

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Liedtke WB, Heller S, editors. TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades. Boca Raton (FL): CRC Press/Taylor & Francis; 2007.

TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades.
Show detailsAbstract
TRPP2 (polycystin-2) is a member of the TRP family of nonselective cation channels that is mutated in human autosomal polycystic kidney disease. TRPP2 has been implicated in Ca2+-dependent mechanosensitive pathways in a variety of biological functions including cell proliferation, sperm fertilization, mating behavior, and asymmetric gene expression. Although its function as a Ca2+-permeable cation channel is well established, its precise role and subcellular localization in the plasma membrane, endoplasmic reticulum (ER), and cilium have remained controversial. The present review summarizes the most pertinent recent evidence regarding the structural and functional properties of TRPP2 channels, focusing on the regulation and physiology of mammalian TRPP2.
INTRODUCING THE TRPP PROTEIN SUBFAMILY
The transient receptor potential (TRP) channel superfamily currently includes 56 related six-transmembrane (TM) domain channels classified in seven subfamilies designated TRPC (“canonical”), TRPV (“vanilloid”), TRPM (“melastatin”), TRPN (“NOMPC,” from no mechanoreceptor potential-C), TRPA (“ankyrin-like with transmembrane domain-1”), TRPML (“mucolipin”), and TRPP (“polycystin”) [1,2]. TRPC, TRPV, and TRPM are related to canonical TRP proteins while TRPN, TRPA, and TRPP are more divergent. TRP channels are linked to a variety of sensory stimuli including phototransduction, thermosensation, and mechanosensation and to multiple integrative cellular functions including Ca2+ and Mg2+ homeostasis and cell cycle.
The TRPP subfamily was named after its founding member polycystin kidney disease-2 (TRPP2 encoded by the PKD2 gene), a gene product mutated in an inherited human disorder known as autosomal dominant polycystic kidney disease (ADPKD). TRPP2 is related to the TRP family of ion channels by virtue of its topological features/structural homology in the sixth TM region and the presence of an ion channel motif.
The polycystin proteins are found in the entire animal kingdom. In humans, the polycystin group contains eight members, which are widely expressed and can be divided structurally into two prototypical subgroups: the PKD1-like proteins and the PKD2 (TRPP2)-like proteins, both having a modest degree of sequence similarity in their C-termini [3–6]. The TRPP subfamily contains three homologous proteins, PKD2, PKD2L1, and PKD2L2, which are currently referred to as TRPP2, TRPP3, and TRPP5 [7]. The mammalian orthologues are highly conserved over the entire length (80–90 percent identity) but show sequence divergence in their cytosolic N-terminal regions. All TRPP2-like proteins are predicted to possess a putative coiled-coil domain at their C-termini, but only TRPP2 and TRPP3 have a Ca2+-binding EF-hand motif. The TRPP2 C-terminus also has a PACS-interacting binding motif, which plays a key role in regulating TRPP2 trafficking between organelles and the cell surface (reference 9 and see below).
TRPP3 was the first PKD2-like protein to be identified as a functional TRP-like cation channel [8]. TRPP2-related channels have large single-channel conductance (80–160 pS) and permeate a number of mono- and divalent cations, including Na+, K+, Ba2+, and Ca2+ [reviewed in reference 10]. Like many TRP channels, TRPP2 activity is blocked by La3+ and reduced by the diuretic amiloride [10]. TRPP3 is usually found to be more permeable to Ca2+ than to other monovalent cations such as Na+ and K+ (PCa/PNa = 4), whereas TRPP2 has a PCa/PNa selectivity ranging from 1 to 3. TRPP2-like cation channels therefore represent relevant routes for calcium entry or release.
PKD1, PKD1L13, and PKDREJ (receptor for egg jelly) are large multidomain proteins and make up the PKD1-like subgroup. Because PKD1-like proteins show very limited sequence similarity with TRP channels, they are not considered members of the TRP superfamily. Each PKD1-like protein contains a large extra-cellular terminal domain, 11 predicted TM segments, and a short intracellular carboxyl terminus. They all possess the combination of REJ (receptor for egg jelly), GPS (G-protein-coupled receptor proteolytic site), and PLAT/LH2 (lipoxygenase homology/polycystin, lipoxygenase, α-toxin) domains that uniquely define them as PKD1 family members. The extracellular region of PKD1 spans more than ~3,000 amino acids and contains a number of adhesive domains that implicate PKD1 in cell–cell and cell–matrix interactions. PKD1 is cleaved at its predicted GPS [11], a feature common to members of the family-B (latrophilin) G-protein-coupled receptors and which may be important for receptor activation or localization. PKD1, PKD1L1, and PKD1L2 encompass a G-protein-interacting site that may be used to regulate at least four different classes of heterotrimeric G-protein activity and multiple downstream effectors, including among others phospholipase C, protein kinase C, adenylyl cyclase, protein kinase A, Janus kinase 2, and nuclear factor of activated T cells (NFAT) [reviewed in reference 4] (Figure 14.1). The intracellular carboxyl terminus of PKD1 also harbors a coiled-coil domain that is involved in physical interaction with TRPP2 [12] and possibly TRPP3a [13].
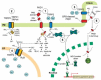
FIGURE 14.1
Signaling and regulation of TRPP2 channels. This illustration depicts the different models for the localization-dependent functions of TRPP2. TRPP2 mediates Ca2+ influx at the plasma membrane (PM) and the ciliary membrane (cilium), where it functions (more...)
