitate cellcell communication through signal transmission.46 The desmosome is divided into three parallel identifiable zones, arranged symmetrically on the cytoplasmic faces of the plasma membranes of bordering cells and separated by the extracellular domain, which in mature desmosomes is bisected by a dense midline. Each desmosomal plaque consists of a thick outer dense plaque and a translucent inner dense plaque. The five major desmosomal components are the desmosomal cadherins, represented by desmogleins (14) and desmocollins (13), the armadillo family members, plakoglobin and the plakophilins (13), and the plakin linker protein desmoplakin, which anchors the intermediate keratin filaments.46
Recent studies using mouse genetic approaches have uncovered a role for desmosomes in tumor suppression, demonstrating that desmosome downregulation occurs before that of adherens junctions to drive tumor development and early invasion, suggesting a two-step model of adhesion dysfunction in cancer progression.47 Studies have shown that an increased expression of desmosome proteins, such as Desmoglein 2 and 3 and PKP3, can be observed in certain cancers of the skin, head and neck, prostate and lung compared with normal tissue, and that this overexpression is associated with enhanced tumor progression.46,48-50
Reduced expression of Desmocollin 2 has been reported in colorectal carcinomas, suggesting that it may play a role in the development and/or progression of colorectal cancer. Kolegraff et al.51 reported that the loss of Desmocollin-2 promotes cell proliferation and enables tumor growth in vivo through the activation of Akt/β-catenin signaling. Inhibition of Akt prevented the increase in β-catenin-dependent transcription and proliferation following Desmocollin-2 knockdown and attenuated the in vivo growth of Desmocollin-2 -deficient cells. This provides evidence that loss of Desmocollin-2 contributes to the growth of colorectal cancer cells and highlights a novel mechanism by which the desmosomal cadherins regulate β-catenin signaling.51
Oral squamous cell carcinomas and pre-malignant dysplasia can be suβ-classified according to their in vitro replicative lifespan, where the immortal dysplasia and carcinoma subsets have p16(ink4a) and p53 dysfunction, telomerase deregulation and genetic instability and the mortal subset do not. It has been demonstrated that desmosomal proteins exhibit a distinct expression pattern in oral mucosa when compared with epidermis in vivo. Microarray data from a large panel of lines shows that the transcript levels of Desmoglein 2 and Desmocollin2/3 are reduced in immortal dysplasia and carcinoma cells.52 Interestingly, Desmoglein 2 was upregulated. Reduction of Desmoglein 3 and upregulation of Desmoglein 2 were found in two independent microarray data sets. Significantly, we demonstrated that reduction of Desmoglein 3 and upregulation of Desmoglein 2 was reversible in vitro by using RNAi-mediated knockdown of Desmoglein 2 in carcinoma cells. The remaining desmosomal proteins were largely disrupted or internalized and associated with retraction of keratin intermediate filaments in oral squamous cell carcinomas lines. These findings suggest dysfunction and loss of desmosomal components are common events in the immortal class of oral squamous cell carcinomas and that these events may precede overt malignancy.52
There are numerous links between the desmosome and the adherens junction. A decrease in the levels of the desmosomal plaque protein, plakophilin3, leads to a decrease in desmosome size and cell-cell adhesion. Gosavi et al.53 investigated whether plakophilin3 is required for desmosome formation. Plakophilin3 knockdown clones showed decreased cell border staining for multiple desmosomal proteins, when compared with vector controls, and did not form desmosomes in a calcium switch assay. Further analysis demonstrated that plakophilin3, plakoglobin and E-cadherin are present at the cell border at low concentrations of calcium. Loss of either plakoglobin or E-cadherin led to a decrease in the levels of plakophilin3 and other desmosomal proteins at the cell border. The results reported here are consistent with the model that plakoglobin and E-cadherin recruit plakophilin 3 to the cell border to initiate desmosome formation.53
Gap Junctions (GJ)
GJ are unique cell-to-cell channels that allow diffusion of small metabolites, second messengers, ions and other molecules between neighboring cells31 (Fig. 1). GJ communication is essential for electrical transduction, signaling and nutrition. The channels can be open or closed, a highly dynamic process regulated at multiple levels, with the integral membrane proteins forming these channels in vertebrates being the connexins of which over 20 family members have now been identified in humans; connexin43 the most abundantly expressed connexin.31 ZO-1 acts as a scaffold in GJ and recruits signaling proteins. Connexins are also known to interact with Occludin and also form complexes with CAR and β-catenin.54
For decades, cancer was associated with GJ defects. However, more recently it appeared that connexins can be re-expressed and participate in cancer cell dissemination during the late stages of tumor progression. Since primary tumors of prostate cancer are known to be connexin deficient, Lamiche et al.55 investigated whether their bone-targeted metastatic behavior could be influenced by the re-expression of the connexin type (connexin43) which is originally present in prostate tissue and highly expressed in bone where it participates in the differentiation of osteoblastic cells. It appeared that Cx43 behaved differently in those cell lines and induced different phenotypes. In LNCaP, connexin43 was functional, localized at the plasma membrane and its high expression was correlated with a more aggressive phenotype both in vitro and in vivo. In particular, those connexin43-expressing LNCaP cells exhibited a high incidence of osteolytic metastases generated by bone xenografts in mice. Interestingly, LNCaP cells were also able to decrease the proliferation of cocultured osteoblastic cells. In contrast, the increased expression of connexin43 in PC-3 cells led to an unfunctional, cytoplasmic localization of the protein and was correlated with a reduction of proliferation, adhesion and invasion of the cells. In conclusion, the localization and the functionality of connexin43 may govern the ability of prostate cancer cells to metastasize in bones.55
In colorectal tumors, loss of connexin43 expression is correlated with significantly shorter relapse-free and overall survival. Connexin43 was further found to negatively regulate growth of colon cancer cells, in part by enhancing apoptosis and was found to colocalize with β-catenin and reduce Wnt signaling.56 This study represents the first evidence that Cx43 acts as a colorectal cancer tumor suppressor and that loss of Cx43 expression during colorectal cancer development is associated with reduced patient survival. Connexin43 was downregulated or aberrantly localized in colon cancer cell lines and colorectal carcinomas, which is associated with loss of gap junction intercellular communication. Such data indicate that Cx43 is a colorectal cancer tumor suppressor protein that predicts clinical outcome.56
Integrins and Selectins
There is accumulating evidence for the role of integrins and selectins in cancer progression of various cancer types, including colon and lung carcinomas and melanomas.57 While selectin-mediated tumor cells arrest and adhesion contribute to metastasis, integrin-mediated interaction from both tumor cells and the surrounding environment further contribute to cancer progression.
Integrins
Integrins are large and complex transmembrane glycoproteins that consist of two distinct chains, α and β-subunits, which form a non-covalent heterodimer and combine to form 24 unique canonical α/β receptors.57 Integrins mediate cell adhesion and directly bind components of the extracellular matrix, such as fibronectin, vitronectin, laminin, or collagen and provide anchorage for cell motility and invasion. Integrins mediate bidirectional signaling where intracellular signals induce alterations in the conformation.57 Integrins participate in multiple cellular processes, including cell adhesion, migration, proliferation, survival, and the activation of growth factor receptors. As many human tumors originate from epithelial cells, integrins expressed on epithelial cells are generally also present in tumor cells and therefore, integrins have become linked with patient survival and metastatic status. Recent studies have shown that expression of αv integrins is elevated in the prostate cancer stem/progenitor cell subpopulation compared with more differentiated, committed precursors. Van den Hoogen et al.58 examined the functional role of αv integrin receptor expression in the acquisition of a metastatic stem/ progenitor phenotype in human prostate cancer. Stable knockdown of αv integrin expression in PC-3M-Pro4 prostate cancer cells coincided with a significant decrease of prostate cancer stem/ progenitor cell characteristics (α2 integrin, CD44, and ALDH(hi)) and decreased expression of invasion-associated genes Snail, Snail2, and Twist. Consistent with these observations, αv-knockdown strongly inhibited the clonogenic and migratory potentials of human prostate cancer cells in vitro and significantly decreased tumorigenicity and metastatic ability in preclinical models of orthotopic growth and bone metastasis. This indicates that integrin αv expression is functionally involved in the maintenance of a highly migratory, mesenchymal cellular phenotype as well as the acquisition of a stem/progenitor phenotype in human prostate cancer cells with metastasis-initiating capacity.58,59
Lu et al.59 investigated the expression of osteopontin and integrin αv (ITGAV, main receptor of the osteopontin) in laryngeal and hypopharyngeal squamous cell carcinoma and any correlation of the expression quantity with tumor biological behavior. The expression quantity of osteopontin and integrin αv in primary and metastatic carcinomas is significantly higher than in normal tissues. The expression of osteopontin and integrin αv in the well-differentiated group was significantly lower than in moderately and poorly differentiated groups; the expression quantity of osteopontin and integrin αv in groups with lymph node metastasis was significantly higher than in groups without lymph node metastasis. The authors conclude that the expression of osteopontin and integrin αv significantly influenced the differentiation and metastasis of the laryngeal and hypopharyngeal squamous cell carcinoma. Overexpression of both proteins may have contributed to invasion and metastasis of the laryngeal and hypopharyngeal squamous cell carcinoma, and therefore, they both may have value as a target for chemotherapy in laryngeal and hypopharyngeal squamous cell carcinoma treatment.59
Selectins
The selectins: E-selectin, P-selectin, and L-selectin are adhesion molecules that are crucial for binding of circulating leukocytes to vascular endothelium during the inflammatory response to injury or infection. Accumulated evidence indicates that selectins regulate adhesion of circulating cancer cells to the walls of blood vessels.60 Selectin ligands are transmembrane glycoproteins expressed on leukocytes and cancer cells that promote bond formations with selectins to mediate inflammatory processes and selectins and their ligands also participate in signal transduction to regulate diverse cellular functions.60
Haematogenous metastasis of small cell lung cancer is still a poorly understood process and represents the life threatening event in this malignancy.61 In particular, the rate-limiting step within the metastatic cascade is not yet clearly defined although, many findings indicate that extravasation of circulating tumor cells is crucially important as most tumor cells within the circulation undergo apoptosis. If extravasation of small cell lung cancer tumor cells mimics leukocyte-endothelial interactions, small cell lung cancer cells should adhere to E- and P-selectins expressed on the luminal surface of activated endothelium. The adhesion to E- and P-selectin under physiological shear stress with regard to adhesive events, rolling behavior and rolling velocity was determined in the human small cell lung cancer cell lines SW2, H69, H82, OH1 and OH3. OH1 SCLC cells adhered best to recombinant human (rh) E-selectin FC-chimeras and human lung endothelial cells (HPMEC), H82 small cell lung cancer cells adhered best to activated human umbilical vein endothelial cells (HUVEC) under physiological shear stress. As OH1 cells had also produced by far the highest number of spontaneous lung metastases when xenografted into pfp/rag2 mice in previous experiments the findings implicate that adhesion of small cell lung cancer cells to E-selectin is of paramount importance in small cell lung cancer metastasis formation.61
Cell-Matrix Interactions
Controlled interaction between the cells and the extracellular matrix is essential for many processes, including normal development, migration and proliferation.31 Interaction between the cell and the matrix can occur through a number of routes; cell adhesion molecules (CAM) including integrins, selectins, cadherins, the Ig superfamily, CD44 and focal adhesions.
