

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Liedtke WB, Heller S, editors. TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades. Boca Raton (FL): CRC Press/Taylor & Francis; 2007.

TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades.
Show detailsCellular function, in particular neuronal activity, is the result of an orchestrated interplay between membrane receptors at the cell surface and intracellular signaling proteins. Membrane receptors transduce external signals into cellular responses by activating cytosolic metabolic pathways. Regulation of the spatial and temporal distribution of membrane receptors begins to be recognized as an important mechanism for controlling the magnitude and time course of cellular signaling [1]. For instance, long-term changes in neuronal plasticity are the result of transcriptional and translational regulation of protein biogenesis and trafficking. Similarly, short-term changes in synaptic function can be achieved by altering the levels of key surface proteins involved in synaptic transmission, such as neurotransmitter receptors and transporters [1]. Thus, modulation of receptor function by trafficking and redistribution plays a central role in synaptic plasticity [1–3]. These processes increase the complexity of the signaling mechanisms in peripheral and central synapses.
Protein trafficking studies, carried out in yeast and mammalian cells, have revealed the underlying mechanism by which proteins travel between intracellular compartments and the plasma membrane. Membrane proteins are synthesized in the endoplasmic reticulum (ER), and ensuing posttranslational modifications such as glycosylation take place in both the ER and the Golgi apparatus. Appropriately folded and assembled membrane proteins are subsequently sorted out into vesicles for trafficking and delivery to the cell surface by a constitutive or regulated pathway of vesicle exocytosis [4]. The constitutive exocytotic route is primarily used by cells for turnover of both membrane proteins and lipids, thus ensuring the removal of aged or damaged membrane components. In contrast, the regulated secretory pathway is used by specialized cells, such as endocrine cells, neurons, acinar cells of the pancreas, mast cells, insulin-sensitive cells, and many other cell types, as a mechanism of cellular communication. Regulated exocytosis is produced only in response to a stimulus that provokes the mobilization, docking, and fusion of an intracellular pool of vesicles stored near the membrane region where they will fuse and release their components [1–5].
Mechanistically, both types of exocytosis use a cascade of protein–protein interactions to ensure the efficient delivery of their vesicle cargo to the membrane [2,5]. At a molecular level, vesicle traffic uses the SNAREs (soluble N-ethylmaleimide-sensitive factor attachment protein receptors), a large family of proteins that are present on all organelles and mediate intracellular vesicle trafficking and secretion (Figure 23.1) [2,5]. The interaction of complementary SNARE proteins found on opposing membranes exemplifies an attractive lock-and-key mechanism, which presumably underlies the remarkable specificity of vesicle docking to target membranes and their consequent fusion [5]. For instance, the interaction of the vesicle membrane SNARE (v-SNARE) synaptobrevin 2, and the corresponding SNAREs in the target membrane (t-SNAREs), syntaxin 1 and SNAP25, results in the formation of a remarkably stable ternary complex that ensures the docking and fusion of vesicles at the active zone upon receiving the stimulus, normally an increase in the intracellular concentration of Ca2+ ([Ca [2]]i).+5 Similarly, molecular interactions of synaptobrevin 2 analogues with syntaxin 1 and SNAP25-like target membrane proteins are thought to be essential for vesicle targeting and fusion in all eukaryotes [2]. It is quite well established that a significant increase in [Ca2+]i is required for triggering the SNARE-mediated fusion of secretory vesicles, although SNARE-dependent exocytosis of vesicles, in particular recycling vesicles, may occur at resting [Ca2+]i by the activation of other signaling pathways [2]. Nonetheless, the rate of vesicle recycling is regulated by changes in the [Ca2+]i
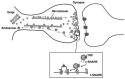
FIGURE 23.1
SNARE-dependent exocytosis of transport and recycling vesicles at a synapse. The two pathways of generating synaptic vesicles containing TRP channels are depicted: a pathway where vesicles are transported from the Golgi to the synaptic terminal, and the (more...)