PKD1 and PKD1L1, but not PKDREJ, PKD1L2, or PKD1L3, have this coiled-coil domain, suggesting that the PKD1-like family could be subdivided into two groups according to their structural domains. Likewise, though PKD1 and PKD1L1 lack a structurally defined surface channel pore domain, recent data suggest that PKD1L2, PKD1L3, and PKDREJ contain strong ion channel signature motifs [14], suggesting their potential functions as pore-forming channel subunits.
MUTATIONS IN TRPP2 AND POLYCYSTIN-1 CAUSE ADPKD
The founding members of the TRPP family were discovered as genes mutated in ADPKD. ADPKD is a common nephropathy affecting 4 to 6 million people worldwide and is a leading cause of end-stage kidney failure. The disease is typically characterized by defects in the polarized phenotype and function of epithelial kidney cells, leading to abnormal renal tubular cell growth and formation of numerous fluid-filled cysts. Eventually these cysts overwhelm the kidney and destroy the parenchyma. This disease is as yet incurable and is often associated with a number of systemic manifestations including hypertension, intracranial aneurysms, and cardiovalvular abnormalities such as mitral valve prolapse.
In the mid-1990s, mutated genes responsible for ADPKD were identified by positional cloning [15]. ADPKD has been shown to result from loss-of-function mutations either of polycystin-1 or of TRPP2, with PKD1 mutations being the most prevalent causes. The dominance of PKD1 and TRPP2 mutations appears to require both a germ-line mutation of PKD1 or TRPP2 and a subsequent somatic mutation of the wild-type allele. This would explain the relatively late development of ADPKD and the focal nature of epithelial cells giving rise to cysts. However, recent studies indicate that various karyotypic changes, not just loss of heterozygosity at the normal PKD allele, are associated with cystogenesis [16], a situation that illustrates the complexity of cyst formation and raises the question as to whether the two-hit mechanism is the only means to generate a cyst.
Consistent with the broad expression of both genes during early organogenesis, mouse models for ADPKD derived from targeted disruption of either PKD genes die in utero or perinatally with cardiac septal defects and severe cystic manifestations in nephrons and pancreatic ducts.
LOCALIZATION AND TRAFFICKING OF TRPP2
TRPP2 is expressed in a variety of tissues including epithelial cells, vascular smooth muscle, cardiac myocytes, adrenal glands, and ovaries [17]. A long-lasting matter of debate has been the subcellular localization of TRPP2. In recombinant cell-based systems and in most native cells, TRPP2 is found to be concentrated in intracellular compartments and most notably in the ER, as judged by immunofluorescence imaging, cofractionation with ER markers, and sensitivity to endoglycosidase H digestion [18]. TRPP2 encompasses an ER retention signal within its C-terminal domain [18], which seems to prevent trafficking to the cell surface when expressed on its own. Deletion mutants for this ER retention signal translocate to the cell surface and can be detected by immunological and electrophysiological means [19]. Opposing these findings are reports that electrogenic TRPP2 activity is present at the plasma membrane of several cell types following treatment with chemical chaperones/proteasome inhibitors [20,21] or upon overexpression [22]. TRPP2 has also been localized to basolateral plasma membranes, lamelopodia, primary cilia, and mitotic spindles [12,18,23–26].
Recent data are clarifying the confusing picture of TRPP2 localization. First, Köttgen et al. [9] have reported that TRPP2 trafficking between the ER, Golgi, and plasma membrane compartments may be directed by the phosphofurin acidic cluster proteins PACS-1 and PACS-2, two sorting proteins that bind to an acidic cluster in the C-ter domain of TRPP2. Binding of these adaptor proteins to TRPP2 is promoted by protein kinase casein kinase-2-dependent phosphorylation of Ser812. TRPP2 accumulates at the plasma membrane only when both PACS-1 and PACS-2 molecules are absent or upon inhibition of protein kinase casein kinase-2 activity. Mutation of Ser812 to alanine or destruction of the acidic cluster (TRPP2 D815–817A) abrogates the interaction between TRPP2 and PACS proteins and increases whole-cell TRPP2 currents. Thus, mechanisms that regulate the interaction of PACS proteins with TRPP2 are likely to play key roles in routing TRPP2 between the ER and the plasma membrane. TRPP3 and TRPP5 lack the PACS-binding acidic cluster in their C-tails, suggesting that their trafficking is regulated differently from that of TRPP2. This may explain why TRPP3 is targeted to the cell surface when overexpressed in oocytes, while TRPP2 is retained in the ER. Second, a recently discovered protein of 14 kDa dubbed PIGEA-14 (polycystin-2 interactor, Golgi- and ER-associated protein) has been shown to interact with the C-ter of TRPP2. Coexpression of both proteins in HeLa cells and in LLC-PK1 induces a redistribution of TRPP2 as well as PIGEA-14 from the ER to an unorthodox trans-Golgi compartment [27], indicating that intracellular trafficking of TRPP2 is regulated both at the levels of the ER and the trans-Golgi network. Trafficking of TRPP2 is therefore rapidly becoming a key issue to understanding TRPP2 function and dysfunction [28].
TRPP2: A CA2+-REGULATED CATION CHANNEL LOCATED IN THE ENDOPLASMIC RETICULUM
TRPP2 may act as a Ca2+-release channel in ER membranes, which amplifies Ca2+ transients initiated by InsP3-generating plasma membrane receptors (Figure 14.1) [29]. This led to the suggestion that TRPP2 represents a new type of intracellular receptor that, along with IP3Rs and RyRs, may be involved in mediating Ca2+-induced Ca2+ release. TRPP2 appears to be directly activated by Ca2+ and displays a bell-shaped dependence on cytoplasmic Ca2+ [29, see 10 for review]. Although it is yet unclear whether the Ca2+-binding EF hand of TRPP2 is involved in Ca2+-dependent modulation of TRPP2, it is noteworthy that pathogenic TRPP2 mutants with premature termination of the peptide chain in their C-ter lost their ability to sense Ca2+. However, TRPP3 C-terminal artificial truncation mutants lacking the EF hand exhibit basal and Ca2+-activated channel activities, suggesting that, at least for TRPP3, the EF hand and other parts of the carboxyl tail are not key determinants of the Ca2+-dependent activation [30].