Integrins
Integrin-mediated adhesions to the extracellular matrix are among the first adhesion junctions where bidirectional signaling occurs.31 At the extracellular side integrins bind directly to the extracellular matrix which includes collagen, fibronectin and laminins etc. Cytoplasmic partners include talins, paxillin, focal adhesion kinase and linkage to α-actinin and actin-stress fibers. These focal adhesion complexes control a variety of signaling pathways regulated by the interplay with the extracellular partners. Substantial cross-talk between the diverse cellcell and cellextracellular matrix junctions has been found, and the architecture of the epithelial monolayer is highly regulated by their concerted actions.31
Cell Adhesion Molecules (CAM)
Cell adhesion molecules (CAM) facilitate cellular processes such as cell proliferation, migration, and differentiation and are essential during development and for maintaining the integrity of tissue architecture in adults.62 CAMs include cadherins, integrins, selectins, and the immunoglobulin superfamily (IgSF). In normal tissue, CAM expression is tightly regulated. However, aberrant expression of CAMs disrupts normal cell-cell and cell-matrix interactions and can facilitate tumor formation and metastasis. A number of IgSF members have been identified as biomarkers for cancer progression and have also been associated with metastatic progression in a range of huma tumors.62
CD44
CD44 is a multifunctional cell surface adhesion molecule that is involved in cell-cell and cell-matrix interaction and has been implicated in tumor cell invasion and metastasis. In humans, the CD44 family is encoded by a single gene located on chromosome 11p13 and comprises at least 20 exons. Exons 15, 1618 and 20, are spliced together to form a CD44 transcript that has become known as the standard isoform (CD44s). At least ten exons can be alternatively spliced and inserted into the standard isoform at an insertion site between exons 5 and 16 to give rise to variant isoforms of CD44. Thus, exons 615 are variant exons and are typically identified as v1v10.63 CD44 is the principal ligand for hyaluronic acid (HA), a major component of the extracellular matrix. However CD44 can also bind to other ECM components including collagen, fibronectin, laminin and non-ECM component such as osteopontin and serglycin. CD44 is expressed on a variety of cells and tissues including T- lymphocytes, B-cells, monocytes, granulocytes, erythrocytes, many epithelial cell types; Keratinocytes, chondrocytes, mesothelial and some endothelial cells. It is also expressed in many cancer cell types and their metastases in particular; high molecular weight forms of CD44 show restricted expression in tumors and may correlate with tumor development and metastasis and have potential diagnostic and prognostic value in some cancers. Additionally, it has been shown in experimental models that CD44 can inhibit tumor growth and metastatic spread. Further investigation is still needed but CD44 may yet prove to be a potential target for cancer therapy.63
The importance of non-coding RNA transcripts in regulating microRNA (miRNA) functions, especially the 3' untranslated region (UTR), has been revealed in recent years. Genes encoding the extracellular matrix normally produce large mRNA transcripts including the 3UTR. How these large transcripts affect miRNA functions and how miRNAs modulate the extracellular matrix protein expression are largely unknown. Jeyapalan and Yang64 demonstrated that the overexpression of the CD44 3UTR results in enhanced cell motility, invasion and cell adhesion in human breast carcinoma cell line MDA-MB-231. They also found that expression of the CD44 3UTR enhances metastasis in vivo. Computational analysis indicated that miRNAs that interact with the CD44 3UTR also have binding sites in other matrix encoding mRNA 3UTRs, including collagen type 1α1 (Col1α1) repressed by miR-328 and fibronectin type 1 (FN1) repressed by miR-5123p, miR-491 and miR-671. Protein analysis demonstrated that expression of CD44, Col1a1, and FN1 were synergistically upregulated in vitro and in vivo upon transfection of the CD44 3UTR. The non-coding 3UTR of CD44 interacts with multiple miRNAs that target extracellular matrix properties and thus can be used to antagonize miRNA activities.64
CD44 is also a causal factor for tumor invasion, metastasis and acquisition of resistance to apoptosis. CD44 knockdown using inducible short hairpin RNA (shRNA) significantly reduces cell growth and invasion. Short hairpin RNA against CD44 and pGFP-V-RS-vector was used for knockdown of CD44 expression in SW620 colon cancer cells. Short hairpin RNA against CD44 reduced the expression of CD44. Cell proliferation, migration and invasion were markedly inhibited and apoptosis was increased in shRNA CD44-transfected cells. Knockdown of CD44 decreased the phosphorylation of PDK1, Akt and GSK3β, and β-catenin levels. Decreased phosphorylated Akt led to an increase in phosphorylated FoxO1 and induced cell cycle arrest in the G0-G1 phase and a decrease in the S phase. The levels of Bcl-2 and Bcl-xL expression were downregulated, while the levels of BAX expression and cleaved caspase-3, -8 and -9 were increased. CD44 knockdown by way of shRNA inhibited cell proliferation and induced cell apoptosis which suggests that it could be used as a therapeutic intervention with the anti-survival/pro-apoptotic machinery in human colon cancer.65
Focal Adhesions
Focal adhesion kinase (FAK), a crucial mediator of integrin and growth factor signaling, is a novel and promising target in cancer therapy. FAK resides within focal adhesions which are contact points between extracellular matrix (ECM) and cytoskeleton, and increased expression of the kinase has been linked with cancer cell migration, proliferation and survival.66 Migration is a coordinated process that involves dynamic changes in the actin cytoskeleton and its interplay with focal adhesions. At the leading edge of a migrating cell, it is the re-arrangement of actin and its attachment to focal adhesions that generates the driving force necessary for movement.67 Signaling by the FAK-Src complex plays a crucial role in regulating the formation of protein complexes at focal adhesions to which the actin filaments are attached. Cortactin, an F-actin associated protein and a substrate of Src kinase interacts with FAK through its SH3 domain and the C-terminal proline-rich regions of FAK. Wang et al.67 showed that the autophosphorylation of Tyr(397) in FAK, which is necessary for FAK activation, was not required for the interaction with cortactin, but was essential for the tyrosine phosphorylation of the associated cortactin. At focal adhesions, cortactin was phosphorylated at tyrosine residues known to be phosphorylated by Src. The tyrosine phosphorylation of cortactin and its ability to associate with the actin cytoskeleton were required in tandem for the regulation of cell motility. Cell motility could be inhibited by truncating the N-terminal F-actin binding domains of cortactin or by blocking tyrosine phosphorylation (Y421/466/475/482F mutation). In addition, the mutant cortactin phosphorylation mimic (Y421/466/475/482E) had a reduced ability to interact with FAK and promoted cell motility. The promotion of cell motility by the cortactin phosphorylation mimic could also be inhibited by truncating its N-terminal F-actin binding domains. This suggests that cortactin acts as a bridging molecule between actin filaments and focal adhesions. The cortactin N-terminus associates with F-actin, while its C-terminus interacts with focal adhesions. The tyrosine phosphorylation of cortactin by the FAK-Src complex modulates its interaction with FAK and increases its turnover at focal adhesions to promote cell motility.67
Clinical Considerations
A number of cell adhesion molecules have now become classed as clinical indicators and there is a clear trend toward using them for prognosis or diagnosis. The number of studies identifying these molecules as biomarkers are legion and cannot be thoroughly reviewed here. Some timely examples are as follows: The TJ transmembrane protein claudin-7 has achieved status as a prognostic indicator in invasive ductal carcinoma of the breast68 and is a candidate expression marker for distinguishing chromophobe renal cell carcinoma from other renal tumor subtypes, including the morphologically similar oncocytoma.69 Moreover, decreased claudin-7 correlated with high tumor grade in prostate cancer70 and is able to regulate the expression of prostate specific antigen.71 When considering potential targets for therapy, claudin-1 has been found to act as a cancer invasion/metastasis suppressor in addition to its use as a prognostic predictor and potential drug treatment target for patients with lung adenocarcinoma.72 E-Cadherin and vimentin have now been described predictive markers of outcome among patients with non-small cell lung cancer treated with erlotinib.73
Epithelial-Mesenchymal Transition
Cell Motility
A major factor shaping the metastatic character of cancer cells lies in their motility. Cell motility and migration is crucial to normal development and is a major component of organogenesis, inflammation and wound healing. However, changes in the signaling pathways directing its regulation can lead to the pathological processes of tumor cell invasion and metastasis.
The development and progression of cell motility is orchestrated by a sequence of specific biophysical, interdependent processes involving cytoskeletal modifications, changes in cell-substrate adhesive properties and alterations in the extracellular matrix. Reacting to a stimulus, a cell will commence polarization and extend protrusions in the direction of migration74 which originates with extension of the leading edge by protrusion of lamellipodia and/or filopodia, driven by actin polymerisation and filament elongation, with frequently associated membrane ruffling,75 which extends the cell body to then produce new, distal adhesion sites. Following protrusion, adhesion is instigated between the cell and substratum at the leading edge accomplished largely by integrin and non-integrin receptors binding to specific extracellular matrix protein domains.74,76 Subsequently, actomyosin-mediated contraction of the cell occurs with resultant forward motion of the cell body, initiated by contractile forces being generated at or near the leading edge, coupled with detachment of the trailing edge from the substratum. In addition, the migrating cell secretes the proteases required to break down the extracellular matrix proteins thus providing a pathway for the advancing cell.
Several molecules have been identified as having important roles to play in the signaling processes leading to cell motility/migration, with the associated loss of epithelial characteristics and gain of a migratory and mesenchymal phenotype. Thus, the acquisition of a mesenchymal-like cell phenotype provides one of the major characteristics of metastatic progression of most carcinomas.
Mechanisms of EMT
There is growing acknowledgment that the detachment and escape of cells from the primary tumor mimics the developmental process known as epithelialmesenchymal transition (EMT) (Fig. 3), a dynamic process permitting polarized epithelial cells to go through multiple biochemical and morphological changes enabling them to assume a mesenchymal phenotype with enhanced migratory and invasive capabilities.77-80
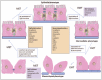
Figure 3.
Schematic description of EMT/MET showing effectors of these processes; dissociation/ association of cell to cell adhesions together with characteristic markers of either epithelial or mesenchymal cells.
Initiation of the process of EMT entails the loss of cell-cell adhesions; activation of transcription factors; alterations in expression of specific cell-surface proteins; reorganization and expression of cytoskeletal proteins; and production of ECM degrading enzymes. Consequently, the course of EMT involves a shift in the characteristic morphology and gene expression pattern of epithelial cells resulting in the acquisition of a characteristic mesenchymal, migratory phenotype.81,82
EMT Progression
Epithelial cells present a highly polarized morphology, intimately linked by cell-cell junctions in the form of TJ, AJ, desmosomes and GJ. Loss of these intercellular connections provides a critical step during EMT allowing for physical detachment of cancer cells from the primary tumor. Thus, EMT is characterized by the combined loss of epithelial cell junction proteins, including E-cadherin, α-catenin, claudins, occludin and ZO-1, an increased expression of mesenchymal markers, such as N-cadherin, vimentin and fibronectin, as well as reorganization of the cytoskeleton, which collectively results in the loss of apical-basal cell polarity and the attainment of a spindle-shaped morphology.77,83
Loss of expression of the cellcell adhesion molecule E-cadherin is a characteristic trait of EMT in development and in the progression of epithelial tumors to invasive, metastatic cancers. The loss of E-cadherin is generally seen to coincide with a gain of expression of the mesenchymal cadherin, N-cadherin in many cancer types; this 'cadherin switch' is thought to be necessary for tumor cells to gain invasive properties and is also a characteristic of EMT.39
It is evident from recent studies that EMT-inducing signals are, in part, initiated by growth factors, including hepatocyte growth factor (HGF), epidermal growth factor (EGF) and transforming growth factor β (TGFβ). These induce downstream activation of a number of EMT-inducing transcription factors including Snail, Slug, Twist and zinc finger E-box binding homeobox 1 (ZEB1).81,84-86
EMT Biomarkers
A number of biomarkers have been found to be useful indicators for EMT (Table 1.).
Table 1.
Biomarkers of EMT .
E-Cadherin
It is essential that weakening of cell-cell adhesion occurs to allow cells to become motile and metastasise and a modification in the adhesive properties of cells is a necessary element of the metastatic process. Cell adhesion molecules (CAMs) regulate cell-cell and cell-matrix adhesion and are implicated in almost all stages of metastasis, therefore alterations in normal levels of CAMs such as E-cadherin will be significant in tumor progression. E-cadherin is a member of a family of Ca2+ dependent CAMs made up of intracellular, extracellular and transmembrane domains. These domains play vital roles in cellular recognition during morphogenesis and development and are responsible for cell-cell adhesion87 thus holding a central role in the maintenance of tissue integrity. E-cadherin and its adhesion complex play an essential function in the adhesion of breast cancer cells, being involved in the control of tumor progression and metastasis. Members of the complex, such as β-catenin, act as regulators of cell adhesion, and also of cell signaling and transcription regulation.88 Studies exploring the expression of E-cadherin and α-catenin in tumor tissues have shown that loss of both molecules is linked to an increased invasiveness of tumor cells.89 Evidence for this comes from in vitro and in vivo studies which demonstrate that E-cadherin expression is inversely correlated with the motile and invasive behavior of tumor cells and also with metastasis in cancer patients.90 Further studies have revealed that the relocalization of β-catenin to the nucleus correlates with the acquisition of the mesenchymal phenotype,91,92 and is associated with the loss of E-cadherin. This reduction of cell surface E-cadherin causes the cells to be receptive to initiation of EMT.93 Numerous reports have indicated that E-cadherin plays a role in meningiomas, tumors of the central nervous system; with upregulation and nuclear localization of β-catenin in 60% of anaplastic memingiomas.94
Transcription Factors in EMT
Important transcription factors shown to be significant in EMT, as they affect the regulation of E-cadherin expression, are Slug and Snail (SNAI1),95 Zeb-185 and Twist.96,97 Importantly, Snail has been identified as having a significant role in the differentiation of epithelial cells into mesenchymal cells during embryonic development98,99 with Slug and Snail effecting the downregulation of E-cadherin expression by binding directly to two proximal E2-boxes of the E-cadherin promoter.84,100 It has been shown that Snail and E-cadherin expression are inversely correlated in squamous cell carcinoma101 and cancer of the breast.102 Snail also represses expression of genes encoding tight junction components, such as claudins and occludins.103
The basic helix-loop-helix protein Twist is also a key transcription factor in EMT and is known to trigger EMT mechanisms possibly by the regulation of the E-cadherin to N-cadherin switch. It is not known if E-cadherin expression can be repressed directly by Twist however, forced N-cadherin expression exerts a dominant effect over E-cadherin in breast cancer cells.104,105 Similarly, expression of N-cadherin in normal epithelial cells results in downregulation of E-cadherin expression.104 Work on glioblastoma (GBM) by Mikheeva et al.106 has shown that TWIST1 promotes GBM invasion through instigation of mesenchymal molecular and cellular changes. This study showed, however, that this effect was not reliant on a cadherin switch as a reduction in levels of E-cadherin and consequent increase in N-cadherin did not occur with TWIST1 overexpression.
Nevertheless many of the genes regulated by TWIST1 in GBM cell lines mirror those which it regulates in cancer metastasis which suggests some overlap with that of TWIST1-mediated EMT in carcinomas.106 In work on medulloblastoma, evidence for a significant role for EMT has been seen with intermittent hypoxic conditions in the tumor microenvironment.107 Hypoxia is recognized as a factor involved in overexpression of the urokinase plasminogen activator (uPA) and its receptor (uPAR) with overexpression promoting uPAR-mediated survival signaling in various cancers.108 Likewise, hypoxia/overexpression of uPAR in cancer cells promotes EMT and thus invasiveness and metastasis. The study by Gupta also showed that when medulloblastoma cells are exposed to intermittent hypoxia this initiates various molecular and phenotypic changes consistent with EMT, as the cell signaling molecules vimentin, N-cadherin, Snail are overexpressed in these medulloblastoma cells with a reduction in the epithelial markers ZO-1 and E-cadherin.
EMT-Related Factors
Bone Morphogenetic Protein (BMP7)
Numerous signaling pathways have been implicated in the initiation of EMT, in particular, TGF-β1 has been identified as a potent initiator of EMT in renal tubular epithelial cells,109 and also in cancer cells, stimulating cell invasion and metastasis.110 However, it has been reported that a member of the TGF-β superfamily, bone morphogenetic protein 7 (BMP-7) reverses TGF-β induced EMT by induction of E-cadherin.111 Indeed, BMP-7 has been shown to regulate epithelial homeostasis in the human mammary gland by preserving the epithelial phenotype.79 Similarly, a decrease in BMP-7 expression in human breast cancer leads to the acquisition of a bone metastatic phenotype,79 with loss of BMP-7 being associated with a more invasive and motile mesenchymal phenotype, in PC-3 prostate cancer cells.112 Furthermore, systemic administration of recombinant BMP-7 to mice with severe renal fibrosis has resulted in reversal of EMT with repair of damaged epithelial structures111 as BMP-7 acts to reverse TGF-β1 induced EMT by upregulating E-cadherin in renal cells. Linked with this, BMP member growth and differentiation factor 9 (GDF-9) has been shown to promote the invasiveness of PC-3 cells together with an induction in the expression of genes including SNAI1, RhoC, ROCK-1 and N-cadherin, while reducing levels of E-cadherin. Thus in PC-3 cells, GDF-9 signaling via ALK-5, promotes cell invasiveness via a complex signaling network working collectively to trigger EMT, thus aiding in the aggressiveness and progression of prostate cancer cells.113
Matrix Metalloproteinases (MMPs)
The matrix metalloproteinases (MMPs) are an important component of cell invasion capable of degrading a range of extracellular matrix proteins allowing cancer cells to migrate and invade. In epithelial ovarian cancer TGFβ and EGF act as inducers of MMP2 production and enhance cell motility,114 while in breast cancer there is an upregulation of MMP9.115
In oral squamous cell carcinoma Snail and Slug are seen to act as regulators of TGFβ triggered EMT, with Snail upregulating MMP2 and MMP9 initiating EMT; while Slug and Snail maintain longer term EMT by stimulating MMP9 expression.116 The MMPs not only function in membrane/ matrix degradation but are also involved in cell adhesion. Treatment of MCF-7 cells with MMP7 results in E-cadherin cleavage producing an 80kDa fraction which is detectable in the serum and urine of cancer patients and has been proposed as a biomarker.117 Similarly, MMP9 appears to cleave the TJ molecule Occludin (personal communication).