Vesicle exocytosis is a highly regulated process counterbalanced by endocytosis of membrane components that ensures constant plasma membrane content. Internalization of receptors and transporters is mediated by a general clathrin-dependent pathway that moves endocytosed proteins to the endosome for appropriate sorting into vesicles, either for release or for transport depending on their final destination [3,4]. The endosome also provides a mechanism to maintain the proper composition of release vesicle proteins by adding newly synthesized vesicle proteins, removing aged polypeptides for degradation, and separating nonsecretory vesicle proteins [3]. Recent evidence suggests that synaptic vesicles are most likely formed at the nerve terminal by endocytosis. This novel notion considers that newly synthesized membrane proteins traffic to the nerve terminal in transport packets and become incorporated into synaptic vesicles at the synapse, thus indicating that synaptic vesicle formation relies primarily on an endocytotic pathway [3,4]. This hypothesis is particularly attractive for the regulation of the surface expression of ion channels, neurotransmitter receptors, and transporters presynaptically due to the lack of biosynthetic pools of membrane proteins available in the synapse [4].
Traditionally, the constitutive exocytotic pathway has been associated to the traffic of membrane proteins to the cell surface, while the regulated secretory pathway has been implicated in the activity-dependent release of neurotransmitters, neuropeptides, and hormones. However, in the past decade emerged the notion that the surface expression of membrane proteins, especially ion channels, receptors, and transporters, may be mediated by a regulated release pathway as well [2–4]. Indeed, cumulative evidence indicates that the function of these plasma membrane proteins can be increased or decreased by accommodating the level of the surface-expressed protein. Indeed, dynamic regulation of the rate of either insertion or retrieval or both of integral membrane proteins in response to stimuli embodies the strategic regulation of their surface expression [3,4]. Examples of receptors and transporters that are modulated by vesicle trafficking are the glucose transporter (GluT4) [6], Aquaporin 2 (AQP2) [7], epithelial sodium channel (ENac) [8], cystic fibrosis transmembrane conductance regulator (CFTR) [8], as well as ionotropic ligand-gated receptors such as -amino-3-hydroxy-5-methylisoxazole-4-propionate (AMPA), N-methyl-D-aspartate (NMDA), gamma-aminobutyrate (GABAA), and the purinergic receptor P2X4 [9]. In addition, several studies have demonstrated that this signaling pathway is also commonly used by members of the transient receptor potential (TRP) family of ion channels for modulation of their physiological function [10].
TRP DEFINITIONS AND FAMILIES
The TRP channels are a superfamily of ion channels that play a wide diversity of physiological functions and are present in many tissues and almost all cell types [11,12]. Most TRP channels are nonselective cation channels with low voltage dependence. TRP channels use a wide variety of activation and regulatory mechanisms and carry out functions as diverse as thermosensation, pheromone reception, magnesium homeostasis, and regulation of vascular tone. Interestingly, these channels are considered molecular gateways in sensory and regulatory systems.
The founder member of the family, the TRP channel, was cloned from the fly Drosophila. The phenotype of the fly, trp, was caused by a sustained response in the electroretinogram in response to continuous light, instead of a transient response, with a concomitant decrease in the light-induced Ca2+ influx. Mutation in the gene TRP caused the defect in a Ca2+ channel required for Ca2+ influx and loss of the transient voltage response [13]. A second gene, trp-like (trpl), with ~40 percent identity with trp, encodes a second class of light-sensitive channel that is responsible for the light-induced current [14].
Since the cloning of the fly TRP, more than 50 different gene products have been reported from fungal genomes to upper mammals. Within the animal kingdom, different TRP genes are well conserved. In mice and humans, 28 TRP homologue genes have been described, whereas in invertebrates there are close to 15 members. Based on their amino acid sequence homology, mammalian TRP channels are divided into six subfamilies, namely the “canonical” TRPC (composed of TRPC1 to TRPC7), “vanilloid” TRPV (with TRPV1 to TRPV6), “melastatin-related” TRPM (with TRPM1 to TRPM8), “polycystin-related” TRPP, “mucolipin-related” TRPML and “ankyrin” TRPA [11,12,15]. An additional family, TRPN, has been described in Drosophila. [15]
TRP proteins have a common topology of six transmembrane segments (S1–S6) with a pore region between the fifth and sixth segment, and cytoplasmic N- and C-termini, reminiscent of voltage-dependent K+ (Kv) channels (Figure 23.2). The charged residues in the putative S4 helix, which usually underlie voltage-dependent gating to Kv channels, are replaced by noncharged amino acids. The length of the cytoplasmic domains varies within subfamilies as well as the structural and functional domains associated to them. As for Kv channels, a functional unit of TRP channel requires tetrameric assembly of homomeric or heteromeric TRP polypeptides. TRPV and TRPC members contain two to four ankyrin domains at the N-terminus that are thought to interact with the cytoskeleton as well as with other cytosolic proteins [11]. In the C-terminus domain, proximal to the S6 transmembrane segment, the TRP domain, a highly conserved segment of 25 amino acids (six of which are referred to as the TRP box), is thought to conform an association domain involved in subunit multimerization to give rise to an active channel [16]. In addition, a putative calmodulin (CaM)-binding domain in the COOH terminal region has been reported that binds CaM in a Ca2+-dependent manner [17,18].