Phosphorylation of Ser812 by a putative casein kinase 2 results in a significant increase in the sensitivity of the TRPP2 channel to calcium stimulation [31]. The S812A substitution, which results in loss of phosphorylation of TRPP2, shifts the Ca2+ dependence such that TRPP2 S812A has a maximum open probability at tenfold higher Ca2+ concentrations (~3 μM [Ca2+]) than normal TRPP2. Thus, the maximum open probability of a wild-type TRPP2 channel occurs at a Ca2+ concentration at which the nonphosphorylated form remains closed. Intracellular TRPP2 is therefore likely to have enhanced Ca2+ sensitivity, because the protein kinase casein kinase-2 is opportunely associated with the ER and most TRPP2 is found to be phosphorylated in vivo [31]. As stated above, phosphorylated TRPP2 fails to escape the ER. Only dephosphorylation of S812 would promote TRPP2 translocation to the plasma membrane. Thus, it can be hypothesized that the dephosphorylated form of TRPP2, possibly the form found in the plasma or the ciliary membranes, is unlikely to be activated by a modest increase in bulk Ca2+.
In line with a role of ER-localized TRPP2 in regulating intracellular Ca2+, TRPP2+/− vascular smooth muscle cells show a lower level of TRPP2 and have altered intracellular Ca2+ homeostasis [32]. Furthermore, TRPP2 has been recently shown to interact functionally and physically with IP3R in oocyte expression systems (Figure 14.1) [33]. The physiological relevance of these results remains to be clarified.
COASSEMBLY OF PKD1 AND TRPP2 RECONSTITUTES A CELL SURFACE CA2+-PERMEABLE CATION CHANNEL WITH MULTIPLE FUNCTIONS
The differential cellular and subcellular pattern of expression of PKD1 and TRPP2 in some systems strongly argues that both proteins have independent functions [25]. Notwithstanding, a central issue surrounding TRPP2 is how much TRPP2 channel activity depends on the presence of PKD1. In a lipid bilayer system, recombinant TRPP2 can reconstitute cation channel activity in the absence of PKD1 [34]. Hanaoka et al. [35] provided a functional substratum for the well-established demonstration that PKD1 and TRPP2 interact physically via their C-termini and form heteromeric complexes in vivo [12]. Coexpression of PKD1 and TRPP2 in CHO cells as well as in sympathetic neurons promotes the translocation of TRPP2 to the plasma membrane and generates a nonselective cation channel with perm-selectivity and sensitivity to di-and trivalent cations very similar to those of homomeric TRPP2 (Figure 14.1) [35,36]. The channel activity is not observed when C-terminal interaction between PKD1 and TRPP2 is not allowed, implying that coassembly of PKD1 and TRPP2 is required for the targeting/retention of TRPP2 to the plasma membrane.
The PKD1/TRPP2 complex has been reconstituted at the cell surface of sympathetic neurons [36]. In this model, TRPP2 is likely to act as the ion-translocating component of the polycystin complex because the pharmacological and permeation properties of the PKD1/TRPP2 channel complex resemble those of recombinant homomeric TRPP2 and because homomeric PKD1 cannot form an ion channel by itself [37]. Within the complex, PKD1 and TRPP2 have reciprocal “stabilizing” effects on each other’s function [34,36]. Indeed, the association of PKD1 appears to repress the constitutive activity of TRPP2 [36], which would be detrimental to the cell if uncontrolled. Conversely, TRPP2 binding to PKD1 represses PKD1’s ability to constitutively activate G proteins possibly by steric/competitive interaction among the different PKD1-binding partners [36,37]. These data favor the view that besides its ion channel function, TRPP2 also regulates the downstream effects of PKD1 on its target effectors and genes. Therefore, the balance between TRPP2 and PKD1 expression, which is manifestly disrupted in ADPKD, may play a critical role in normal PKD1–TRPP2 signaling. TRPP2 repression of G-protein activation by PKD1 has been confirmed in the case of PKD1-mediated NFAT (nuclear factor of activated T-cells) activation [38]; more recently, TRPP2 has been shown to impair the nuclear translocation of the PKD1 C-terminus by binding to, and sequestering of, the latter (Figure 14.1) [39].
TRPP2, in conjunction with PKD1, has been shown to regulate messages to the nucleus by preventing the pro-proliferative helix-loop-helix protein Id2 from entering the nucleus (Figure 14.1) [65]. Id2 is known to associate with E proteins and blocks their ability to turn on growth-suppressive genes. Id2 is normally prevented from translocating to the nucleus through its association with the serine-phosphorylated C-terminal domain of TRPP2, which is promoted by PKD1. These data predict that loss-of-function mutations in either TRPP2 or PKD1, or disruption of their functional interaction, cause Id2 to enter the nucleus and turn off growth-suppressive genes, making a case for the involvement of such a mechanism in the pathogenesis of ADPKD.