Epithelial Protein Lost in Neoplasm (EPLIN)
The cytoskeletal protein EPLIN has been identified as a key molecule linking the cadherin-catenin complex to F-actin and stabilizing the Zona Adherens in MDCK and DLD-1 cells.118 It is an actin cross linking protein that bundles actin in the cells and stabilizes the cytoskeletal filaments. By doing so, EPLIN protein inhibits cell motility, and has been found to be downregulated in a number of oral, breast and prostate cancer cell lines. Forced expression of EPLIN in the EPLIN-α negative breast cancer cell line, MDA MB-231 has been shown to reduce migration and invasion in these cells so reducing their aggressiveness.119 Similarly, overexpression of EPLIN in the PC-3 cell line results in a reduction in both in vivo and in vitro growth potential together with a reduction in cell invasiveness and ability to adhere to extracellular matrix.120
Thus, EPLIN could be seen to be acting as a tumor suppressor. Recently, biochemical and functional evidence has exposed EPLIN as a negative regulator of EMT and invasiveness in prostate cancer cells. Evidence has emerged to show that a downregulation of EPLIN significantly disrupts epithelial structures, initiates actin cytoskeleton remodelling via the EPLIN link between actin filaments and β-catenin, affects explicit gene expression profiles and triggers a pro-EMT program.121
A great deal of energy has been focused, over the last four decades, on the elucidation of the molecular mechanisms governing EMT/MET since the concepts were first defined by Hay (1968).122 Evidence has emerged that the process of EMT can be classified into three different subtypes; type 1 associated with implantation, embryo formation, and organ development; type 2 EMT associated with wound healing, tissue regeneration, and organ fibrosis and type 3 EMT which arises in neoplastic cells in relation to tumor growth and cancer progression, occurring in cells that have gone through epigenetic changes in genes that support the instigation of localized tumors. Many investigators have found that applying the principles of carcinoma EMT to their studies has aided in the understanding of tumor cell invasion in various cancer types and pinpointed many of the genes specifically associated with EMT in relation to tumor growth and metastasis. Continued studies will hopefully provide significantly more information concerning the molecular mechanisms that drive EMT, in relation to the effects of EMT on the progression of carcinomas and will possibly offer new approaches and targets to prevent the most fatal characteristic of tumorigenesis-metastasis.
Angiogenesis and Lymphangiogenesis in Cancer Metastasis
Introduction to Angiogenesis and Lymphangiogenesis
The growth of new blood or lymphatic vessels from pre-existing vessels (the process of angiogenesis or lymphangiogenesis) is essential in physiological events such as reproduction, development, wound-healing and immunity. However, imbalance or manipulation of these essential processes is seen in a number of disease states and these processes are frequently involved in cancer progression and metastasis.123,124
Angiogenic and Lymphangiogenic Cascade
The angiogenic process is made up of a complex multi-step cascade, which is tightly regulated through the balance of a number of pro- and anti- angiogenic factors. Tumor cells frequently tip this balance in favor of blood vessel production through the secretion of pro-angiogenic factors as summarized in Fig. 4. The production of angiogenic factors from a source tissue or tumor bind to and activate endothelial cells of a neighboring blood vessel. Following activation, the endothelial cells begin to produce enzymes that break down the basement membrane of the blood vessel creating tiny pores. Endothelial cells then proliferate and migrate through these pores, toward the angiogenic source, a mechanism that involves a variety of adhesion molecules to aid movement of the new blood vessel toward the source and also the production of various enzymes, such as matrix metalloproteinases, at the sprouting tip, to facilitate this movement through the extrα-cellular matrix. Endothelial cells of the new vessel then undergo a tubule formation phase, where these cells roll to form a tube like structure before establishment of a blood vessel loop between the source and the existing vessel. Finally, structural stabilization of this loop is obtained through recruitment of additional cell types, such as smooth muscle cells, providing support to the vessel and allowing blood flow to the angiogenic source.125
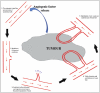
Figure 4.
Summary of key steps involved in the angiogenic cascade
While the vasculature system and lymphatic system are structurally different, the process of lymphangiogenesis shares similarities with the angiogenesis process. New lymphatic vessel growth can be stimulated by a variety of factors such as members of the vascular endothelial growth factor (VEGF) family (e.g., VEGF-C and VEGF-D), which induce sprouting of new vessels and proliferation of lymphatic endothelial cells (LEC),126,127 a process which, similar to angiogenesis, is utilized by metastasising tumor cells. Key angiogenic and lymphangiogenic factors are summaried in Table 2.
Table 2.
Key angiogenic and lymphangiogenic factors .
Therapeutic Potential of Angiogenesis and Lymphangiogenesis in Targeting Cancer Metastasis
While lymphangiogenesis and angiogenesis are essential in numerous physiological processes they are also commonly involved in disease states, in particular the progression of cancer and metastasis.
Angiogenesis and Anti-Angiogenesis Strategies in Cancer
The importance of angiogenesis in advanced tumor development has been known for many years. Without their own vasculature, tumors are unable to grow beyond a size of approximately 23 mm and are limited by their reliance on simple diffusion to obtain required resources.3,128 To overcome this, cancer cells often secrete certain factors to encourage new blood vessel growth to the tumor (tumor angiogenesis). These new blood vessels provide the required resources for advanced and rapid development of the tumor and also provide direct links with the vascular system to the tumor, facilitating metastatic invasion into this system and dissemination around the body.
There are a number of factors that have been demonstrated to enhance angiogenesis such as vascular endothelial growth factor (VEGF) and hepatocyte growth factor (HGF) and, given the importance of tumor angiogenesis in facilitating advanced tumor growth and metastatic spread, research into effective targeting of tumor angiogenesis has been a key area of interest in the scientific community, employing various strategies to disrupt or block new blood vessel growth to the developing tumor.
VEGF is perhaps one of the best known and established angiogenesis regulators to date and given its major role in angiogenesis, it has been subjected to vast scientific study. The VEGF family itself consists of several members, which signal through a number of VEGF receptors, however, the main angiogenesis regulator in normal physiology and cancer appears to be VEGF (also known as VEGF-A) and the VEGF receptor-2 (VEGFR-2 or FLK).129,130 Early research established the importance of VEGF in regulating endothelial proliferation and survival and its ability to promote angiogenesis using in vitro models.131 Given its vital role in tumor angiogenesis, specific targeting of VEGF signaling has been one of the key avenues in developing anti-angiogenic therapies. One such strategy has employed the development and use of a VEGF neutralising antibody termed Bevacizumab (also known as Avastin). This therapy has been approved for use in a variety of cancer types, such as non-squamous non-small-cell lung cancer and colorectal cancer.130 Scientific research into the benefits of Bevacizumab is ongoing, with studies examining and demonstrating the potential of Bevacizumab in additional cancer types such as epithelial ovarian cancer, where previous trials have yielded promising results.132
HGF represents another potential target for the treatment of cancer progression and angiogenesis. The role of HGF in contributing to cancer progression has been well demonstrated within the literature. This is largely due to the ability of HGF to promote pro-metastatic traits such as motogenesis, morphogenesis, mitogenesis and angiogenesis.133 HGF has the capacity to enhance angiogenesis both directly and in-directly, either through its motogenic or morphogenic effects on endothelial cells or through its capacity to enhance other pro-angiogenic factors such as VEGF and its receptor.133,134 Several earlier studies conducted in our labs have highlighted the potential anti-angiogenic application for targeting HGF. HGF treatment in vivo was found to enhance the expression of tumor endothelial markers (TEMs) in tumors obtained from the inoculation of PC-3 prostate cancer cells into CD1 athymic nude mice. However, the addition of NK4, a HGF antagonist, to the treatment was able to prevent the elevation of these TEMs in the tumors.135 Similarly, in a breast cancer in vivo model HGF treatment was found to enhance vessel formation in tumors arising from MDA-MB-231 inoculation into CD1 athymic nude mice using immunohistochemical staining (IHC) analysis of resulting tumor tissues. In keeping with its role, addition of NK4 again prevented the enhanced angiogenesis seen in HGF treatment groups.136 In both studies HGF treatment caused enhanced tumor development, whereas co-treatment could suppress these increases in tumor growth.135,136
Given its involvement in the processes of angiogenesis and tumor progression, inhibitors to the cMET tyrosine kinase receptor of HGF have been developed as treatment regimes. Strategies such as Foretinib, an oral multikinase inhibitor targeting a variety of proteins including cMET and the VEGF receptor have been developed and are being assessed for their efficacy.137
Lymphangiogenesis and Anti-Lymphangiogenesis in Cancer
The area of lymphangiogenesis and the potential of anti-lymphangiogenic therapies in the treatment of cancer has been somewhat over-shadowed by research into anti-angiogenic strategies and the relative lack of pro-lymphangiogenic markers. However, the last 15 -20 years has seen the identification of lymphangiogenic markers and markers of lymphatic endothelial cells, such as lymphatic vessel endothelial hyaluronan receptor-1 LYVE-1138 and vascular endothelial growth factor receptor-3 (VEFGR-3).139 Studies such as these have aided in the progression of this field of research and demonstrated its importance in cancer metastasis.
Lymphatic metastases are common, with a number of cancers first metastasising to regional lymph nodes. The determination of lymph node involvement is an important factor in determining the aggressive nature of a particular cancer, with lymphatic metastasis commonly being associated with a poorer patient outlook.140 Scientific research, examining the role of VEGF-C and D in mouse models has demonstrated the potential of these factors to enhance tumor lymphatics and promote metastatic spread of tumor cells.141,142 In keeping with this, a number of recent studies have reported the association of lymphatic factors such as VEGF-C and D and the VEGFR3 receptor with lymph node metastasis and patient survival.143-145 Taken together, these studies highlight the importance of tumor lymphangiogenesis in cancer spread and survival and demonstrate the potential for anti-lymphatic therapies, targeting factors such as VEGF-C, -D or the VEGFR3 receptor, to limit cancer spread and enhance survival rates.
In summary, anti-angiogenesis and anti-lympangiogenesis therapies hold great potential in combating the ongoing problem of cancer metastasis and the poor survival rates associated with cancer spread. Research and development of drugs in this area have so far begun to yield positive results with therapies such as Bevacizumab being implemented in the treatment of several cancer types. However, resistance to these anti-angiogenic strategies are possible and thus further research into new and multi target inhibitors of angiogenesis and lymphangiogenesis is essential in the ongoing fight against cancer spread.
Organ Specific Metastasis
Cancer metastases are responsible for the majority of cancer-related deaths. From a primary tumor to a distant site and eventually developing a secondary tumor, cancerous cells need to proceed along a series of interrelated and sequential steps, including invasion through extracellular matrix, intravasation, survival in the circulation, extravasation into a distant site, and progressive growth at that site. The metastatic procedure is an inefficient process whereby the vast majority of circulating tumor cells are not able to progressively grow at distant sites. A latent period may exist between infiltration of cancer cells at a distant site and colonization leading progressively to the growth of a secondary tumor. Such a period can be as long as a couple of years seen in some metastases of breast cancer after initial management, and it can also be as short as a few months in lung cancer which may develop a metastasis rapidly within a few months of diagnosis. The cellular origin, intrinsic properties of the tumor, tissue affinities and circulation patterns determine not only the sites of tumor spread, but also the temporal course and severity of metastasis to vital organs. In addition to the above aspects of metastases, certain metastatic cells exhibit tissue tropism, preferring to grow in certain organs (Table 3). In breast cancer, for example, metastasis affects the bone and the lung, and less frequently the liver, brain, and adrenal medulla. Although the genetic and epigenetic basis of these metastatic properties is yet to be fully established, acquisition of the ability to complete each step involved in metastasis is thought to be driven by the accumulation of genetic mutations and epigenetic events that may result in a cells acquisition of metastatic traits during the process of developing a secondary tumor.
Table 3.
Common metastatic sites of certain solid tumors .
The organs mostly assaulted by metastases are lung, liver, brain and bone146 (Fig. 5). The lungs are the commonest site of metastases for many primary tumors. However, there is a great difference in propensity between the malignancies. It is just as high as 90% in melanomas at autopsy. The lungs serve as first filter for tumor cells spreading through blood circulation in malignancies whose venous drainage flows directly into the lungs. The tumors of testis, melanoma, osteosarcoma, and head and neck tumors have the highest incidence of pulmonary metastases.146 The liver is one of the most common sites for metastatic disease, accounting for 25% of all metastases to solid organs.147 In the United States and Europe, secondary liver neoplasms are far more common than primary hepatic neoplasms. In the adult oncology patient, most are metastatic carcinomas, of which adenocarcinomas are the predominant subtype, followed by squamous cell carcinomas and neuroendocrine carcinomas. Other tumor types that metastasize to the liver include melanomas, lymphomas, and rarely sarcomas. The most frequent metastasis to the brain occurs in patients with lung, breast, melanoma, renal, and colorectal tumors.148 In 2700 cases from the Memorial Sloan-Kettering Cancer Center in New York, the distribution of primary cancers was as follows: 48% lung, 15% breast, 9% melanoma, 1% lymphoma (mainly non-Hodgkin), 3% GI (3% colon and 2% pancreatic), 11% genitourinary (21% kidney, 46% testes, 5% cervix, 5% ovary), 10% osteosarcoma, 5% neuroblastoma, and 6% head and neck tumor . Once metastasis to the brain is diagnosed, the median survival of untreated patients is 12 mo. Bone metastases are most commonly seen in prostate, breast and lung cancer, which are leading malignancies in female and/or male having the highest incidence and mortality rates.149-151 Bone metastasis usually leads to severe morbidities, which always persist until the death of patients, including bone pain, hypercalcemia, pathological fracture, spinal cord compression and consequent paralysis. In the following part, we generally reviewed the process and molecular mechanisms of organ specific metastases with a focus on bone metastasis.
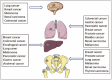
Figure 5.
Organ specific metastases from primary tumors.
Metastatic Course, Routes and Steps
At an early stage, cancerous cells are confined to the primary site within the boundary of certain surrounding tissues. As the disease progresses, some cancer cells, as the result of genetic/ epigenetic predisposition, environmental interaction/stimulation, and indeed the combination of these elements, become more aggressive and begin to breach the surrounding structure. These cells would either directly invade the surrounding tissue, or disseminate via lymphatic and hematogenous routes. Direct invasion may result in the spreading of cancer cells to surrounding tissues and neighboring organs. For example, the local invasion of prostate cancer, can affect the erectile nerves, seminal vesicles, bladder and rectum nearby the prostate. The lymphatic and vascular routes differ from cancer to cancer according to their primary sites, however, frequently result in the systemic spread of cancer cells to distant organs, including bones, lung, and liver. For example, the primary lymphatic drainage of the prostate is via the internal iliac, perivesical, external iliac, obturator, and presacral nodes. The secondary lymphatic drainage includes the inguinal, common iliac and parα-aortic nodes. These nodes are therefore prime locations when one searches for the involved positive lymph nodes. Since the end of last century, a new technique, sentinel lymph node dissection has been developed and introduced in the detection, staging and management of lymph node involvement in cancer. The detection of a positive sentinel node indicates the need for a wide dissection of lymph nodes during surgery.