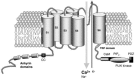
FIGURE 23.2
Molecular model of the TRP receptor subunit. The figure displays a membrane domain composed of six transmembrane segments (S1–S6) with an amphipathic region between the fifth and sixth segment that forms the channel conductive pore. The protein (more...)
Mammalian TRPCs are closest in sequence to Drosophila TRP and TRPL and were the first to be cloned. Initially, these channels were proposed to function as store-operated Ca2+ channels (SOCs) [19]. SOC channels represent the primordial Ca2+entry pathway. They activate upon Ca2+ release from internal stores either by the Ca2+-ATPase inhibitor thapsigargin, or by the presence of ionomycin in Ca2+-free medium [19,20]. Store depletion causes gating of plasma membrane Ca2+-permeable channels, resulting in Ca2+ influx. SOCs are believed to replenish and maintain ER Ca2+ levels and to generate prolonged Ca2+ signals. The most intensely studied SOC is the Ca2+ release–activated Ca2+ current (ICRAC). ICRAC Ca2+ is a nonvoltage-activated and selective channel, inwardly rectifying at negative voltages, first described in mast cells [20]. ICRAC is not the only SOC channel because other store-operated influx have been described with different biophysical properties in other tissues such as epidermal, endothelial, vascular smooth muscle, and neuronal cells. Although the molecular identity of SOC channels is yet unknown, it has been proposed that they are TRP proteins. Specifically, several TRPC channels have been shown to mediate Ca2+ entry with functional and biophysical properties that mirror those of SOC channels [21,22]. In addition, the TRPV6 channel has a high Ca2+ selectivity that suggests a functional similarity to the CRAC channel, and in some studies activates in response to store depletion [23,24], although this conclusion is still a matter of discussion [25,26]. Thus, despite all these findings, the role of TRP channels in mediating Ca2+ entry in response to Ca2+ store depletion and receptor activation remains uncertain and controversial.
TRP TRANSLOCATION AS AN ACTIVATION AND REGULATORY MECHANISM
TRP channels use a wide variety of activation and regulatory mechanisms to carry their functions. For instance, channel activity is affected by different physical stimuli such as light, temperature, osmolarity, mechanical force, or by different chemical or biochemical factors such as pH and external and internal ligands. These stimuli may directly activate the channels such as with TRPV1, which is gated by heat, acid, and vanilloids, or the stimuli may open the channels through a phospholipase C-inositol trisphosphate-mediated pathway [11,12]. For instance, TRPC channels activate primarily in response to PLC-coupled receptors and interact with intracellular InsP3 receptors, suggesting that they can receive information directly from Ca2+ stores, although there is also evidence that TRPC channels can function independently of stores [11,12,15]. Some members of the TRP channel family appear to be constitutively active channels because they are not gated by an identified stimulus.
Cumulative evidence substantiates the tenet that regulated translocation of TRP proteins to the plasma membrane may be a potentially common, but poorly understood, mode of activation of these channels (Figure 23.1) [10]. This mode of activation has been particularly proposed for those TRP channels that are constitutively active, and whose overactivity may lead to Ca2+ overload, although the members of the TRP family that are regulated by this mechanism is augmenting. Regulated translocation of TRP-containing vesicles could be envisioned as a strategy for brief, controlled increases in Ca2+ influx. More generally, fast activity-dependent incorporation of TRP channels from a cytosolic vesicle pool to the plasma membrane, followed by endocytotic uptake, could exemplify an elegant mechanism of ion channel regulation [4]. This process has been demonstrated for several members of the TRP channel family as described next.