ACTIVATION OF THE PKD1–TRPP2 COMPLEX
That PKD1 and TRPP2 are interacting partners within a heteromultimeric polycystin complex has been intuited from the observation that mutations in PKD1 or TRPP2 produce virtually identical clinical presentations, irrespective of the causative gene [15]. Reconstituted PKD1–TRPP2 complexes can be activated by applying antibodies directed against the extracellular REJ domain of PKD1 [36]. This activates bidirectional signaling events, concordantly enhancing TRPP2 activity and stimulating heterotrimeric G-protein pathways. Thus, PKD1 and TRPP2 form functionally associated subunits of a receptor-ion channel signaling complex in which PKD1 acts as a “receptor/regulator” that controls TRPP2 activity and G proteins (Figure 14.1) [3,34,36]. The activation of TRPP2 and G proteins appears to proceed through a structural rearrangement of the polycystin complex that requires both proteins to have intact C-termini. This mechanism may mimic a yet-to-be-determined (mechanical? ligand?) extracellular signal that activates the polycystin complex.
This proposed mechanism may be paradigmatic for the function of other polycystin orthologues in a variety of tissues [3]. For example, PKD1 signaling has fascinating functional parallels with the acrosome reaction (AR) in sea urchin spermatozoa, a prerequisite for sperm-egg fusion [40]. AR requires the activation of suREJ1 or suREJ3, two PKD1 orthologues harboring REJ modules; each binds components of the egg jelly [41]. Importantly, antibodies directed against the REJ domain of suREJs induce the AR by opening Ca2+-permeant channels [42]. Recently, suREJ3 has been shown to physically bind to the sea urchin sperm orthologue of TRPP2 in the acrosome plasma membrane [43], raising the possibility that suTRPP2 may be involved in the Ca2+-regulated AR. Collectively, these findings add further weight to the primary importance of the REJ domain in activating the polycystin complex.
Other early evidence in support of the idea that PKD1 and TRPP2 form heteromultimeric complexes came from studies of C. elegans orthologues of ADPKD genes, lov-1 (location of vulva) and pkd-2 [44]. Lov-1, however, has an unrelated extracellular domain to PKD1 in that it lacks the REJ domain and harbors instead several mucin domains [3]. Mutation analysis shows identical male sensory behavioral defects in single or double lov-1 and pkd-2 mutants, indicating that both proteins act together in a single sensory pathway necessary for normal mating behavior [45]. Lov-1 and pkd-2 concentrate in cilia and cell bodies of male-specific sensory neurons, consistent with their functions as associated subunits of a receptor-ion channel complex involved in mechanosensitive signaling.
TRPP2: A BONA FIDE MECHANOSENSITIVE CHANNEL?
Recent studies have provided evidence that PKD1 and TRPP2 colocalize in primary cilia of renal epithelial cells, where they may function in transducing sensory information, such as shear fluid stress [23]. The primary cilium of renal epithelial cells is a solitary nonmotile structure of a few micrometers that arises from the basal body or centriole and projects into the lumen of the tubule. Its central role in cyst formation has been suggested from the primary observation that defects in proteins necessary for the assembly or function of primary cilia such as cystin, polaris, inversin, and kinesin-II cause polycystic kidney diseases. The cilium is proposed to serve as a flow sensor because it can reversibly bend in response to fluid flow rates comparable to those observed in renal tubules and because it was shown to be essential for Madin-Darby canine kidney (MDCK) cells’ ability to sense flow, because deciliated cells are irresponsive to changes in flow rate [46,47]. Fluid shear-force bending of the cilium causes Ca2+ influx through mechanically sensitive channels. Although it is not known whether these mechanosensitive channels reside at the base of cilia or throughout the cilium membrane, one could predict that they localize in the intervening bilayer regions that increase in tension during cilium bending.
A calcium signal through mechanically activated channels is then amplified by Ca2+ release from IP3R stores in MDCK cells and spreads to neighboring cells through gap junctions [46]. These data conflict, however, with a study in murine embryonic kidney epithelial (MEK) cells, where ryanodine receptors instead of IP3Rs have been implicated in Ca2+ amplification [23]. Thus, the cilium acts as the mechanosensor of changes in laminar fluid flow and transduces stimulus energy into change in membrane permeability. In this model, the PKD1–TRPP2 polycystin complex may be envisioned as a mechanotransducer, which is used to signal relevant intratubular information such as flow rates, directing attention to the regulation of Ca2+ influx as a crucial misstep that initiates cystogenesis. However, whether the PKD1–TRPP2 complex is mechanosensitive still awaits direct experimental evidence.
Recently, TRPP2 has been shown to play a central role in establishing the left-right (LR) asymmetry of visceral organs [48], which occurs during early embryonic development when the nodal gene, initially expressed throughout the node, becomes limited to the left margin of the node. The node is a triangular-shaped structure at the distal tip of ~E7 embryos, consisting of endodermally derived cells, each carrying a single cilium on their apical surface. The monocilia located at the center of the node express dynein, a microtubular motor protein, and are motile, producing rotational movement that creates a leftward fluid flow across the node. This leftward nodal flow is critical for the sideness of asymmetric gene expression because mice with immotile cilia develop laterality defects. In contrast, monocilia located in the periphery of the node are immobile (lacking dynein) and act as sensors of directional nodal flow by generating an asymmetric Ca2+ signal [48]. TRPP2 is expressed in both motile and immotile monocilia, yet a perinodal Ca2+ signal is absent in TRPP2−/− mice embryos, suggesting that TRPP2 functions as a mechanotransducer in immotile monocilia and transduces leftward nodal flow into an increase in Ca2+ at the left border of the node. This function would be key for the establishment of a morphogenic gradient at the embryonic node and consistent with the observation that targeted disruption in TRPP2 causes situs inversus in addition to the hallmark cardiac and kidney defects [49]. The lack of laterality defects in PKD1 knockout embryos correlates with the absence of PKD1 in cilia [50], favoring the idea that TRPP2 may be involved in mechanosensation in the absence of PKD1. In the same vein, the orthologue of TRPP2 encoded by the amo gene (almost there) in Drosophila melanogaster is localized to the distal tip of the sperm flagella and, apparently in the absence of the PKD1 orthologue, plays a critical role for directional movement inside the female reproductive tract [51,52]. This suggests that TRPP2 in Drosophila sperm is part of a signaling pathway involved in detecting directional cues that are necessary for entry into the female storage organs, perhaps supporting a common role for Ca2+-dependent TRPP2 signals in both motile and immotile axonemal-based structures.