Both lymphatic and hematogenous dissemination frequently occur, even during early stages of the disease, and are seen in a vast majority of the patients who have an advanced cancer. To determine if systemic spread 'occurred' or not is a highly controversial topic, a conclusion of which is dependent on a wide variety of factors, from the type of samples to test, location and timing of sampling, techniques to detect cancer cells, to the interpretation of the presence of cancer cells or a cancer cell in a sample. Nonetheless, brain, bone, lung and liver are the most leading hematogenous sites from certain solid tumors.152-155
The process of metastasis is complex and arduous, which incorporates multiple cells, factors and stages. During the development and progression of primary tumors, certain clones of tumor cells will have the required genotypic and phenotypic characteristics to enable themselves to interact with the local microenvironment. For example, tumor cells release VEGF to initiate angiogenesis, thus enhancing the blood supply to the tumor. The stromal cells are rich sources of protein factors that directly act on cancer cells thus driving the growth of tumors and dissemination of cancer cells. On the other hand, some of the stromal cell derived factors will directly induce angiogenesis thus supporting the growth and spread of an aggressive tumor. A good example of these stromα-derived protein factors is hepatocyte growth factor (HGF), a cytokine secreted by the stroma cells, which has been implicated in the angiogenesis and the dissemination of tumor cells.133 The disruption of intercellular adhesion in the tumor causes some tumor cells to detach from the tumor mass (detachment), followed by these cells invading through the extracellular matrix, a process so-called invasion which incorporates the motility, migration of tumor cells and breakdown of extracellular matrix. Some tumor cells will penetrate the blood vessels, thus entering the circulation (intravasation). From this point, these tumor cells move away from the primary site and circulate in the blood circulation where, they would encounter resistance by the immune system and the mechanical stresses of blood flow. Some tumor cells will eventually survive and adopt a process to leave the blood circulation, known as extravasation, in which cells adhere and penetrate the blood vessel again (a virtual reversal of the intravasation process). Once the tumor cells escape from the circulation, they will have to survive and finally develop a secondary tumor at the other site, in this case in bone. This complex process also needs the integration of multiple factors and events, such as invasion of tumor, angiogenesis and the interaction between tumor cells and the local microenvironment at a distant site/organ.
Metastasis Regulators
The interrelated and sequential multi-steps of metastasis require certain transformations of cancer cells at each step, from primary site to metastatic site. Numerous genes and molecules have been implicated into this dynamic and adaptable evolution of metastatic cancer cells, including suppressors and promoters of metastasis which may be altered genetically or epigenetically in accordance with the requirements at each step. Initiating factors for tumor progression and metastasis are critical and essential, particularly for dissociation and invasion which allow cancer cells to leave primary sites. The genes that determine these activities have been defined as metastasis initiation genes.156,157 These genes could promote cell motility, epithelial mesenchymal transition (EMT), extracellular matrix degradation, angiogenesis or evasion of the immune system. For example, EMT is mediated by developmental programmes that are under the control of aberrantly regulated transcription factors, such as Twist1, Snai1 and Snai2 (also known as Slug). Other determinants of invasion are components and modulators of certain pathways which include hepatocyte growth factor (HGF), VEGF and ERK pathways. Metastatic growth is also initiated by the suppression of non-coding RNAs, such as miR-126 and miR-335 in breast and gastric carcinomas.158,159 Some of the initiating factors that allow transformed cells to invade the surrounding tissue and attract a supportive stroma facilitate the dissemination of cancer cells and probably continue to do so after cancer cells infiltrate distant tissues. This is why some prognosis signatures of a malignancy can also be utilized as a signature to predict metastases.153
Metastasis suppressor genes are defined by their ability to inhibit metastasis at any step of the metastatic cascade. These metastasis suppressor genes inhibit metastasis of cancer cells, in vivo, without blocking tumorigenicity. To date, some metastasis suppressor genes have been identified, such as nonmetastatic gene 23 (NM23), Kangai 1 (KAI1), KISS1, mitogen-activated protein kinase 4 (MKK4), breast cancer metastasis suppressor 1 (BRMS1), Rho GDP dissociation inhibitor 2 (RhoGDI2), cofactor required for Sp1 transcriptional activation subunit 3 (CRSP3) and Vitamin D3 upregulated protein 1 (VDUP1). Deregulation of these metastasis suppressor genes has been indicated in certain solid tumors.160-162
'Seeds' and 'Soil' Crosstalk between Cancer Cells and the Microenvironment during Bone Metastasis
Bone metastasis has been characterized as either osteolytic or osteoblastic. This classification actually represents two extremes of a continuum in which dysregulation of the normal bone remodelling process occurs. Patients can have both osteolytic and osteoblastic metastases or mixed lesions containing both elements. Most metastatic bone tumors from breast cancer have predominantly osteolytic lesions. In contrast, the metastatic lesions from prostate cancer are predominantly osteoblastic. During osteoblastic bone metastases, the balance between bone resorption and bone formation is tipped in favor of the latter. Patients suffer severe bone pain and the poor quality of bone produced in osteoblastic bone metastases frequently leads to bone fractures. Models to investigate osteoblastic metastases are rather rare, compared with models of osteolytic metastasis. Mechanisms, by which a metastatic lesion becomes osteoblastic or osteolytic remain unclear. However, a number of factors produced by cancer cells, such as platelet-derived growth factor (PDGF), insulin-like growth factors (IGFs), fibroblast growth factors (FGFs), VEGF, Wingless and NT-1 (WNT1), parathyroid hormone related protein (PTHrP), urokinase-type plasminogen activator (uPA), prostate specific antigen (PSA), endothelin-1 (ET-1) and BMPs, have been implicated in osteoblastic lesions.
The question of why the bone is the most preferred metastatic site of some solid tumors (breast, prostate and lung cancer) has aroused intense interest. One would first contemplate the anatomical characteristics of the organs at primary sites. The blood supply to the organs may provide a shortcut for the hematogenous dissemination of tumor cells from primary tumor to certain bones. For example, a rich venous plexus surrounds the prostate and connects to the venous drainage of the spine: this collection of veins (Batson's plexus) is potentially one of the reasons why the lumbosacral spinal metastases are common in advanced prostate cancer.163 However, the anatomical explanation is not able to explain why the other axial skeleton, skull and ribs may also be involved in the bone metastasis from prostate cancer.
The 'seed and soil' theory proposed by Paget may provide some clue from a different standpoint.164 Osteotropic 'seeds' (tumor cells) may be developed during the progression of prostate cancer. These tumor cells may have acquired specific genetic phenotype, or activation of specific cytokine and proteases. These features direct the metastasis to bone. For example, elevated expression of BMPs and TGF-β in prostate cancer cells have been implicated in bone metastasis.165-168 The “seeds” may also attach to the bone endothelium more effectively than to the endothelia of other organs.169 It has been suggested that the protease-activated receptor (PAR1, thrombin receptor) and integrin αVβ3 which are highly expressed in primary prostate cancer cell lines and metastatic prostate cancer cells derived from bone metastasis, may contribute to the bone metastases through facilitating the attachment of tumor cells to blood vessel walls and the process of extravasation.170-173 The vascular endothelial growth factor (VEGF) secreted by the tumor cells may also contribute to the bone metastasis due to both the promotion of angiogenesis and the activation of osteoblasts.174-176
On the other hand, bone also provides a fertile “soil” for the “seeds”. The bone matrix synthesized by osteoblasts has a particular abundance of cytokines and non-collagen proteins, which may attract prostate cancer cells and allow them to survive and proliferate in the bone matrix. For example, BMPs and TGF-β enriched in bone matrix can facilitate the development of bone metatstasis. Osteonectin, osteopontin, osteocalcin, and bone sialoprotein can also modulate the properties of prostate cancer cells and facilitate the spreading and growth, including promoting their migration, invasion and proliferation.177-182 Bone turnover, as a characteristic of the adult bone, occurs most often in the bones rich in trabecular bone, such as the vertebrae, proximal femur, calcaneous, and ultradistal radius. During the bone turnover, cytokines and NCPs released or synthesized through bone resorption and bone formation thus generate a fertile 'soil'. This may supplement the explanation of the favorite locations in bone metastases.
During the development of bone metastasis from prostate cancer, the interactions among tumor cells, bone cells and bone matrix constitute a “vicious cycle” of osteoblast/ osteoclast-mediated bone metastasis. For example, during the osteoblastic bone metastases of prostate cancer, cancer cells produce osteogenic factors such as ET-1, BMPs and PDGF, to activate osteoblasts. The osteoblasts differentiated from their progenitor cells deposit new matrix for bone formation. However, this unmineralised new matrix provides a more fertile soil to tumor cells, which is enriched with growth factors and NCPs. These factors help prostate cancer cells survive and proliferate in the bone microenvironment. The prostate cancer cells then further activate osteoblasts. In addition to this vicious cycle, at certain stages, both tumor-derived factors and osteoblasts expressing RANKL can activate osteoclasts, leading to some level of bone resorption, and subsequently generate bigger space for dominant osteoblastic lesion. The cytokines and NCPs released from bone matrix during bone resorption can also enhance this “vicious cycle” through facilitating proliferation of both prostate cancer cells and osteoblasts.
Conclusion
Metastasis, the leading cause of mortality in patients with cancer, is receiving increasing attention in both scientfic and clinical research. Yet the mechanisms remain poorly understood and methods in combatting metastasis remain limited. It is however pleasing to observe some of the major progresses in this vital area of cancer research. With the increasing knowledge in gene expression, cellular behavior, biological events in the spread paths of cancer cells, there are now new prospects of taking some of the observations into the diagnosis, prognosis and treatment in the metastatic disease. For example, new knowledge on barrier function and paracelluar permeability may allow one to devise new direction in controlling the trepassing cancer cells and their entry into the destination tissues and organs. New biomarkers in areas such as epithelial to mesenchymal transtion offer new opportunities in predictive methods of metastatic potential of a primary tumor and new target for therapy. Angiogenesis has already been a fruitful area in new therapies and the organ specific spread of a solid tumor may allow new method of detection and a new way of targeting metastatic tumor cells. Although enormous challenges remain, it is anticipated that these lines of research will steadily find their into clinical practice.
Acknowledgment
The authors wish to thank Cancer Research Wales, the Albert Hung Foundation, the Breast Cancer Hope Foundation, and the Welsh Assembly Government for supporting their work.
References
- 1.
- DeVita VT Jr., Young RC, Canellos GP. Combination versus single agent chemotherapy: a review of the basis for selection of drug treatment of cancer. Cancer. 1975;35:98–110. http://dx.doi.org/10.1002/1097-0142(197501)35:1<98::AID-CNCR2820350115>3.0.CO;2-B . [PubMed: 162854]
- 2.
- Martin TA, Jiang WG. Loss of tight junction barrier function and its role in cancer metastasis. Biochim Biophys Acta. 2009;1788:872–91. http://dx.doi.org/10.1016/j.bbamem.2008.11.005 . [PubMed: 19059202]
- 3.
- Brooks PC. Cell adhesion molecules in angiogenesis. Cancer Metastasis Rev. 1996;15:187–94. http://dx.doi.org/10.1007/BF00437471 . [PubMed: 8842490]
- 4.
- Folkman J, Shing Y. Angiogenesis. J Biol Chem. 1992;267:10931–4. [PubMed: 1375931]
- 5.
- Folkman J. Fighting cancer by attacking its blood supply. Sci Am. 1996;275:150–4. http://dx.doi.org/10.1038/scientificamerican0996-150 . [PubMed: 8701285]
- 6.
- Ono M, Torisu H, Fukushi J, Nishie A, Kuwano M. Biological implications of macrophage infiltration in human tumor angiogenesis. Cancer Chemother Pharmacol. 1999;43(Suppl):S69–71. http://dx.doi.org/10.1007/s002800051101 . [PubMed: 10357562]
- 7.
- Jiang WG, Bryce RP, Horrobin DF, Mansel RE. Regulation of tight junction permeability and occludin expression by polyunsaturated fatty acids. Biochem Biophys Res Commun. 1998;244:414–20. http://dx.doi.org/10.1006/bbrc.1998.8288 . [PubMed: 9514943]
- 8.
- Jiang WG, Martin TA, Matsumoto K, Nakamura T, Mansel RE. Hepatocyte growth factor/scatter factor decreases the expression of occludin and transendothelial resistance (TER) and increases paracellular permeability in human vascular endothelial cells. J Cell Physiol. 1999;181:319–29. http://dx.doi.org/10.1002/(SICI)1097-4652(199911)181:2<319::AID-JCP14>3.0.CO;2-S . [PubMed: 10497311]
- 9.
- Tsukita S, Furuse M. Occludin and claudins in tight-junction strands: leading or supporting players? Trends Cell Biol. 1999;9:268–73. http://dx.doi.org/10.1016/S0962-8924(99)01578-0 . [PubMed: 10370242]
- 10.
- Wong V, Gumbiner BM. A synthetic peptide corresponding to the extracellular domain of occludin perturbs the tight junction permeability barrier. J Cell Biol. 1997;136:399–409. http://dx.doi.org/10.1083/jcb.136.2.399 . [PMC free article: PMC2134825] [PubMed: 9015310]
- 11.
- Hollande F, Blanc EM, Bali JP, Whitehead RH, Pelegrin A, Baldwin GS, et al. HGF regulates tight junctions in new nontumorigenic gastric epithelial cell line. Am J Physiol Gastrointest Liver Physiol. 2001;280:G910–21. [PubMed: 11292600]
- 12.
- Martin TA, Mansel RE, Jiang WG. Antagonistic effect of NK4 on HGF/SF induced changes in the transendothelial resistance (TER) and paracellular permeability of human vascular endothelial cells. J Cell Physiol. 2002;192:268–75. http://dx.doi.org/10.1002/jcp.10133 . [PubMed: 12124772]
- 13.
- Ren J, Hamada J, Takeichi N, Fujikawa S, Kobayashi H. Ultrastructural differences in junctional intercellular communication between highly and weakly metastatic clones derived from rat mammary carcinoma. Cancer Res. 1990;50:358–62. [PubMed: 2295075]
- 14.