Activation of SOC Channels by Protein Translocation to the Plasma Membrane
Although the molecular identity of SOC channels is yet elusive, there is evidence that TRP channels may underlie SOC-mediated Ca2+ influx. These channels are activated by store depletion through a yet unknown mechanism. Three hypotheses have been proposed: [19] (1) the existence of a diffusible factor released from the empty stores; (2) direct coupling of SOC channels in the plasma membrane with a protein sensor in the ER; and (3) vesicle-mediated translocation of SOC channels from a readily available, sub-plasmalemmal vesicle pool. Yao et al. [27], in a study of SOC activity in Xenopus oocytes, documented that SOC activity could be measured in the absence of a small messenger molecule. More interestingly, these authors showed that SOC function was affected by treatments that specifically abrogated vesicle trafficking to and fusion with the plasma membrane. In particular, regulation of Rho A significantly influenced the magnitude of the SOC-mediated ionic currents in oocytes [27]. More important, SOC-dependent currents were inhibited 50 percent by treatment with botulinum neurotoxin A (BoNTA) and by a dominant-negative mutant of SNAP-25, thus indicating that SOC currents are due to translocation of channels to the cell surface by SNARE-mediated exocytosis. Similarly, BoNTA and tetanus neurotoxin (TeNT) reduced cyclopiazonic-induced, SOC-mediated Ca2+ entry into embryonic kidney cells [28], further substantiating the involvement of a regulated secretory pathway for SOC activation. However, the vesicular delivery hypothesis was questioned by Scott et al. [29], who reported that SOC-dependent Ca2+ fluxes were insensitive to BoNTA, TeNT, and a dominant-negative mutant of NSF, suggesting that SOC channels and the machinery necessary to activate them are present in the plasma membrane prior to activation. Taken together, these results imply that trafficking of SOC channels to the plasma membrane may contribute at least in part to channel gating, although further experimental evidence is needed to demonstrate this mechanism of activation.
Functional Modulation of TRPL and TRP3 Channels
Important evidence that a TRP channel undergoes dynamic translocation in a native system was obtained in fly photoreceptor cells [30]. In Drosophila, light regulates the subcellular distribution of TRPL channels between the rhabdomers and the plasma membrane, and it induces long-term adaptation to luminosity that modifies the light-induced, TRPL-mediated ionic currents. A pivotal finding was that the TRPL channel is translocated back and forth between the signaling membrane and the intracellular compartment by a light-regulated mechanism, resulting in a high-level rhabdomeral TRPL in the dark and a low level in the light [30]. A physiological consequence of regulating the subcellular distribution of TRPL is that photoreceptors of flies kept in the dark are sensitive to dim backgrounds, which allows photoreceptors enriched in TRPL to function better in darkness and faint background illumination.
In Caenorhabditis elegans, the TRPC channel TRP-3, which is present in intracellular vesicles of spermatids, translocates to the plasma membrane during sperma activation, thus contributing to the acrosomal reaction [31]. Akin to the TRPL, the regulated translocation of TRP-3 channels to the plasma membrane has profound physiological consequences. The molecular mechanisms underlying the regulated exocytosis of both TRPL and TRP-3 channels are yet elusive, although they may involve vesicle recycling and SNARE-dependent exocytosis.