Despite the widespread utilization of mechanosensitive channels in a variety of physiological processes including the detection of touch, hearing function, blood pressure control, and osmotic pressure, little is known about the molecular structure and organization of vertebrate mechanotransducers. In this respect, the recent identification of TRP channels as core components of mechanoreceptors in C. elegans, Drosophila melanogaster, and vertebrates may offer clues to the conservative mechanoreceptive structural elements of mechanotransducers [53,54].
In the fruit fly, mechanoelectrical responses in bristle sensory neurons occur rapidly upon deflection of the bristle hair shaft and result from the opening—among others—of the NOMPC channel, a member of the TRPN subfamily. NOMPC has a particularly long intracellular amino-terminal tail harboring 29 ankyrin repeats, which are considered to anchor the channel to the cytoskeleton and may mediate the protein–protein interaction of a tethered mechanism that might be required for mechanical gating.
The nematode C. elegans senses nose touch by stimulating ciliated nociceptive sensory neurons, which detect, among others, mechanical and osmotic stimuli. The OSM-9 channel is thought to be part of the mechanosensitive channel because OSM-9 mutants are defective in osmotic avoidance and in sensitivity to nose touch. OSM-9 is a homologue to members of the TRPV channels, with three ankyrin-repeat domains at its amino-terminal intracellular domain. TRPV2 and TRPV4, its invertebrate TRP counterparts, have multiple ankyrin-repeat domains and are implicated in vertebrate mechanosensation in that they can sense membrane stretch [55] and hypo-osmotic stress [56], respectively. With regard to mechanical gating, it is noteworthy that TRPV4 requires the amino-terminal domain with the three ankyrin-repeats to sense physical challenges [57]. More recently, Corey et al. [58] have shown that TRPA1 (also called ANKTM1), which harbors 17 ankyrin domains, constitutes or is a component of the mechanosensitive transduction channel of vertebrate hair cells. Although the mechanism of activation by mechanical force is not yet established, an ankyrin repeat has been hypothesized to form a springlike gating structure, consistent with a “tethered channel” model [4].
Neither PKD1 nor TRPP2 display ankyrin-repeats that would allow tight interactions between the channel complex and the cytoskeleton. TRPP2, however, has been shown to connect indirectly with the actin cytoskeletal network, though it remains to be shown whether these actin-based elements play a role in cilium mechanotransduction, given that the cilium is primarily the domain of microtubules rather than actin filaments [3]. On this last issue, the extracellular domain of PKD1 has been shown to display a dynamic extensibility whereby its length might be regulated through unfolding/refolding its Ig-like domains [59]. Although these mechanical properties of PKD1 are important in the context of mechanosensation, they seem more appropriate to provide structural support in cell–cell or cell–matrix interactions at basolateral membranes than mechanosensation in the solitary cilium.
At this point, the available information does not entirely support the candidacy of a PKD1–TRPP2 complex as the core component of the primary cilium’s mechanosensitive apparatus. This would seem ample reason to consider alternative models in which mechanical force is transmitted indirectly to the protein complex or via an auxiliary subunit. On the one hand, this may imply that the TRPP2 channel acts as a nonmechanosensitive amplifier of a true mechanically gated channel, with cytosolic Ca2+ acting as a suitable activator of TRPP2. A critical issue to be established is the polymodal nature of TRPP2 regulation (i.e., whereby channel opening is not only dependent on mechanical stimuli but also modulated by PKD1, Ca2+, H+, and phosphorylation). Thus, the basic distinction between physical and chemical mechanisms of PKD1–TRPP2 mechanotransduction is not yet made. On the other hand, it is also conceivable that TRPP2 coassembles with other TRP channels to form a mechanosensitive channel as many invertebrate TRP-related channels do. A tantalizing link points to TRPV4, because it is expressed in renal epithelial cells, particularly in the distal nephron and collecting ducts, which are flow-sensitive segments [60]. In this regard, preliminary evidence by Walz’s group indicate that TRPV4 and TRPP2 functionally interact and colocalize in the primary cilium [61]. TRPC1 has also been recently found to be a component of the vertebrate mechanosensitive cation channel [62]. In contrast to TRPA1, TRPC1 is gated by tension developed in the lipid bilayer. Interestingly, TRPC1 is known to interact with TRPP2 in expression systems [63] and to form functional heterotetramers with TRPP2 [64], suggesting that TRPC1 as well may contribute to the mechanosensory TRPP2 apparatus (Figure 14.1).
CONCLUDING REMARKS
In the past five years, considerable progress has been achieved in evaluating the distribution, pathophysiology, and functional characteristics of TRPP proteins. Most notably, TRPP2 channels have been shown to traffic to different subcellular compartments and to display specific subcellular functions. In renal primary cilia, TRPP2 interacts with PKD1, a process that may be essential functionally for the regulation of mechanosensation and the cell cycle. In the ER, TRPP2 serves as an intracellular Ca2+ release channel. It would be important to identify ligands for PKD1 that affect the function of the PKD1–TRPP2 complex and to identify the factors interacting with TRPP2 that determine and regulate its compartment-specific functions. These studies will contribute not only to a better understanding of TRPP physiological functions but also to the development of new strategies for targeted therapeutic intervention.