- Satoh H, Zhong Y, Isomura H, Saitoh M, Enomoto K, Sawada N, et al. Localization of 7H6 tight junction-associated antigen along the cell border of vascular endothelial cells correlates with paracellular barrier function against ions, large molecules, and cancer cells. Exp Cell Res. 1996;222:269–74. http://dx.doi.org/10.1006/excr.1996.0034 . [PubMed: 8598213]
- 15.
- Hoevel T, Macek R, Mundigl O, Swisshelm K, Kubbies M. Expression and targeting of the tight junction protein CLDN1 in CLDN1-negative human breast tumor cells. J Cell Physiol. 2002;191:60–8. http://dx.doi.org/10.1002/jcp.10076 . [PubMed: 11920682]
- 16.
- Martinez-Paloma A. Ultrastructural modifications of intercellular junctions in some epithelial tumors Lab Invest 1970. 22 605 14. [PubMed: 4193941]
- 17.
- Inoue T, Shimono M, Yamamura T, Saito I, Watanabe O, Kawahara H. Acinic cell carcinoma arising in the glossopalatine glands: a report of two cases with electron microscopic observations. Oral Surg Oral Med Oral Pathol. 1984;57:398–407. http://dx.doi.org/10.1016/0030-4220(84)90159-2 . [PubMed: 6584836]
- 18.
- Mullin JM, O'Brien TG. Effects of tumor promoters on LLC-PK1 renal epithelial tight junctions and transepithelial fluxes. Am J Physiol. 1986;251:C597–602. [PubMed: 3464217]
- 19.
- Bornholdt J, Friis S, Godiksen S, Poulsen SS, Santoni-Rugiu E, Bisgaard HC, et al. The level of claudin-7 is reduced as an early event in colorectal carcinogenesis. BMC Cancer. 2011;11:65. http://dx.doi.org/10.1186/1471-2407-11-65 . [PMC free article: PMC3045986] [PubMed: 21310043]
- 20.
- Nakagawa S, Miyoshi N, Ishii H, Mimori K, Tanaka F, Sekimoto M, et al. Expression of CLDN1 in colorectal cancer: a novel marker for prognosis. Int J Oncol. 2011;39:791–6. [PubMed: 21720707]
- 21.
- Wang X, Tully O, Ngo B, Zitin M, Mullin JM. Epithelial tight junctional changes in colorectal cancer tissues. ScientificWorldJournal. 2011;11:826–41. http://dx.doi.org/10.1100/ tsw.2011.86 . [PMC free article: PMC5720014] [PubMed: 21479352]
- 22.
- Takai E, Tan X, Tamori Y, Hirota M, Egami H, Ogawa M. Correlation of translocation of tight junction protein Zonula occludens-1 and activation of epidermal growth factor receptor in the regulation of invasion of pancreatic cancer cells. Int J Oncol. 2005;27:645–51. [PubMed: 16077912]
- 23.
- Zhou L, Tan X, Wang W, Wang B, Dai X, Liu J. Analysis of invasion-metastasis in pancreatic cancer: Correlation between the expression and arrangement of tight junction protein-2 and cell dissociation in pancreatic cancer cells. Mol Med Report. 2010;3:149–53. [PubMed: 21472214]
- 24.
- Kojima T, Takasawa A, Kyuno D, Ito T, Yamaguchi H, Hirata K, et al. Downregulation of tight junction-associated MARVEL protein marvelD3 during epithelial-mesenchymal transition in human pancreatic cancer cells. Exp Cell Res. 2011;317:2288–98. http://dx.doi.org/10.1016/j. yexcr.2011.06.020 . [PubMed: 21763689]
- 25.
- Martin TA, Mansel RE, Jiang WG. Loss of occludin leads to the progression of human breast cancer. Int J Mol Med. 2010;26:723–34. http://dx.doi.org/10.3892/ijmm_00000519 . [PubMed: 20878095]
- 26.
- Myal Y, Leygue E, Blanchard AA. Claudin 1 in breast tumorigenesis: revelation of a possible novel “claudin high” subset of breast cancers. J Biomed Biotechnol. 2010;2010:956897. http://dx.doi.org/10.1155/2010/956897 . [PMC free article: PMC2871677] [PubMed: 20490282]
- 27.
- Wu Q, Liu Y, Ren Y, Xu X, Yu L, Li Y, et al. Tight junction protein, claudin-6, downregulates the malignant phenotype of breast carcinoma. Eur J Cancer Prev. 2010;19:186–94. http://dx.doi.org/10.1097/CEJ.0b013e328337210e . [PubMed: 20215972]
- 28.
- Martin TA, Watkins G, Mansel RE, Jiang WG. Hepatocyte growth factor disrupts tight junctions in human breast cancer cells. Cell Biol Int. 2004a;28:361–71. http://dx.doi. org/10.1016/j.cellbi.2004.03.003 . [PubMed: 15193279]
- 29.
- Utoguchi N, Mizuguchi H, Dantakean A, Makimoto H, Wakai Y, Tsutsumi Y, et al. Effect of tumour cell-conditioned medium on endothelial macromolecular permeability and its correlation with collagen. Br J Cancer. 1996;73:24–8. http://dx.doi.org/10.1038/bjc.1996.5 . [PMC free article: PMC2074298] [PubMed: 8554978]
- 30.
- Martin TA, Watkins G, Mansel RE, Jiang WG. Loss of tight junction plaque molecules in breast cancer tissues is associated with a poor prognosis in patients with breast cancer. Eur J Cancer. 2004b;40:271725. http://dx.doi.org/10.1016/j.ejca.2004.08.008 . [PubMed: 15571953]
- 31.
- Giepmans BN, van Ijzendoorn SC. Epithelial cell-cell junctions and plasma membrane domains. Biochim Biophys Acta. 2009;1788:820–31. http://dx.doi.org/10.1016/j.bbamem.2008.07.015 . [PubMed: 18706883]
- 32.
- Farquhar MG, Palade GE. Junctional complexes in various epithelia. J Cell Biol. 1963;17:375–412. http://dx.doi.org/10.1083/jcb.17.2.375 . [PMC free article: PMC2106201] [PubMed: 13944428]
- 33.
- Meng W, Takeichi M. Adherens junction: molecular architecture and regulation. Cold Spring Harb Perspect Biol. 2009;1:a002899. http://dx.doi.org/10.1101/cshperspect.a002899 . [PMC free article: PMC2882120] [PubMed: 20457565]
- 34.
- Yonemura S, Itoh M, Nagafuchi A, Tsukita S. Cell-to-cell adherens junction formation and actin filament organization: similarities and differences between non-polarized fibroblasts and polarized epithelial cells. J Cell Sci. 1995;108:127–42. [PubMed: 7738090]
- 35.
- Uchida N, Honjo Y, Johnson KR, Wheelock MJ, Takeichi M. The catenin/cadherin adhesion system is localized in synaptic junctions bordering transmitter release zones. J Cell Biol. 1996;135:767–79. http://dx.doi.org/10.1083/jcb.135.3.767 . [PMC free article: PMC2121068] [PubMed: 8909549]
- 36.
- Gooding JM, Yap KL, Ikura M. The cadherin-catenin complex as a focal point of cell adhesion and signalling: new insights from three-dimensional structures. Bioessays. 2004;26:497–511. http://dx.doi.org/10.1002/bies.20033 . [PubMed: 15112230]
- 37.
- Reynolds AB, Daniel J, McCrea PD, Wheelock MJ, Wu J, Zhang Z. Identification of a new catenin: the tyrosine kinase substrate p120cas associates with E-cadherin complexes. Mol Cell Biol. 1994;14:8333–42. [PMC free article: PMC359372] [PubMed: 7526156]
- 38.
- Togashi H, Miyoshi J, Honda T, Sakisaka T, Takai Y, Takeichi M. Interneurite affinity is regulated by heterophilic nectin interactions in concert with the cadherin machinery. J Cell Biol. 2006;174:141–51. http://dx.doi.org/10.1083/jcb.200601089 . [PMC free article: PMC2064171] [PubMed: 16801389]
- 39.
- Cavallaro U, Christofori G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev Cancer. 2004;4:118–32. http://dx.doi.org/10.1038/nrc1276 . [PubMed: 14964308]
- 40.
- Vleminckx K, Vakaet L Jr., Mareel M, Fiers W, van Roy F. Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell. 1991;66:107–19. http://dx.doi.org/10.1016/0092-8674(91)90143-M . [PubMed: 2070412]
- 41.
- Luo J, Lubaroff DM, Hendrix MJ. Suppression of prostate cancer invasive potential and matrix metalloproteinase activity by E-cadherin transfection. Cancer Res. 1999;59:3552–6. [PubMed: 10446959]
- 42.
- Hsu MY, Meier FE, Nesbit M, Hsu JY, Van Belle P, Elder DE, et al. E-cadherin expression in melanoma cells restores keratinocyte-mediated growth control and down-regulates expression of invasion-related adhesion receptors. Am J Pathol. 2000;156:1515–25. http://dx.doi.org/10.1016/ S0002-9440(10)65023-7 . [PMC free article: PMC1876923] [PubMed: 10793063]
- 43.
- Kashiwagi S, Yashiro M, Takashima T, Aomatsu N, Ikeda K, Ogawa Y, et al. Advantages of adjuvant chemotherapy for patients with triple-negative breast cancer at Stage II: usefulness of prognostic markers E-cadherin and Ki67. Breast Cancer Res. 2011;13:R122. http://dx.doi.org/10.1186/ bcr3068 . [PMC free article: PMC3326564] [PubMed: 22126395]
- 44.
- Morrogh M, Andrade VP, Giri D, Sakr RA, Paik W, Qin LX, et al. Cadherin-catenin complex dissociation in lobular neoplasia of the breast. Breast Cancer Res Treat. 2012;132:641–52. http://dx.doi.org/10.1007/s10549-011-1860-0 . [PMC free article: PMC4349355] [PubMed: 22080244]
- 45.
- Garrod D, Chidgey M. Desmosome structure, composition and function. Biochim Biophys Acta. 2008;1778:572–87. http://dx.doi.org/10.1016/j.bbamem.2007.07.014 . [PubMed: 17854763]
- 46.
- Brooke MA, Nitoiu D, Kelsell DP. Cell-cell connectivity: desmosomes and disease. J Pathol. 2012;226:158–71. http://dx.doi.org/10.1002/path.3027 . [PubMed: 21989576]
- 47.
- Dusek RL, Attardi LD. Desmosomes: new perpetrators in tumour suppression. Nat Rev Cancer. 2011;11:317–23. http://dx.doi.org/10.1038/nrc3051 . [PMC free article: PMC3799918] [PubMed: 21508970]
- 48.
- Furukawa C, Daigo Y, Ishikawa N, Kato T, Ito T, Tsuchiya E, et al. Plakophilin 3 oncogene as prognostic marker and therapeutic target for lung cancer. Cancer Res. 2005;65:7102–10. http://dx.doi.org/10.1158/0008-5472.CAN-04-1877 . [PubMed: 16103059]
- 49.
- Brennan D, Mahoney MG. Increased expression of Dsg2 in malignant skin carcinomas: A tissuemicroarray based study. Cell Adh Migr. 2009;3:148–54. http://dx.doi.org/10.4161/ cam.3.2.7539 . [PMC free article: PMC2679873] [PubMed: 19458482]
- 50.
- Breuninger S, Reidenbach S, Sauer CG, Strbel P, Pfitzenmaier J, Trojan L, et al. Desmosomal plakophilins in the prostate and prostatic adenocarcinomas: implications for diagnosis and tumor progression. Am J Pathol. 2010;176:2509–19. http://dx.doi.org/10.2353/ajpath.2010.090737 . [PMC free article: PMC2861115] [PubMed: 20348237]
- 51.
- Kolegraff K, Nava P, Helms MN, Parkos CA, Nusrat A. Loss of desmocollin-2 confers a tumorigenic phenotype to colonic epithelial cells through activation of Akt/b-catenin signaling. Mol Biol Cell. 2011;22:1121–34. http://dx.doi.org/10.1091/mbc.E10-10-0845 . [PMC free article: PMC3078068] [PubMed: 21325624]
- 52.
- Teh MT, Parkinson EK, Thurlow JK, Liu F, Fortune F, Wan H. A molecular study of desmosomes identifies a desmoglein isoform switch in head and neck squamous cell carcinoma. J Oral Pathol Med. 2011;40:67–76. http://dx.doi.org/10.1111/j.1600-0714.2010.00951.x . [PubMed: 20923451]
- 53.
- Gosavi P, Kundu ST, Khapare N, Sehgal L, Karkhanis MS, Dalal SN. E-cadherin and plakoglobin recruit plakophilin3 to the cell border to initiate desmosome assembly. Cell Mol Life Sci. 2011;68:1439–54. http://dx.doi.org/10.1007/s00018-010-0531-3 . [PubMed: 20859650]
- 54.
- Derangeon M, Spray DC, Bourmeyster N, Sarrouilhe D, Herv JC. Reciprocal influence of connexins and apical junction proteins on their expressions and functions. Biochim Biophys Acta. 2009;1788:76878. http://dx.doi.org/10.1016/j.bbamem.2008.10.023 . [PMC free article: PMC2666103] [PubMed: 19046940]
- 55.
- Lamiche C, Clarhaut J, Strale PO, Crespin S, Pedretti N, Bernard FX, et al. The gap junction protein Cx43 is involved in the bone-targeted metastatic behaviour of human prostate cancer cells. Clin Exp Metastasis. 2012;29:111–22. http://dx.doi.org/10.1007/s10585-011-9434-4 . [PubMed: 22080401]
- 56.
- Sirnes S, Bruun J, Kolberg M, Kjenseth A, Lind GE, Svindland A, et al. Connexin43 acts as a colorectal cancer tumor suppressor and predicts disease outcome. Int J Cancer. 2011. http://dx.doi.org/10.1002/ijc.26392 . [PubMed: 21866551]
- 57.
- Bendas G, Borsig L. Cancer cell adhesion and metastasis: selectins, integrins, and the inhibitory potential of heparins. Int J Cell Biol. 2012;2012:676731. http://dx.doi. org/10.1155/2012/676731 . [PMC free article: PMC3296185] [PubMed: 22505933]
- 58.
- van den Hoogen C, van der Horst G, Cheung H, Buijs JT, Pelger RC, van der Pluijm G. Integrin αv expression is required for the acquisition of a metastatic stem/progenitor cell phenotype in human prostate cancer. Am J Pathol. 2011;179:2559–68. http://dx.doi.org/10.1016/j. ajpath.2011.07.011 . [PMC free article: PMC3204028] [PubMed: 21907176]
- 59.
- Lu JG, Li Y, Li L, Kan X. Overexpression of osteopontin and integrin αv in laryngeal and hypopharyngeal carcinomas associated with differentiation and metastasis. J Cancer Res Clin Oncol. 2011;137:1613–8. http://dx.doi.org/10.1007/s00432-011-1024-y . [PubMed: 21853313]
- 60.