Agonist-Induced Translocation of TRPC Channels
Three members of the TRPC subfamily of ion channels—TRPC3, TRPC5, and TRPC6—have been found to be regulated by an agonist-activated, SNARE-dependent vesicle translocation to the plasma membrane [32–34]. In a nice study, Singh et al. [32] found that VAMP-dependent, Ca2+-independent exocytosis regulates plasma membrane insertion of TRPC3 channels and contributes to agonist-stimulated Ca2+ influx. These authors found that TRPC3-containing vesicles were localized immediately below the plasma membrane, and they demonstrated that agonist-induced TRPC3 translocation appears to be receptor specific because thapsigargin, a compound that releases Ca2+from the ER, did not change the surface expression of the channel. Protein translocation to the plasma membrane, but not channel trafficking, was abrogated by cleavage of VAMP2 with TeNT, indicating a SNARE-dependent exocytosis [32]. In contrast, although [Ca2+]i at or below resting modulated trafficking of the TRPC3 vesicle to the plasma membrane, the agonist-induced surface expression of the protein was not produced by a rise in [Ca2+]i. In a similar study, Cayouette et al. [33] found in a cell line stably expressing the TRPC6 channel that agonist-induced Ca2+ influx was preceded by exocytotic insertion of TRPC6 proteins into the plasma membrane. Notably, they observed that externalization of TRPC6 channels occurred at an agonist concentration lower than that required for Ca2+ entry, implying that a significant rise in [Ca2+]i is not implied. However, at variance with TRPC3, depletion of Ca2+ stores by thapsigargin caused the translocation of the channel to the cell surface, although it did not activate the channel [33]. The molecular mechanism involved in the exocytosis of TRPC6 was not addressed in the study, although, akin to the TRPC3 channel, it may be mediated by SNARE proteins. Taken together, these findings provide pivotal information for understanding the molecular mechanism involved in agonist-stimulated, G-protein-coupled Ca2+ influx through these TRP channels.
Both TRPC3 and TRPC6 studies provide compelling evidence of the existence of ligand-induced translocation of channels in the cell surface. These findings are further substantiated by the results reported for TRPC5. The work of Bezzerides et al. [34] clearly demonstrated that stimulation of HEK293 cells expressing TRPC5 channels with EGF initiated the rapid incorporation of the channel from a plasmalemma pool of vesicles to the cell surface. This process was coined with the term rapid vesicular insertion of TRP (RiVIT) [34]. These authors were also able to uncover the molecular mechanism of channel translocation and found that the signaling pathway involved phosphatidylinositide 3-kinase, the Rho GTPase Rac1, and phosphatidylinositol-4 phosphate 5-kinase (PIP[5]K). Most remarkable, translocation of TRPC5 from the vesicular pool was observed in hippocampal neurons in culture. In this in vivo system, nerve growth factor (NGF), brain-derived growth factor (BDNF), and insulin growth factor (IGF) promoted the rapid exocytosis of TRPC5 into the neuronal membrane [34]. Because TRPC5 interacts with VAMP and sinaptotagmin I in synaptic vesicles [35], it is reasonable to conclude that these proteins target the TRPC5-containing vesicles to the plasma membrane, thus mediating the SNARE-dependent exocytotic insertion of the channel. Physiologically, the stimulus-induced increase of TRPC5 channels in the neuronal surface notably affected neurite extension [34,35], presumably by modulating the extent of Ca2+ influx at the synaptic terminal. Thus, this elegant study demonstrates that regulated exteriorization of ion channels provides a mechanism for transient, local, self-limiting increases of Ca2+ through TRP channels. The insertion-retrieval of channels from the cell surface represents a clever strategy to avoid the consequences of Ca2+ overload that could occur through TRP channels that are constitutively active.
Modulation of TRPV Channel Activity
TRPV channels have also been reported to be inserted into the plasma membrane in response to a stimulus. Indeed, the first evidence suggesting exocytotic mechanism–mediated TRP channel activity was the demonstration that IGF induced the translocation of TRPV2 to the plasma membrane in cells heterologously overexpressing the channel [36,37]. However, a study of hippocampal neurons in culture failed to observe IGF-induced translocation of TRPV2 to the plasma membrane [34]. Thus, whether regulated exocytosis occurs in vivo is still under debate.
The TRPV5 and TRPV6 channels are members of the TRPV channels that appear constitutively active and very selective for Ca2+. Therefore, akin to the TRPC family, it seems reasonable that the activity of these channels is also controlled by their regulated insertion into the plasma membrane when needed. Although no direct evidence for the secretion mechanism is available yet for TRPV5 and TRPV6 exocytosis, their interaction with the S100A-annexin 2 complex, which is associated with the cortical cytoskeleton, clearly suggests this sort of mechanism [38].