Acknowledgments
ACKNOWLEDGMENTS
This work was supported by the Centre National de la Recherche Scientifique (CNRS), and by grants from the Agence Nationale de la Recherche (ANR Cardiovasculaire, obésité et diabète, N° ANR-05-PCOD-029-02), la Fondation Schlumberger pour l’Education et la Recherche, the French Ministère délégué à la Recherche (ACI Jeune Chercheurs, N° 5294), and a fellowship from the French Research Ministry and the University of Méditerranée Marseille.
REFERENCES
- 1.
- Nilius B, Voets T. TRP channels: a TR(I)P through a world of multifunctional cation channels. Pflügers Arch. 2005;451:1. [PubMed: 16012814]
- 2.
- Montell C, et al. A unified nomenclature for the superfamily of TRP cation channels. Mol Cell. 2002;9:229. [PubMed: 11864597]
- 3.
- Delmas P. Polycystins: from mechanosensation to gene regulation. Cell. 2004;118:145. [PubMed: 15260985]
- 4.
- Delmas P. Polycystins: polymodal receptor/ion-channel cellular sensors. Pflügers Arch. 2005;451:264. [PubMed: 15889307]
- 5.
- Igarashi P, Somlo S. Genetics and pathogenesis of polycystic kidney disease. J Am Soc Nephrol. 2002;13:2384. [PubMed: 12191984]
- 6.
- Nauli SM, Zhou J. Polycystins and mechanosensation in renal and nodal cilia. Bioessays. 2004;26:844. [PubMed: 15273987]
- 7.
- Montell C. The TRP superfamily of cation channels. Sci STKE. 2005:re3. [PubMed: 15728426]
- 8.
- Chen XZ, et al. Polycystin-L is a calcium-regulated cation channel permeable to calcium ions. Nature. 1999;401:383. [PubMed: 10517637]
- 9.
- Köttgen M, et al. Trafficking of TRPP2 by PACS proteins represents a novel mechanism of ion channel regulation. EMBO J. 2005;24:705. [PMC free article: PMC549624] [PubMed: 15692563]
- 10.
- Delmas P, et al. Polycystins, calcium signaling, and human diseases. Biochem Biophys Res Commun. 2004;322:1374. [PubMed: 15336986]
- 11.
- Qian F, et al. Cleavage of polycystin-1 requires the receptor for egg jelly domain and is disrupted by human autosomal-dominant polycystic kidney disease 1-associated mutations. Proc Natl Acad Sci USA. 2002;99:16981. [PMC free article: PMC139255] [PubMed: 12482949]
- 12.
- Newby LJ, et al. Identification, characterization, and localization of a novel kidney polycystin-1–polycystin-2 complex. J Biol Chem. 2002;277:20763. [PubMed: 11901144]
- 13.
- Murakami M, et al. Genomic organization and functional analysis of murine PKD2L1. J Biol Chem. 2005;280:5626. [PubMed: 15548533]
- 14.
- Li A, Tian X, Sung SW, Somlo S. Identification of two novel polycystic kidney disease-1-like genes in human and mouse genomes. Genomics. 2003;81:596. [PubMed: 12782129]
- 15.
- Sutters M, Germino GG. Autosomal dominant polycystic kidney disease: molecular genetics and pathophysiology. J Lab Clin Med. 2003;141:91. [PubMed: 12577044]
- 16.
- Gogusev J, et al. Molecular cytogenetic aberrations in autonomal dominant polycystic kidney disease tissue. J Am Soc Nephrol. 2003;14:359. [PubMed: 12538736]
- 17.
- Ong AC. Polycystin expression in the kidney and other tissues: complexity, consensus and controversy. Exp Nephrol. 2000;8:208. [PubMed: 10940718]
- 18.
- Cai Y, et al. Identification and characterization of polycystin-2, the PKD2 gene product. J Biol Chem. 1999;274:28557. [PubMed: 10497221]
- 19.
- Chen XZ, et al. Transport function of the naturally occurring pathogenic polycystin-2 mutant, R742X. Biochem Biophys Res Commun. 2001;282:1251. [PubMed: 11302751]
- 20.
- Luo Y, et al. Native polycystin 2 functions as a plasma membrane Ca2+-permeable cation channel in renal epithelia. Mol Cell Biol. 2003;23:2600. [PMC free article: PMC150742] [PubMed: 12640140]
- 21.
- Vassilev PM. Polycystin-2 is a novel cation channel implicated in defective intra-cellular Ca2+ homeostasis in polycystic kidney disease. Biochem Biophys Res Commun. 2001;282:341. [PubMed: 11264013]
- 22.
- Gonzalez-Perrett S. Polycystin-2, the protein mutated in autosomal dominant polycystic kidney disease (ADPKD), is a Ca2+-permeable nonselective cation channel. Proc Natl Acad Sci USA. 2001;98:1182. [PMC free article: PMC14729] [PubMed: 11252306]
- 23.
- Nauli SM. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33:129. [PubMed: 12514735]
- 24.
- Yoder BK, Hou X, Guay-Woodford LM. The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris and cystin, are co-localized in renal cilia. J Am Soc Nephrol. 2002;13:2508. [PubMed: 12239239]
- 25.
- Foggensteiner L, et al. Cellular and subcellular distribution of polycystin-2, the protein product of the PKD2 gene. J Am Soc Nephrol. 2000;11:814. [PubMed: 10770959]
- 26.