- St Hill CA. Interactions between endothelial selectins and cancer cells regulate metastasis. [Elite Ed] Front Biosci. 2012;17:3233–51. [PubMed: 21622232]
- 61.
- Richter U, Schrder C, Wicklein D, Lange T, Geleff S, Dippel V, et al. Adhesion of small cell lung cancer cells to E- and P-selectin under physiological flow conditions: implications for metastasis formation. Histochem Cell Biol. 2011;135:499–512. http://dx.doi.org/10.1007/s00418-0110804-4 . [PubMed: 21544708]
- 62.
- Wai Wong C, Dye DE, Coombe DR. The role of immunoglobulin superfamily cell adhesion molecules in cancer metastasis. Int J Cell Biol. 2012;2012:340296. http://dx.doi. org/10.1155/2012/340296 . [PMC free article: PMC3261479] [PubMed: 22272201]
- 63.
- Martin TA, Harrison G, Mansel RE, Jiang WG. The role of the CD44/ezrin complex in cancer metastasis. Crit Rev Oncol Hematol. 2003;46:165–86. http://dx.doi.org/10.1016/ S1040-8428(02)00172-5 . [PubMed: 12711360]
- 64.
- Jeyapalan Z., Yang B. B. The non-coding 3'UTR of CD44 induces metastasis by regulating extracellular matrix functions. J Cell Sci. 2012 [PubMed: 22637644]
- 65.
- Park YS, Huh JW, Lee JH, Kim HR. shRNA against CD44 inhibits cell proliferation, invasion and migration, and promotes apoptosis of colon carcinoma cells. Oncol Rep. 2012;27:339–46. [PubMed: 22076607]
- 66.
- Schwock J, Dhani N, Hedley DW. Targeting focal adhesion kinase signaling in tumor growth and metastasis. Expert Opin Ther Targets. 2010;14:77–94. http://dx.doi. org/10.1517/14728220903460340 . [PubMed: 20001212]
- 67.
- Wang W, Liu Y, Liao K. Tyrosine phosphorylation of cortactin by the FAK-Src complex at focal adhesions regulates cell motility. BMC Cell Biol. 2011;12:49. http://dx.doi.org/10.1186/14712121-12-49 . [PMC free article: PMC3245448] [PubMed: 22078467]
- 68.
- Bernardi MA, Logullo AF, Pasini FS, Nonogaki S, Blumke C, Soares FA, et al. Prognostic significance of CD24 and claudin-7 immunoexpression in ductal invasive breast cancer. Oncol Rep. 2012;27:28–38. [PubMed: 21956537]
- 69.
- Hornsby CD, Cohen C, Amin MB, Picken MM, Lawson D, Yin-Goen Q, et al. Claudin-7 immunohistochemistry in renal tumors: a candidate marker for chromophobe renal cell carcinoma identified by gene expression profiling. Arch Pathol Lab Med. 2007;131:1541–6. [PubMed: 17922590]
- 70.
- Sheehan GM, Kallakury BV, Sheehan CE, Fisher HA, Kaufman RP Jr., Ross JS. Loss of claudins-1 and -7 and expression of claudins-3 and -4 correlate with prognostic variables in prostatic adenocarcinomas. Hum Pathol. 2007;38:564–9. http://dx.doi.org/10.1016/j.humpath.2006.11.007 . [PubMed: 17306334]
- 71.
- Zheng JY, Yu D, Foroohar M, Ko E, Chan J, Kim N, et al. Regulation of the expression of the prostate-specific antigen by claudin-7. J Membr Biol. 2003;194:187–97. http://dx.doi. org/10.1007/s00232-003-2038-4 . [PubMed: 14502431]
- 72.
- Chao YC, Pan SH, Yang SC, Yu SL, Che TF, Lin CW, et al. Claudin-1 is a metastasis suppressor and correlates with clinical outcome in lung adenocarcinoma. Am J Respir Crit Care Med. 2009;179:12333. http://dx.doi.org/10.1164/rccm.200803-456OC . [PubMed: 18787218]
- 73.
- Richardson F, Young GD, Sennello R, Wolf J, Argast GM, Mercado P, et al. The evaluation of E-Cadherin and vimentin as biomarkers of clinical outcomes among patients with non-small cell lung cancer treated with erlotinib as second- or third-line therapy. Anticancer Res. 2012;32:537–52. [PubMed: 22287743]
- 74.
- Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, et al. Cell migration: integrating signals from front to back. Science. 2003;302:1704–9. http://dx.doi.org/10.1126/science.1092053 . [PubMed: 14657486]
- 75.
- Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–10. http://dx.doi.org/10.1016/0092-8674(92)90164-8 . [PubMed: 1643658]
- 76.
- Faassen AE, Schrager JA, Klein DJ, Oegema TR, Couchman JR, McCarthy JB. A cell surface chondroitin sulfate proteoglycan, immunologically related to CD44, is involved in type I collagen-mediated melanoma cell motility and invasion. J Cell Biol. 1992;116:521–31. http://dx.doi.org/10.1083/ jcb.116.2.521 . [PMC free article: PMC2289300] [PubMed: 1730766]
- 77.
- Huber MA, Kraut N, Beug H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol. 2005;17:548–58. http://dx.doi.org/10.1016/j.ceb.2005.08.001 . [PubMed: 16098727]
- 78.
- Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–42. http://dx.doi.org/10.1038/nrm1835 . [PubMed: 16493418]
- 79.
- Buijs JT, Henriquez NV, van Overveld PG, van der Horst G, ten Dijke P, van der Pluijm G. TGF-beta and BMP7 interactions in tumour progression and bone metastasis. Clin Exp Metastasis. 2007;24:60917. http://dx.doi.org/10.1007/s10585-007-9118-2 . [PubMed: 18008174]
- 80.
- Guarino M. Epithelial-mesenchymal transition and tumour invasion. Int J Biochem Cell Biol. 2007;39:2153–60. http://dx.doi.org/10.1016/j.biocel.2007.07.011 . [PubMed: 17825600]
- 81.
- Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–54. http://dx.doi.org/10.1038/nrc822 . [PubMed: 12189386]
- 82.
- Thiery JP. Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2003;15:740–6. http://dx.doi.org/10.1016/j.ceb.2003.10.006 . [PubMed: 14644200]
- 83.
- Trimboli AJ, Fukino K, de Bruin A, Wei G, Shen L, Tanner SM, et al. Direct evidence for epithelial-mesenchymal transitions in breast cancer. Cancer Res. 2008;68:937–45. http://dx.doi.org/10.1158/0008-5472.CAN-07-2148 . [PubMed: 18245497]
- 84.
- Cano A, Prez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83. http://dx.doi.org/10.1038/35000025 . [PubMed: 10655586]
- 85.
- Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–28. http://dx.doi. org/10.1038/nrc2131 . [PubMed: 17508028]
- 86.
- Medici D, Hay ED, Olsen BR. Snail and Slug promote epithelial-mesenchymal transition through betacatenin-T-cell factor-4-dependent expression of transforming growth factor-beta3. Mol Biol Cell. 2008;19:4875–87. http://dx.doi.org/10.1091/mbc.E08-05-0506 . [PMC free article: PMC2575183] [PubMed: 18799618]
- 87.
- Takeichi M. Cadherin cell adhesion receptors as a morphogenetic regulator. Science. 1991;251:1451–5. http://dx.doi.org/10.1126/science.2006419 . [PubMed: 2006419]
- 88.
- Jiang WG, Mansel RE. E-cadherin complex and its abnormalities in human breast cancer. Surg Oncol. 2000;9:151–71. http://dx.doi.org/10.1016/S0960-7404(01)00010-X . [PubMed: 11476987]
- 89.
- Zschiesche W, Schnborn I, Behrens J, Herrenknecht K, Hartveit F, Lilleng P, et al. Expression of E-cadherin and catenins in invasive mammary carcinomas. Anticancer Res. 1997;17(1B):561–7. [PubMed: 9066580]
- 90.
- Jiang WG. E-cadherin and its associated protein catenins, cancer invasion and metastasis. Br J Surg. 1996;83:437–46. http://dx.doi.org/10.1002/bjs.1800830404 . [PubMed: 8665230]
- 91.
- Gottardi CJ, Wong E, Gumbiner BM. E-cadherin suppresses cellular transformation by inhibiting betacatenin signaling in an adhesion-independent manner. J Cell Biol. 2001;153:1049–60. http://dx.doi.org/10.1083/jcb.153.5.1049 . [PMC free article: PMC2174337] [PubMed: 11381089]
- 92.
- Stockinger A, Eger A, Wolf J, Beug H, Foisner R. E-cadherin regulates cell growth by modulating proliferation-dependent beta-catenin transcriptional activity. J Cell Biol. 2001;154:1185–96. http://dx.doi.org/10.1083/jcb.200104036 . [PMC free article: PMC2150811] [PubMed: 11564756]
- 93.
- Kim K, Lu Z, Hay ED. Direct evidence for a role of beta-catenin/LEF-1 signaling pathway in induction of EMT. Cell Biol Int. 2002;26:463–76. http://dx.doi.org/10.1006/cbir.2002.0901 . [PubMed: 12095232]
- 94.
- Pecina-Slaus N, Cicvara-Pecina T, Kafka A. Epithelial-to-mesenchymal transition: possible role in meningiomas [Elite Ed] Front Biosci (Elite Ed). 2012;4:889–96. http://dx.doi. org/10.2741/427 . [PubMed: 22201922]
- 95.
- Martin TA, Goyal A, Watkins G, Jiang WG. Expression of the transcription factors snail, slug, and twist and their clinical significance in human breast cancer. Ann Surg Oncol. 2005;12:488–96. http://dx.doi.org/10.1245/ASO.2005.04.010 . [PubMed: 15864483]
- 96.
- Rosivatz E, Becker I, Specht K, Fricke E, Luber B, Busch R, et al. Differential expression of the epithelial-mesenchymal transition regulators snail, SIP1, and twist in gastric cancer. Am J Pathol. 2002;161:1881–91. http://dx.doi.org/10.1016/S0002-9440(10)64464-1 . [PMC free article: PMC1850763] [PubMed: 12414534]
- 97.
- Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–39. http://dx.doi.org/10.1016/j.cell.2004.06.006 . [PubMed: 15210113]
- 98.
- Leptin M, Grunewald B. Cell shape changes during gastrulation in Drosophila. Development. 1990;110:73–84. [PubMed: 2081472]
- 99.
- Smith DE, Franco del Amo F, Gridley T. Isolation of Sna, a mouse gene homologous to the Drosophila genes snail and escargot: its expression pattern suggests multiple roles during postimplantation development. Development. 1992;116:1033–9. [PubMed: 1295727]
- 100.
- Batlle E, Sancho E, Franc C, Domnguez D, Monfar M, Baulida J, et al. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2:84–9. http://dx.doi.org/10.1038/35000034 . [PubMed: 10655587]
- 101.
- Yokoyama K, Kamata N, Hayashi E, Hoteiya T, Ueda N, Fujimoto R, et al. Reverse correlation of E-cadherin and snail expression in oral squamous cell carcinoma cells in vitro. Oral Oncol. 2001;37:6571. http://dx.doi.org/10.1016/S1368-8375(00)00059-2 . [PubMed: 11120485]
- 102.
- Blanco MJ, Moreno-Bueno G, Sarrio D, Locascio A, Cano A, Palacios J, et al. Correlation of Snail expression with histological grade and lymph node status in breast carcinomas. Oncogene. 2002;21:32416. http://dx.doi.org/10.1038/sj.onc.1205416 . [PubMed: 12082640]
- 103.
- Ikenouchi J, Matsuda M, Furuse M, Tsukita S. Regulation of tight junctions during the epithelium-mesenchyme transition: direct repression of the gene expression of claudins/occludin by Snail. J Cell Sci. 2003;116:1959–67. http://dx.doi.org/10.1242/jcs.00389 . [PubMed: 12668723]
- 104.
- Islam S, Carey TE, Wolf GT, Wheelock MJ, Johnson KR. Expression of N-cadherin by human squamous carcinoma cells induces a scattered fibroblastic phenotype with disrupted cell-cell adhesion. J Cell Biol. 1996;135:1643–54. http://dx.doi.org/10.1083/jcb.135.6.1643 . [PMC free article: PMC2133960] [PubMed: 8978829]
- 105.
- Nieman MT, Prudoff RS, Johnson KR, Wheelock MJ. N-cadherin promotes motility in human breast cancer cells regardless of their E-cadherin expression. J Cell Biol. 1999;147:631–44. http://dx.doi.org/10.1083/jcb.147.3.631 . [PMC free article: PMC2151177] [PubMed: 10545506]
- 106.
- Mikheeva SA, Mikheev AM, Petit A, Beyer R, Oxford RG, Khorasani L, et al. TWIST1 promotes invasion through mesenchymal change in human glioblastoma. Mol Cancer. 2010;9:194. http://dx.doi.org/10.1186/1476-4598-9-194 . [PMC free article: PMC2920263] [PubMed: 20646316]
- 107.
- Gupta R, Chetty C, Bhoopathi P, Lakka S, Mohanam S, Rao JS, et al. Downregulation of uPA/uPAR inhibits intermittent hypoxia-induced epithelial-mesenchymal transition (EMT) in DAOY and D283 medulloblastoma cells. Int J Oncol. 2011;38:733–44. [PubMed: 21181094]
- 108.
- Lester RD, Jo M, Montel V, Takimoto S, Gonias SL. uPAR induces epithelial-mesenchymal transition in hypoxic breast cancer cells. J Cell Biol. 2007;178:425–36. http://dx.doi.org/10.1083/ jcb.200701092 . [PMC free article: PMC2064849] [PubMed: 17664334]
- 109.
- Strutz F, Zeisberg M, Ziyadeh FN, Yang CQ, Kalluri R, Mller GA, et al. Role of basic fibroblast growth factor-2 in epithelial-mesenchymal transformation. Kidney Int. 2002;61:1714–28. http://dx.doi.org/10.1046/j.1523-1755.2002.00333.x . [PubMed: 11967021]
- 110.
- Akhurst RJ, Derynck R. TGF-beta signaling in cancer--a double-edged sword. Trends Cell Biol. 2001;11:S44–51. [PubMed: 11684442]
- 111.
- Zeisberg M, Hanai J, Sugimoto H, Mammoto T, Charytan D, Strutz F, et al. BMP-7 counteracts TGFbeta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med. 2003;9:964–8. http://dx.doi.org/10.1038/nm888 . [PubMed: 12808448]
- 112.
- Ye L, Lewis-Russell JM, Kynaston H, Jiang WG. Endogenous bone morphogenetic protein-7 controls the motility of prostate cancer cells through regulation of bone morphogenetic protein antagonists. J Urol. 2007;178:1086–91. http://dx.doi.org/10.1016/j.juro.2007.05.003 . [PubMed: 17644136]
- 113.