Similar to other TRP channels, TRPV1 is arranged in major molecular complexes establishing high-order signaling networks that notably determine the response to external stimuli. These proteins’ networks are in turn the target of intracellular signaling pathways that modulate their composition, structure, and function. A yeast-two hybrid screen of a rat brain library using the N-terminus of TRPV1 as bait identified two synaptic vesicle proteins that were interacting partners of TRPV1: Snapin and Synaptotagmin IX [39]. These proteins bind to the SNARE proteins and participate in neuronal exocytosis, suggesting that surface delivery of TRPV1 channels is a highly regulated, Ca2+-dependent exocytotic process (Figure 23.3). Indeed, the interaction of both vesicular proteins with TRPV1 appears temporal and seems uninvolved in the formation of the molecular complexes at the cell surface. However, these interactions are pivotal for the trafficking and surface expression of TRPV1 channels in response to activation of intracellular pathways by inflammatory mediators [39,40]. In support of this tenet, PKC-induced TRPV1 trafficking to the plasma membrane was blocked by botulinum neurotoxins, indicating that PKC-induced sensitization of TRPV1 receptors is due at least in part to the regulated exocytosis of channels located in a reserve pool of cytosolic vesicles (Figure 23.3) [39,40]. A recent observation that botulinum neurotoxin attenuates heat hyperalgesia substantiates this hypothesis [41], although additional in vivo experiments are needed to determine the precise role of receptor exocytosis to the onset and maintenance of neurogenic inflammation. Therefore, regulation of TRPV1 surface density in nociceptor peripheral terminals could be an important mechanism for both the development and preservation of inflammatory hyperalgesia.
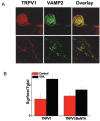
FIGURE 23.3
TRPV1 is translocated to the cell surface by regulated exocytosis. (A) TRPV1 colocalizes in neuronal vesicles with the v-SNARE protein VAMP2, as evidenced from the immunocytochemistry [39]. (B) BoNTA abrogates the PKC-induced translocation of TRPV1 to (more...)
CONCLUDING REMARKS
Regulated translocation of ion channels to the plasma membrane is a widespread strategy aimed at the spatiotemporal control of channel activity. Whether this sort of mechanism underlies the activation of TRP channels is still an open question that requires further documentation. However, it is becoming clear that agonist-induced exocytosis of integral membrane proteins is important to finely tune the cellular response to environmental signals. The number of channels and receptors that experience regulated exocytosis in vivo may increase in the near future, as this novel mechanism becomes general for regulating ion channel activity.
ACKNOWLEDGMENTS
We thank all members of our group and colleagues of collaboration groups for their fundamental contributions to the results herein presented. We are indebted to financial support from the MEC, GVA, Fundació La Caixa, and Fundación Ramón Areces.
REFERENCES
- 1.
- Inoue A, Okabe S. The dynamic organization of postsynaptic proteins: translocating molecules regulate synaptic function. Curr Opin Neurobiol. 2003;13:332–340. [PubMed: 12850218]
- 2.
- Kavalali ET. SNARE interactions in membrane trafficking: a perspective from mammalian central synapses. BioEssays. 2002;24:926–936. [PubMed: 12325125]
- 3.
- Buckley KM, et al. Regulation of neuronal function by protein trafficking: a role for the endosomal pathway. J Physiol. 2000;525(1):11–19. [PMC free article: PMC2269916] [PubMed: 10811720]
- 4.
- Royle SJ, Murrel-Lagnado RD. Constitutive cycling: a general mechanism to regulate cell surface proteins. BioEssays. 2002;25:39–46. [PubMed: 12508281]
- 5.
- Jahn R, Lang T, Sudhof TC. Membrane fusion. Cell. 2003;112:519–533. [PubMed: 12600315]
- 6.
- Watson RT, Kanzaki M, Pessin JE. Regulated membrane trafficking of the insulin responsive glucose transporter 4 in adipocytes. Endocrine Rev. 2004;25:177–204. [PubMed: 15082519]
- 7.
- Valenti G, et al. Aquaporin 2 trafficking. Endocrinology. 2005 Epub ahead of print. [PubMed: 16150901]
- 8.
- Peters KW, et al. Role of SNARE proteins in CFTR and ENaC trafficking. Pflügers Arch. 2001;443:S65–S69. [PubMed: 11845306]
- 9.