- Rundle DR, Gorbsky GJ, Tsiokas L. PKD2 interacts and co-localizes with mDia1 to mitotic spindles of dividing cells: role of mDia1 in PKD2 localization to mitotic spindles. J Biol Chem. 2004;279:29728. [PubMed: 15123714]
- 27.
- Hidaka S, Könecke V, Osten L, Witzgall R. PIGEA-14, a novel coiled-coil protein affecting the intracellular distribution of polycystin-2. J Biol Chem. 2004;279:35009. [PubMed: 15194699]
- 28.
- Köttgen M, Walz G. Subcellular localization and trafficking of polycystins. Pflügers Arch. 2005;451:286. [PubMed: 15895248]
- 29.
- Koulen P. Polycystin-2 is an intracellular calcium release channel. Nat Cell Biol. 2002;4:191. [PubMed: 11854751]
- 30.
- Li Q, Liu Y, Zhao W, Chen XZ. The calcium-binding EF-hand in polycystin-L is not a domain for channel activation and ensuing inactivation. FEBS Lett. 2002;516:270. [PubMed: 11959145]
- 31.
- Cai Y, et al. Calcium dependence of polycystin-2 channel activity is modulated by phosphorylation at Ser812. J Biol Chem. 2004;279:19987. [PubMed: 14742446]
- 32.
- Qian Q, et al. Pkd2 haploinsufficiency alters intracellular calcium regulation in vascular smooth muscle cells. Hum Mol Genet. 2003;12:1875. [PubMed: 12874107]
- 33.
- Li Y, et al. Polycystin 2 interacts with type I inositol 1,4,5-triphosphate receptor to modulate intracellular Ca2+ signaling. J Biol Chem. 2005;280:41298. [PubMed: 16223735]
- 34.
- Xu GM, et al. Polycystin-1 activates and stabilizes the polycystin-2 channel. J Biol Chem. 2003;278:1457. [PubMed: 12407099]
- 35.
- Hanaoka K, et al. Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature. 2000;408:990. [PubMed: 11140688]
- 36.
- Delmas P, et al. Gating of the polycystin ion channel signaling complex in neurons and kidney cells. FASEB J. 2004;18:740. [PubMed: 14766803]
- 37.
- Delmas P, et al. Constitutive activation of G-proteins by polycystin-1 is antagonized by polycystin-2. J Biol Chem. 2002;277:11276. [PubMed: 11786542]
- 38.
- Puri S, et al. Polycystin-1 activates the calcineurin/NFAT (nuclear factor of activated T-cells) signaling pathway. J Biol Chem. 2004;279:55455. [PubMed: 15466861]
- 39.
- Chauvet V, et al. Mechanical stimuli induce cleavage and nuclear translocation of the polycystin-1 C-terminus. J Clin Invest. 2004;114:1433. [PMC free article: PMC525739] [PubMed: 15545994]
- 40.
- Mengerink KJ, Moy GW, Vacquier VD. suREJ proteins: new signalling molecules in sea urchin spermatozoa. Zygote. 2000;8:S28. [PubMed: 11191296]
- 41.
- Hirohashi N, Vacquier VD. High molecular mass egg fucose sulfate polymer is required for opening both Ca2+ channels involved in triggering the sea urchin sperm acrosome reaction. J Biol Chem. 2002;277:1182. [PubMed: 11700311]
- 42.
- Moy GW, et al. The sea urchin sperm receptor for egg jelly is a modular protein with extensive homology to the human polycystic kidney disease protein, PKD1. J Cell Biol. 1996;133:809. [PMC free article: PMC2120838] [PubMed: 8666666]
- 43.
- Neill AT, Moy GW, Vacquier VD. Polycystin-2 associates with the poly-cystin-1 homolog, suREJ3, and localizes to the acrosomal region of sea urchin spermatozoa. Mol Reprod Dev. 2004;67:472. [PubMed: 14991739]
- 44.
- Barr MM, Sternberg PW. A polycystic kidney-disease gene homologue required for male mating behaviour in. C elegans Nature. 1999;401:386. [PubMed: 10517638]
- 45.
- Barr MM, et al. The Caenorhabditis elegans autosomal dominant polycystic kidney disease gene homologs lov-1 and pkd-2 act in the same pathway. Curr Biol. 2001;11:1341. [PubMed: 11553327]
- 46.
- Praetorius HA, Spring KR. Bending the MDCK cell primary cilium increases intracellular calcium. J Membr Biol. 2001;184:71. [PubMed: 11687880]
- 47.
- Praetorius HA, Spring KR. Removal of the MDCK cell primary cilium abolishes flow sensing. J Membr Biol. 2003;191:69. [PubMed: 12532278]
- 48.
- McGrath J, et al. Two populations of node monocilia initiate left-right asymmetry in the mouse. Cell. 2003;114:61. [PubMed: 12859898]
- 49.
- Pennekamp P, et al. The ion channel polycystin-2 is required for left-right axis determination in mice. Curr Biol. 2002;12:938. [PubMed: 12062060]
- 50.
- Karcher C, et al. Lack of laterality phenotype in Pkd1 knock-out embryos correlates with the absence of polycystin-1 in nodal cilia. Differentiation. 2005;73:425. [PubMed: 16316413]
- 51.
- Gao Z, Ruden DM, Lu X. PKD2 cation channel is required for directional sperm movement and male fertility. Curr Biol. 2003;13:2175. [PubMed: 14680633]
- 52.
- Watnick TJ, et al. A flagellar polycystin-2 homolog required for male fertility in. Drosophila Curr Biol. 2003;13:2179. [PubMed: 14680634]
- 53.