- Bokobza SM, Ye L, Kynaston H, Jiang WG. Growth and differentiation factor 9 (GDF-9) induces epithelial-mesenchymal transition in prostate cancer cells. Mol Cell Biochem. 2011;349:33–40. http://dx.doi.org/10.1007/s11010-010-0657-5 . [PubMed: 21116689]
- 114.
- Xu Z, Jiang Y, Steed H, Davidge S, Fu Y. TGFβ and EGF synergistically induce a more invasive phenotype of epithelial ovarian cancer cells. Biochem Biophys Res Commun. 2010;401:376–81. http://dx.doi.org/10.1016/j.bbrc.2010.09.059 . [PubMed: 20854793]
- 115.
- Wang X, Lu H, Urvalek AM, Li T, Yu L, Lamar J, et al. KLF8 promotes human breast cancer cell invasion and metastasis by transcriptional activation of MMP9. Oncogene. 2011;30:1901–11. http://dx.doi.org/10.1038/onc.2010.563 . [PMC free article: PMC3952074] [PubMed: 21151179]
- 116.
- Qiao B, Johnson NW, Gao J. Epithelial-mesenchymal transition in oral squamous cell carcinoma triggered by transforming growth factor-beta1 is Snail family-dependent and correlates with matrix metalloproteinase-2 and -9 expressions. Int J Oncol. 2010;37:663–8. [PubMed: 20664935]
- 117.
- Lynch CC, Vargo-Gogola T, Matrisian LM, Fingleton B. Cleavage of E-Cadherin by Matrix Metalloproteinase-7 Promotes Cellular Proliferation in Nontransformed Cell Lines via Activation of RhoA. J Oncol. 2010;2010:530745. http://dx.doi.org/10.1155/2010/530745 . [PMC free article: PMC2902104] [PubMed: 20628524]
- 118.
- Abe K, Takeichi M. EPLIN mediates linkage of the cadherin catenin complex to F-actin and stabilizes the circumferential actin belt. Proc Natl Acad Sci U S A. 2008;105:13–9. http://dx.doi.org/10.1073/pnas.0710504105 . [PMC free article: PMC2224173] [PubMed: 18093941]
- 119.
- Jiang WG, Martin TA, Lewis-Russell JM, Douglas-Jones A, Ye L, Mansel RE. Eplin-alpha expression in human breast cancer, the impact on cellular migration and clinical outcome. Mol Cancer. 2008;7:71. http://dx.doi.org/10.1186/1476-4598-7-71 . [PMC free article: PMC2553413] [PubMed: 18796137]
- 120.
- Sanders AJ, Martin TA, Ye L, Mason MD, Jiang WG. EPLIN is a negative regulator of prostate cancer growth and invasion. J Urol. 2011;186:295–301. http://dx.doi.org/10.1016/j.juro.2011.03.038 . [PubMed: 21600601]
- 121.
- Zhang S, Wang X, Osunkoya AO, Iqbal S, Wang Y, Chen Z, et al. EPLIN downregulation promotes epithelial-mesenchymal transition in prostate cancer cells and correlates with clinical lymph node metastasis. Oncogene. 2011;30:4941–52. http://dx.doi.org/10.1038/onc.2011.199 . [PMC free article: PMC3165108] [PubMed: 21625216]
- 122.
- Hay ED. Organization and fine structure of epithelium and mesenchyme in the developing chick embryo. In: Epithelial-mesenchymal interactions. In: Fleischmajer R, Billingham RE, editors. Baltimore: Williams & Wilkins; 1968. pp. 31–55.
- 123.
- Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 2007;6:273–86. http://dx.doi.org/10.1038/nrd2115 . [PubMed: 17396134]
- 124.
- Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146:87387. http://dx.doi.org/10.1016/j.cell.2011.08.039 . [PubMed: 21925313]
- 125.
- Pandya NM, Dhalla NS, Santani DD. Angiogenesis--a new target for future therapy. Vascul Pharmacol. 2006;44:265–74. http://dx.doi.org/10.1016/j.vph.2006.01.005 . [PubMed: 16545987]
- 126.
- Al-Rawi MA, Jiang WG. Lymphangiogenesis and cancer metastasis. Front Biosci. 2011;16:723–39. http://dx.doi.org/10.2741/3715 . [PubMed: 21196198]
- 127.
- Albrecht I, Christofori G. Molecular mechanisms of lymphangiogenesis in development and cancer. Int J Dev Biol. 2011;55:483–94. http://dx.doi.org/10.1387/ijdb.103226ia . [PubMed: 21858772]
- 128.
- Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–6. http://dx.doi.org/10.1056/NEJM197111182852108 . [PubMed: 4938153]
- 129.
- Klagsbrun M, D'Amore PA. Vascular endothelial growth factor and its receptors. Cytokine Growth Factor Rev. 1996;7:259–70. http://dx.doi.org/10.1016/S1359-6101(96)00027-5 . [PubMed: 8971481]
- 130.
- Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. http://dx.doi.org/10.1038/nature10144 . [PMC free article: PMC4049445] [PubMed: 21593862]
- 131.
- Ferrara N, Davis-Smyth T. The biology of vascular endothelial growth factor. Endocr Rev. 1997;18:425. http://dx.doi.org/10.1210/er.18.1.4 . [PubMed: 9034784]
- 132.
- Sato S, Itamochi H. Bevacizumab and ovarian cancer. Curr Opin Obstet Gynecol. 2012;24:8–13. http://dx.doi.org/10.1097/GCO.0b013e32834daeed . [PubMed: 22123222]
- 133.
- Jiang WG, Martin TA, Parr C, Davies G, Matsumoto K, Nakamura T. Hepatocyte growth factor, its receptor, and their potential value in cancer therapies. Crit Rev Oncol Hematol. 2005;53:35–69. http://dx.doi.org/10.1016/j.critrevonc.2004.09.004 . [PubMed: 15607934]
- 134.
- Wojta J, Kaun C, Breuss JM, Koshelnick Y, Beckmann R, Hattey E, et al. Hepatocyte growth factor increases expression of vascular endothelial growth factor and plasminogen activator inhibitor-1 in human keratinocytes and the vascular endothelial growth factor receptor flk-1 in human endothelial cells. Lab Invest. 1999;79:427–38. [PubMed: 10211995]
- 135.
- Davies G, Mason MD, Martin TA, Parr C, Watkins G, Lane J, et al. The HGF/SF antagonist NK4 reverses fibroblast- and HGF-induced prostate tumor growth and angiogenesis in vivo. Int J Cancer. 2003;106:348–54. http://dx.doi.org/10.1002/ijc.11220 . [PubMed: 12845672]
- 136.
- Martin TA, Parr C, Davies G, Watkins G, Lane J, Matsumoto K, et al. Growth and angiogenesis of human breast cancer in a nude mouse tumour model is reduced by NK4, a HGF/SF antagonist. Carcinogenesis. 2003;24:1317–23. http://dx.doi.org/10.1093/carcin/bgg072 . [PubMed: 12807719]
- 137.
- Eder JP, Shapiro GI, Appleman LJ, Zhu AX, Miles D, Keer H, et al. A phase I study of foretinib, a multi-targeted inhibitor of c-Met and vascular endothelial growth factor receptor 2. Clin Cancer Res. 2010;16:3507–16. http://dx.doi.org/10.1158/1078-0432.CCR-10-0574 . [PubMed: 20472683]
- 138.
- Banerji S, Ni J, Wang SX, Clasper S, Su J, Tammi R, et al. LYVE-1, a new homologue of the CD44 glycoprotein, is a lymph-specific receptor for hyaluronan. J Cell Biol. 1999;144:789–801. http://dx.doi.org/10.1083/jcb.144.4.789 . [PMC free article: PMC2132933] [PubMed: 10037799]
- 139.
- Kaipainen A, Korhonen J, Mustonen T, van Hinsbergh VW, Fang GH, Dumont D, et al. Expression of the fms-like tyrosine kinase 4 gene becomes restricted to lymphatic endothelium during development. Proc Natl Acad Sci U S A. 1995;92:3566–70. http://dx.doi.org/10.1073/pnas.92.8.3566 . [PMC free article: PMC42208] [PubMed: 7724599]
- 140.
- McAllaster JD, Cohen MS. Role of the lymphatics in cancer metastasis and chemotherapy applications. Adv Drug Deliv Rev. 2011;63:867–75. http://dx.doi.org/10.1016/j.addr.2011.05.014 . [PubMed: 21699937]
- 141.
- Skobe M, Hawighorst T, Jackson DG, Prevo R, Janes L, Velasco P, et al. Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat Med. 2001;7:192–8. http://dx.doi.org/10.1038/84643 . [PubMed: 11175850]
- 142.
- Stacker SA, Caesar C, Baldwin ME, Thornton GE, Williams RA, Prevo R, et al. VEGF-D promotes the metastatic spread of tumor cells via the lymphatics. Nat Med. 2001;7:186–91. http://dx.doi.org/10.1038/84635 . [PubMed: 11175849]
- 143.
- Feng Y, Wang W, Hu J, Ma J, Zhang Y, Zhang J. Expression of VEGF-C and VEGF-D as significant markers for assessment of lymphangiogenesis and lymph node metastasis in non-small cell lung cancer. Anat Rec (Hoboken). 2010;293:802–12. http://dx.doi.org/10.1002/ar.21096 . [PubMed: 20225197]
- 144.
- Kilvaer TK, Valkov A, Sorbye S, Smeland E, Bremnes RM, Busund LT, et al. Profiling of VEGFs and VEGFRs as prognostic factors in soft tissue sarcoma: VEGFR-3 is an independent predictor of poor prognosis. PLoS One. 2010;5:e15368. http://dx.doi.org/10.1371/journal. pone.0015368 . [PMC free article: PMC3001883] [PubMed: 21179485]
- 145.
- Raica M, Cimpean AM, Ceausu R, Ribatti D. Lymphatic microvessel density, VEGF-C, and VEGFR-3 expression in different molecular types of breast cancer. Anticancer Res. 2011;31:1757–64. [PubMed: 21617236]
- 146.
- Debois JM. 2002. TxNxM1: The Anatomy and Clinics of Metastatic Cancer, Kluwer Academic Publisher.
- 147.
- Abbruzzese JL, Abbruzzese MC, Lenzi R, Hess KR, Raber MN. Analysis of a diagnostic strategy for patients with suspected tumors of unknown origin. J Clin Oncol. 1995;13:2094–103. [PubMed: 7636553]
- 148.
- Schouten LJ, Rutten J, Huveneers HA, Twijnstra A. Incidence of brain metastases in a cohort of patients with carcinoma of the breast, colon, kidney, and lung and melanoma. Cancer. 2002;94:2698–705. http://dx.doi.org/10.1002/cncr.10541 . [PubMed: 12173339]
- 149.
- Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer. 2002;2:584–93. http://dx.doi.org/10.1038/nrc867 . [PubMed: 12154351]
- 150.
- Hess KR, Varadhachary GR, Taylor SH, Wei W, Raber MN, Lenzi R, et al. Metastatic patterns in adenocarcinoma. Cancer. 2006;106:1624–33. http://dx.doi.org/10.1002/cncr.21778 . [PubMed: 16518827]
- 151.
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–49. http://dx.doi.org/10.3322/caac.20006 . [PubMed: 19474385]
- 152.
- Bubendorf L, Schpfer A, Wagner U, Sauter G, Moch H, Willi N, et al. Metastatic patterns of prostate cancer: an autopsy study of 1,589 patients. Hum Pathol. 2000;31:578–83. http://dx.doi.org/10.1053/hp.2000.6698 . [PubMed: 10836297]
- 153.
- Minn AJ, Kang Y, Serganova I, Gupta GP, Giri DD, Doubrovin M, et al. Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J Clin Invest. 2005;115:44–55. [PMC free article: PMC539194] [PubMed: 15630443]
- 154.
- Schlter K, Gassmann P, Enns A, Korb T, Hemping-Bovenkerk A, Hlzen J, et al. Organ-specific metastatic tumor cell adhesion and extravasation of colon carcinoma cells with different metastatic potential. Am J Pathol. 2006;169:1064–73. http://dx.doi.org/10.2353/ ajpath.2006.050566 . [PMC free article: PMC1698818] [PubMed: 16936278]
- 155.
- Nguyen DX, Bos PD, Massagu J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9:274–84. http://dx.doi.org/10.1038/nrc2622 . [PubMed: 19308067]
- 156.
- Nguyen DX, Massagu J. Genetic determinants of cancer metastasis. Nat Rev Genet. 2007;8:341–52. http://dx.doi.org/10.1038/nrg2101 . [PubMed: 17440531]
- 157.
- Chiang AC, Massagu J. Molecular basis of metastasis. N Engl J Med. 2008;359:2814–23. http://dx.doi.org/10.1056/NEJMra0805239 . [PMC free article: PMC4189180] [PubMed: 19109576]
- 158.
- Feng R, Chen X, Yu Y, Su L, Yu B, Li J, et al. miR-126 functions as a tumour suppressor in human gastric cancer. Cancer Lett. 2010;298:50–63. http://dx.doi.org/10.1016/j.canlet.2010.06.004 . [PubMed: 20619534]
- 159.
- Yang R, Dick M, Marme F, Schneeweiss A, Langheinz A, Hemminki K, et al. Genetic variants within miR-126 and miR-335 are not associated with breast cancer risk. Breast Cancer Res Treat. 2011;127:54954. http://dx.doi.org/10.1007/s10549-010-1244-x . [PubMed: 21046227]
- 160.
- Steeg PS. Metastasis suppressors alter the signal transduction of cancer cells. Nat Rev Cancer. 2003;3:55–63. http://dx.doi.org/10.1038/nrc967 . [PubMed: 12509767]
- 161.
- Malik FA, Sanders AJ, Jones AD, Mansel RE, Jiang WG. Transcriptional and translational modulation of KAI1 expression in ductal carcinoma of the breast and the prognostic significance. Int J Mol Med. 2009;23:273–8. [PubMed: 19148553]
- 162.
- Malik FA, Sanders AJ, Kayani MA, Jiang WG. Effect of expressional alteration of KAI1 on breast cancer cell growth, adhesion, migration and invasion. Cancer Genomics Proteomics. 2009;6:205–13. [PubMed: 19656997]
- 163.
- Batson OV. The function of the vertebral veins and their role in the spread of metastases. 1940. Clin Orthop Relat Res. 1995:4–9. [PubMed: 7634616]
- 164.
- Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989;8:98–101. [PubMed: 2673568]
- 165.
- Autzen P, Robson CN, Bjartell A, Malcolm AJ, Johnson MI, Neal DE, et al. Bone morphogenetic protein 6 in skeletal metastases from prostate cancer and other common human malignancies. Br J Cancer. 1998;78:1219–23. http://dx.doi.org/10.1038/bjc.1998.658 . [PMC free article: PMC2062993] [PubMed: 9820184]
- 166.