- Barry MF, Ziff EB. Receptor trafficking and the plasticity of excitatory synapses. Curr Opin Neurobiol. 2002;12:279–286. [PubMed: 12049934]
- 10.
- Montell C. Exciting trips for TRPs. Nat Cell Biol. 2004;6:690–692. [PubMed: 15303095]
- 11.
- Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–524. [PubMed: 14654832]
- 12.
- Montell C, Birnbaumer L, Flockerzi V. The TRP channels, a remarkably functional family. Cell. 2002;108:595–598. [PubMed: 11893331]
- 13.
- Cosens DJ, Manning A. Abnormal retinogram from a Drosophila mutant. Nature. 1969;224:285–287. [PubMed: 5344615]
- 14.
- Niemeyer BA, et al. The Drosophila light-activated conductance is composed of the two channels TRP and TRPL. Cell. 1996;85:651–659. [PubMed: 8646774]
- 15.
- Moran MM, Xu H, Clapham DE. TRP ion channels in the nervous system. Curr Opin Neurobiol. 2004;14:362–369. [PubMed: 15194117]
- 16.
- Garcia-Sanz N, et al. Identification of a tetramerization domain in the C-terminus of the vanilloid receptor. J Neurosci. 2004;24:5307–5314. [PMC free article: PMC6729306] [PubMed: 15190102]
- 17.
- Phillips AM, Bull A, Kelly LE. Identification of a Drosophila gene encoding a calmodulin-binding protein with homology to the trp phototransduction gene. Neuron. 1992;8:631–642. [PubMed: 1314616]
- 18.
- Chevesich J, Kreuz AJ, Montell C. Requirement for the PDZ domain protein, INAD, for localization of the TRP store-operated channel to a signaling complex. Neuron. 1997;18:95–105. [PubMed: 9010208]
- 19.
- Parekh AB, Putney JW Jr. Store-operated calcium channels. Physiol Rev. 2005;85:757–810. [PubMed: 15788710]
- 20.
- Hoth M, Penner R. Calcium release–activated calcium current in rat mast cells. J Physiol. 1993;465:359–386. [PMC free article: PMC1175434] [PubMed: 8229840]
- 21.
- Venkatachalam K, et al. The cellular and molecular basis of store-operated calcium entry. Nat Cell Biol. 2002;4:E263–E272. [PubMed: 12415286]
- 22.
- Liu X, et al. Trp1, a candidate protein for the store-operated Ca2+ influx mechanism in salivary gland cells. J Biol Chem. 2000;275:3403–3411. [PubMed: 10652333]
- 23.
- Yue L, et al. CaT1 manifests the pore properties of the calcium release–activated calcium channel. Nature. 2001;410:705–709. [PubMed: 11287959]
- 24.
- Schindl R, et al. Store depletion–activated CaT1 currents in rat basophilic leukaemia mast cells are inhibited by 2-amino ethoxydiphenyl borate. Evidence for a regulatory component that controls activation of both CaT1 and CRAC (Ca2+ release–activated Ca2+ current) channels. J Biol Chem. 2002;277:26950–26958. [PubMed: 12011062]
- 25.
- Voets T, et al. CaT1 and the calcium release–activated calcium channel manifest distinct pore properties. J Biol Chem. 2001;276:477767–47770. [PubMed: 11687570]
- 26.
- Kahr H, et al. CaT1 knock-down fail to affect CRAC channels in mucosal-type mast cells. J Physiol. 2004;557:121–132. [PMC free article: PMC1665038] [PubMed: 15020691]
- 27.
- Yao Y, et al. Activation of store-operated Ca2+ currents in Xenopus oocytes requires SNAP-25 but not a diffusible messenger. Cell. 1999;98:475–485. [PubMed: 10481912]
- 28.
- Alderton JM, et al. Evidence for a vesicle-mediated maintenance of store-operated calcium channels in a human embryonic kidney cell line. Cell Calcium. 2000;28:161–169. [PubMed: 11020378]
- 29.
- Scott CC, et al. Activation of store-operated calcium channels. Assessment of the role of SNARE-mediated vesicular transport. J Biol Chem. 2003;278:30534–30539. [PubMed: 12764154]
- 30.
- Bahner M, et al. Light-regulated subcellular translocation of Drosophila TRPL channels induces long-term adaptation and modifies the light-induced current. Neuron. 2002;34:83–93. [PubMed: 11931743]
- 31.