- Pedersen SF, Owsianik G, Nilius B. TRP channels: an overview. Cell Calcium. 2005;38:233. [PubMed: 16098585]
- 54.
- O’Neil RG, Heller S. The mechanosensitive nature of TRPV channels. Pflügers Arch. 2005;451:193. [PubMed: 15909178]
- 55.
- Muraki K, et al. TRPV2 is a component of osmotically sensitive cation channels in murine aortic myocytes. Circ Res. 2003;93:829. [PubMed: 14512441]
- 56.
- Alessandri-Haber N, et al. Hypotonicity induces TRPV4-mediated nociception in rat. Neuron. 2003;39:497. [PubMed: 12895423]
- 57.
- Liedtke W, et al. Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell. 2000;103:525. [PMC free article: PMC2211528] [PubMed: 11081638]
- 58.
- Corey DP, et al. TRPA1 is a candidate for the mechanosensitive transduction channel of vertebrate hair cells. Nature. 2004;432:723. [PubMed: 15483558]
- 59.
- Qian F, et al. The nanomechanics of polycystin-1 extracellular region. J Biol Chem. 2005;280:40723. [PMC free article: PMC2703997] [PubMed: 16219758]
- 60.
- Tian W, et al. Renal expression of osmotically responsive cation channel TRPV4 is restricted to water-impermeant nephron segments. Am J Physiol. 2004;287:F17. [PubMed: 15026302]
- 61.
- Köttgen M, et al. Polycystin-2 and TRPV4 form a functional heteromultimeric complex that might act as a cilial mechanosensor. J Am Soc Nephrol. 2005;16:TH-FC116.
- 62.
- Maroto R, et al. TRPC1 forms the stretch-activated cation channel in vertebrate cells. Nature Cell Biol. 2005;7:179. [PubMed: 15665854]
- 63.
- Tsiokas L, et al. Specific association of the gene product of PKD2 with the TRPC1 channel. Proc Natl Acad Sci USA. 1999;9:3934. [PMC free article: PMC22398] [PubMed: 10097141]
- 64.
- Delmas P. Assembly and gating of TRPC channels in signalling microdomains. Novartis Found Symp. 2004;258:75. [PubMed: 15104177]
- 65.
- Li X, et al. Polycystin-1 and polycystin-2 regulate the cell cycle through the helix-loop-helix inhibitor Id2. Nature Cell Biol. 2005;7:1202. [PubMed: 16311606]
- Abstract
- INTRODUCING THE TRPP PROTEIN SUBFAMILY
- MUTATIONS IN TRPP2 AND POLYCYSTIN-1 CAUSE ADPKD
- LOCALIZATION AND TRAFFICKING OF TRPP2
- TRPP2: A CA2+-REGULATED CATION CHANNEL LOCATED IN THE ENDOPLASMIC RETICULUM
- COASSEMBLY OF PKD1 AND TRPP2 RECONSTITUTES A CELL SURFACE CA2+-PERMEABLE CATION CHANNEL WITH MULTIPLE FUNCTIONS
- ACTIVATION OF THE PKD1–TRPP2 COMPLEX
- TRPP2: A BONA FIDE MECHANOSENSITIVE CHANNEL?
- CONCLUDING REMARKS
- Acknowledgments
- REFERENCES
- Review TRPP2: Ca2+-permeable cation channel and more.[Cell Mol Biol (Noisy-le-grand)...]Review TRPP2: Ca2+-permeable cation channel and more.Giamarchi A, Padilla F, Crest M, Honore E, Delmas P. Cell Mol Biol (Noisy-le-grand). 2006 Dec 30; 52(8):105-14. Epub 2006 Dec 30.
- Review The versatile nature of the calcium-permeable cation channel TRPP2.[EMBO Rep. 2006]Review The versatile nature of the calcium-permeable cation channel TRPP2.Giamarchi A, Padilla F, Coste B, Raoux M, Crest M, Honoré E, Delmas P. EMBO Rep. 2006 Aug; 7(8):787-93.
- Review TRPP2 and autosomal dominant polycystic kidney disease.[Biochim Biophys Acta. 2007]Review TRPP2 and autosomal dominant polycystic kidney disease.Köttgen M. Biochim Biophys Acta. 2007 Aug; 1772(8):836-50. Epub 2007 Jan 17.
- Review Cellular and molecular function of mucolipins (TRPML) and polycystin 2 (TRPP2).[Pflugers Arch. 2005]Review Cellular and molecular function of mucolipins (TRPML) and polycystin 2 (TRPP2).Qian F, Noben-Trauth K. Pflugers Arch. 2005 Oct; 451(1):277-85. Epub 2005 Jun 22.
- Review Polycystin-2 (TRPP2): Ion channel properties and regulation.[Gene. 2022]Review Polycystin-2 (TRPP2): Ion channel properties and regulation.Cantero MDR, Cantiello HF. Gene. 2022 Jun 15; 827:146313. Epub 2022 Mar 18.
- Activation Mechanisms and Functional Roles of TRPP2 Cation Channels - TRP Ion Ch...Activation Mechanisms and Functional Roles of TRPP2 Cation Channels - TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades
- Genetics Can Be Painless: Molecular Genetic Analysis of Nociception in Drosophil...Genetics Can Be Painless: Molecular Genetic Analysis of Nociception in Drosophila - TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades
- The Ca2+-Activated TRP Channels: TRPM4 and TRPM5 - TRP Ion Channel Function in S...The Ca2+-Activated TRP Channels: TRPM4 and TRPM5 - TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades
Your browsing activity is empty.
Activity recording is turned off.
See more...