- De Pinieux G, Flam T, Zerbib M, Taupin P, Bellahcne A, Waltregny D, et al. Bone sialoprotein, bone morphogenetic protein 6 and thymidine phosphorylase expression in localized human prostatic adenocarcinoma as predictors of clinical outcome: a clinicopathological and immunohistochemical study of 43 cases. J Urol. 2001;166:1924–30. http://dx.doi.org/10.1016/S00225347(05)65722-9 . [PubMed: 11586262]
- 167.
- Shariat SF, Shalev M, Menesses-Diaz A, Kim IY, Kattan MW, Wheeler TM, et al. Preoperative plasma levels of transforming growth factor beta(1) (TGF-beta(1)) strongly predict progression in patients undergoing radical prostatectomy. J Clin Oncol. 2001;19:2856–64. [PubMed: 11387358]
- 168.
- Masuda H, Fukabori Y, Nakano K, Takezawa Y, CSuzuki T, Yamanaka H. Increased expression of bone morphogenetic protein-7 in bone metastatic prostate cancer. Prostate. 2003;54:268–74. http://dx.doi.org/10.1002/pros.10193 . [PubMed: 12539225]
- 169.
- Lehr JE, Pienta KJ. Preferential adhesion of prostate cancer cells to a human bone marrow endothelial cell line. J Natl Cancer Inst. 1998;90:118–23. http://dx.doi.org/10.1093/jnci/90.2.118 . [PubMed: 9450571]
- 170.
- Chay CH, Cooper CR, Gendernalik JD, Dhanasekaran SM, Chinnaiyan AM, Rubin MA, et al. A functional thrombin receptor (PAR1) is expressed on bone-derived prostate cancer cell lines. Urology. 2002;60:760–5. http://dx.doi.org/10.1016/S0090-4295(02)01969-6 . [PubMed: 12429291]
- 171.
- Cooper CR, Chay CH, Pienta KJ. The role of alpha(v)beta(3) in prostate cancer progression. Neoplasia. 2002;4:191–4. http://dx.doi.org/10.1038/sj.neo.7900224 . [PMC free article: PMC1531692] [PubMed: 11988838]
- 172.
- Cooper CR, Chay CH, Gendernalik JD, Lee HL, Bhatia J, Taichman RS, et al. Stromal factors involved in prostate carcinoma metastasis to bone. Cancer. 2003;97(Suppl):739–47. http://dx.doi.org/10.1002/cncr.11181 . [PubMed: 12548571]
- 173.
- Kumar CC. Integrin alpha v beta 3 as a therapeutic target for blocking tumor-induced angiogenesis. Curr Drug Targets. 2003;4:123–31. http://dx.doi.org/10.2174/1389450033346830 . [PubMed: 12558065]
- 174.
- Krupski T, Harding MA, Herce ME, Gulding KM, Stoler MH, Theodorescu D. The role of vascular endothelial growth factor in the tissue specific in vivo growth of prostate cancer cells. Growth Factors. 2001;18:287–302. http://dx.doi.org/10.3109/08977190109029117 . [PubMed: 11519827]
- 175.
- Chen J, De S, Brainard J, Byzova TV. Metastatic properties of prostate cancer cells are controlled by VEGF. Cell Commun Adhes. 2004;11:1–11. http://dx.doi.org/10.1080/15419060490471739 . [PubMed: 15500293]
- 176.
- Dai J, Kitagawa Y, Zhang J, Yao Z, Mizokami A, Cheng S, et al. Vascular endothelial growth factor contributes to the prostate cancer-induced osteoblast differentiation mediated by bone morphogenetic protein. Cancer Res. 2004;64:994–9. http://dx.doi.org/10.1158/0008-5472.CAN03-1382 . [PubMed: 14871830]
- 177.
- Jacob K, Webber M, Benayahu D, Kleinman HK. Osteonectin promotes prostate cancer cell migration and invasion: a possible mechanism for metastasis to bone. Cancer Res. 1999;59:4453–7. [PubMed: 10485497]
- 178.
- Rosol TJ. Pathogenesis of bone metastases: role of tumor-related proteins. J Bone Miner Res. 2000;15:844–50. http://dx.doi.org/10.1359/jbmr.2000.15.5.844 . [PubMed: 10804013]
- 179.
- Denhardt DT, Giachelli CM, Rittling SR. Role of osteopontin in cellular signaling and toxicant injury. Annu Rev Pharmacol Toxicol. 2001;41:723–49. http://dx.doi.org/10.1146/annurev. pharmtox.41.1.723 . [PubMed: 11264474]
- 180.
- Fizazi K, Yang J, Peleg S, Sikes CR, Kreimann EL, Daliani D, et al. Prostate cancer cells-osteoblast interaction shifts expression of growth/survival-related genes in prostate cancer and reduces expression of osteoprotegerin in osteoblasts. Clin Cancer Res. 2003;9:2587–97. [PubMed: 12855635]
- 181.
- Zhang JH, Tang J, Wang J, Ma W, Zheng W, Yoneda T, et al. Over-expression of bone sialoprotein enhances bone metastasis of human breast cancer cells in a mouse model. Int J Oncol. 2003;23:1043–8. [PubMed: 12963984]
- 182.
- Khodavirdi AC, Song Z, Yang S, Zhong C, Wang S, Wu H, et al. Increased expression of osteopontin contributes to the progression of prostate cancer. Cancer Res. 2006;66:883–8. http://dx.doi.org/10.1158/0008-5472.CAN-05-2816 . [PubMed: 16424021]
- 183.
- Resnick MB, Konkin T, Routhier J, Sabo E, Pricolo VE. Claudin-1 is a strong prognostic indicator in stage II colonic cancer: a tissue microarray study. Mod Pathol. 2005;18:511–8. http://dx.doi.org/10.1038/modpathol.3800301 . [PubMed: 15475928]
- 184.
- Montgomery E, Mamelak AJ, Gibson M, Maitra A, Sheikh S, Amr SS, et al. Overexpression of claudin proteins in esophageal adenocarcinoma and its precursor lesions. Appl Immunohistochem Mol Morphol. 2006;14:24–30. http://dx.doi.org/10.1097/01.pai.0000151933.04800.1c . [PubMed: 16540726]
- 185.
- Sommers CL, Heckford SE, Skerker JM, Worland P, Torri JA, Thompson EW, et al. Loss of epithelial markers and acquisition of vimentin expression in adriamycin- and vinblastine-resistant human breast cancer cell lines. Cancer Res. 1992;52:5190–7. [PubMed: 1382837]
- 186.
- Zajchowski DA, Bartholdi MF, Gong Y, Webster L, Liu HL, Munishkin A, et al. Identification of gene expression profiles that predict the aggressive behavior of breast cancer cells. Cancer Res. 2001;61:5168–78. [PubMed: 11431356]
- 187.
- Ponnusamy MP, Lakshmanan I, Jain M, Das S, Chakraborty S, Dey P, et al. MUC4 mucin-induced epithelial to mesenchymal transition: a novel mechanism for metastasis of human ovarian cancer cells. Oncogene. 2010;29:5741–54. http://dx.doi.org/10.1038/onc.2010.309 . [PMC free article: PMC3005772] [PubMed: 20697346]
- 188.
- Wang J, Chen L, Li Y, Guan XY. Overexpression of cathepsin Z contributes to tumor metastasis by inducing epithelial-mesenchymal transition in hepatocellular carcinoma. PLoS One. 2011;6:e24967. http://dx.doi.org/10.1371/journal.pone.0024967 . [PMC free article: PMC3178578] [PubMed: 21966391]
- 189.
- Sarri D, Rodriguez-Pinilla SM, Hardisson D, Cano A, Moreno-Bueno G, Palacios J. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 2008;68:989–97. http://dx.doi.org/10.1158/0008-5472.CAN-07-2017 . [PubMed: 18281472]
- 190.
- Davies BR, Barraclough R, Davies MP, Rudland PS. Production of the metastatic phenotype by DNA transfection in a rat mammary model. Cell Biol Int. 1993;17:871–9. http://dx.doi. org/10.1006/cbir.1993.1150 . [PubMed: 8220314]
- 191.
- Andersen K, Mori H, Fata J, Bascom J, Oyjord T, Mlandsmo GM, et al. The metastasis-promoting protein S100A4 regulates mammary branching morphogenesis. Dev Biol. 2011;352:181–90. http://dx.doi.org/10.1016/j.ydbio.2010.12.033 . [PMC free article: PMC3643517] [PubMed: 21195708]
- 192.
- Cavallaro U. N-cadherin as an invasion promoter: a novel target for antitumor therapy? Curr Opin Investig Drugs. 2004;5:1274–8. [PubMed: 15648948]
- 193.
- Cme C, Magnino F, Bibeau F, De Santa Barbara P, Becker KF, Theillet C, et al. Snail and slug play distinct roles during breast carcinoma progression. Clin Cancer Res. 2006;12:5395–402. http://dx.doi.org/10.1158/1078-0432.CCR-06-0478 . [PubMed: 17000672]
- 194.
- Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, et al. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell. 2001;7:1267–78. http://dx.doi.org/10.1016/S1097-2765(01)00260-X . [PubMed: 11430829]
- 195.
- Blissett AR, Garbellini D, Calomeni EP, Mihai C, Elton TS, Agarwal G. Regulation of collagen fibrillogenesis by cell-surface expression of kinase dead DDR2. J Mol Biol. 2009;385:902–11. http://dx.doi.org/10.1016/j.jmb.2008.10.060 . [PMC free article: PMC2677101] [PubMed: 18996394]
- 196.
- Pea C, Garca JM, Silva J, Garca V, Rodrguez R, Alonso I, et al. E-cadherin and vitamin D receptor regulation by SNAIL and ZEB1 in colon cancer: clinicopathological correlations. Hum Mol Genet. 2005;14:3361–70. http://dx.doi.org/10.1093/hmg/ddi366 . [PubMed: 16203744]
- 197.
- Spoelstra NS, Manning NG, Higashi Y, Darling D, Singh M, Shroyer KR, et al. The transcription factor ZEB1 is aberrantly expressed in aggressive uterine cancers. Cancer Res. 2006;66:3893–902. http://dx.doi.org/10.1158/0008-5472.CAN-05-2881 . [PubMed: 16585218]
- 198.
- Conn G, Soderman DD, Schaeffer MT, Wile M, Hatcher VB, Thomas KA. Purification of a glycoprotein vascular endothelial cell mitogen from a rat glioma-derived cell line. Proc Natl Acad Sci U S A. 1990;87:1323–7. http://dx.doi.org/10.1073/pnas.87.4.1323 . [PMC free article: PMC53467] [PubMed: 2406719]
- 199.
- Gerber HP, Dixit V, Ferrara N. Vascular endothelial growth factor induces expression of the antiapoptotic proteins Bcl-2 and A1 in vascular endothelial cells. J Biol Chem. 1998;273:13313–6. http://dx.doi.org/10.1074/jbc.273.21.13313 . [PubMed: 9582377]
- 200.
- Dimmeler S, Dernbach E, Zeiher AM. Phosphorylation of the endothelial nitric oxide synthase at ser-1177 is required for VEGF-induced endothelial cell migration. FEBS Lett. 2000;477:258–62. http://dx.doi.org/10.1016/S0014-5793(00)01657-4 . [PubMed: 10908731]
- 201.
- Khurana R, Simons M. Insights from angiogenesis trials using fibroblast growth factor for advanced arteriosclerotic disease. Trends Cardiovasc Med. 2003;13:116–22. http://dx.doi. org/10.1016/S1050-1738(02)00259-1 . [PubMed: 12691676]
- 202.
- DAmore PA, Smith SR. Growth factor effects on cells of the vascular wall: a survey. Growth Factors. 1993;8:61–75. http://dx.doi.org/10.3109/08977199309029135 . [PubMed: 7680568]
- 203.
- Otrock ZK, Mahfouz RA, Makarem JA, Shamseddine AI. Understanding the biology of angiogenesis: review of the most important molecular mechanisms. Blood Cells Mol Dis. 2007;39:212–20. http://dx.doi.org/10.1016/j.bcmd.2007.04.001 . [PubMed: 17553709]
- 204.
- Falcone DJ, McCaffrey TA, Haimovitz-Friedman A, Garcia M. Transforming growth factor-beta 1 stimulates macrophage urokinase expression and release of matrix-bound basic fibroblast growth factor. J Cell Physiol. 1993;155:595–605. http://dx.doi.org/10.1002/jcp.1041550317 . [PubMed: 7684044]
- 205.
- Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9:653–60. http://dx.doi.org/10.1038/nm0603-653 . [PubMed: 12778163]
- 206.
- Thurston G. Role of Angiopoietins and Tie receptor tyrosine kinases in angiogenesis and lymphangiogenesis. Cell Tissue Res. 2003;314:61–8. http://dx.doi.org/10.1007/ s00441-003-0749-6 . [PubMed: 12915980]
- 207.
- Breiteneder-Geleff S, Soleiman A, Kowalski H, Horvat R, Amann G, Kriehuber E, et al. Angiosarcomas express mixed endothelial phenotypes of blood and lymphatic capillaries: podoplanin as a specific marker for lymphatic endothelium. Am J Pathol. 1999;154:385–94. http://dx.doi. org/10.1016/S0002-9440(10)65285-6 . [PMC free article: PMC1849992] [PubMed: 10027397]
- 208.
- Wigle JT, Harvey N, Detmar M, Lagutina I, Grosveld G, Gunn MD, et al. An essential role for Prox1 in the induction of the lymphatic endothelial cell phenotype. EMBO J. 2002;21:1505–13. http://dx.doi.org/10.1093/emboj/21.7.1505 . [PMC free article: PMC125938] [PubMed: 11927535]
- 209.
- He Y, Karpanen T, Alitalo K. Role of lymphangiogenic factors in tumor metastasis. Biochim Biophys Acta. 2004;1654:3–12. [PubMed: 14984763]
- Cancer Invasion and Metastasis: Molecular and Cellular Perspective - Madame Curi...Cancer Invasion and Metastasis: Molecular and Cellular Perspective - Madame Curie Bioscience Database
- Lipoxins as an Immune-Escape Mechanism - Madame Curie Bioscience DatabaseLipoxins as an Immune-Escape Mechanism - Madame Curie Bioscience Database
- Slits and Their Receptors - Madame Curie Bioscience DatabaseSlits and Their Receptors - Madame Curie Bioscience Database
- FOXP Genes, Neural Development, Speech and Language Disorders - Madame Curie Bio...FOXP Genes, Neural Development, Speech and Language Disorders - Madame Curie Bioscience Database
- Regulation of Paracellular Transport across Tight Junctions by the Actin Cytoske...Regulation of Paracellular Transport across Tight Junctions by the Actin Cytoskeleton - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...