- Xu ZZ, Sternberg PW. A C. elegans sperm TRP protein required for sperm–egg interactions during fertilization. Cell. 2003;114:285–297. [PubMed: 12914694]
- 32.
- Singh BB, et al. VAMP2-dependent exocytosis regulates plasma membrane insertion of TRPC3 channels and contributes to agonist-stimulated Ca2+ influx. Molecular Cell. 2004;15:635–646. [PubMed: 15327778]
- 33.
- Cayouette S, et al. Exocytotic insertion of TRPC6 channel into the plasma membrane upon protein-coupled receptor activation. J Biol Chem. 2004;279Gq:7241–7246. [PubMed: 14662757]
- 34.
- Bezzerides V, et al. Rapid vesicular translocation and insertion of TRP channels. Nat Cell Biol. 2004;6:709–720. [PubMed: 15258588]
- 35.
- Greka A, et al. TRPC5 is a regulator of hippocampal neurite length and growth cone morphology. Nat Neurosci. 2003;6:837–845. [PubMed: 12858178]
- 36.
- Kanzaki M, et al. Translocation of a calcium-permeable cation channel induced by insulin-like growth factor-1. Nat Cell Biol. 1999;1:165–170. [PubMed: 10559903]
- 37.
- Boels K, et al. The neuropeptide head activator induces activation and translocation of the growth-factor-regulated Ca2+-permeable channel GRC. J Cell Sci. 2001;114:3599–3606. [PubMed: 11707512]
- 38.
- van de Graaf SFJ, et al. Functional expression of the epithelial Ca2+ channels (TRPV5 and TRPV6) requires association of the S100A10-annexin 2 complex. EMBO J. 2003;22:1478–1487. [PMC free article: PMC152906] [PubMed: 12660155]
- 39.
- Morenilla-Palao C, et al. Regulated exocytosis contributes to protein kinase C potentiation of vanilloid receptor activity. J Biol Chem. 2004;279:25665–25679. [PubMed: 15066994]
- 40.
- Van Buren JJ, et al. Sensitization and translocation of TRPV1 by insulin and IGF-1. Mol Pain. 2005;1:1–11. [PMC free article: PMC1142339] [PubMed: 15857517]
- 41.
- Cui M, Khanijou S, Aoki KR. Subcutaneous administration of botulinum toxin A reduces formalin-induced pain. Pain. 2004;107:125–133. [PubMed: 14715398]
- Review Regulation and Role of Store-Operated Ca(2+) Entry in Cellular Proliferation.[Calcium Entry Channels in Non-...]Review Regulation and Role of Store-Operated Ca(2+) Entry in Cellular Proliferation.Hodeify R, Yu F, Courjaret R, Nader N, Dib M, Sun L, Adap E, Hubrack S, Machaca K. Calcium Entry Channels in Non-Excitable Cells. 2018
- Review SNARE interactions in membrane trafficking: a perspective from mammalian central synapses.[Bioessays. 2002]Review SNARE interactions in membrane trafficking: a perspective from mammalian central synapses.Kavalali ET. Bioessays. 2002 Oct; 24(10):926-36.
- Review v- and t-SNAREs in neuronal exocytosis: a need for additional components to define sites of release.[Neuropharmacology. 1995]Review v- and t-SNAREs in neuronal exocytosis: a need for additional components to define sites of release.Galli T, Garcia EP, Mundigl O, Chilcote TJ, De Camilli P. Neuropharmacology. 1995 Nov; 34(11):1351-60.
- Review Exocytosis and synaptic vesicle function.[Compr Physiol. 2014]Review Exocytosis and synaptic vesicle function.Shin OH. Compr Physiol. 2014 Jan; 4(1):149-75.
- Review Core proteins of the secretory machinery.[Handb Exp Pharmacol. 2008]Review Core proteins of the secretory machinery.Lang T, Jahn R. Handb Exp Pharmacol. 2008; (184):107-27.
- TRP Channel Trafficking - TRP Ion Channel Function in Sensory Transduction and C...TRP Channel Trafficking - TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades
Your browsing activity is empty.
Activity recording is turned off.
See more...