

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Liedtke WB, Heller S, editors. TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades. Boca Raton (FL): CRC Press/Taylor & Francis; 2007.

TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades.
Show detailsThe endothelium is a highly specialized multifunctional cell monolayer between blood and tissue. It regulates a variety of vascular functions such as the passage of macromolecules and oxygen supply to organs and tissues, of immune responses, of angiogenesis, and of vascular remodeling. An additional and overall important role of the arterial endothelium is the control of the contractile state of the vascular smooth muscle and thus systemic blood pressure by the release of vasoactive factors. Moreover, endothelial dysfunction contributes to several cardiovascular pathologies such as arteriosclerosis, restenosis disease, vasculitis, or hypertension.
The crucial role of the endothelium in the control of vascular tone was first demonstrated by Furchgott and Zawadzki in 1980 [1]. In this pioneer work, they demonstrated that following stimulation with acetylcholine, the endothelium releases a short-lived factor that relaxes the vascular smooth muscle. This factor was later identified as nitric oxide. In addition to humoral stimulation, the endothelium also controls vasodilatation in response to increased hemodynamic forces (i.e., increased shear stress exerted by streaming blood) [2]. This is an important mechanism by which the endothelium controls adequate organ perfusion and protects vessel walls against mechanical damage.
CA2+ AND ENDOTHELIAL FUNCTION
In response to classical agonists such as acetylcholine and bradykinin as well as to hemodynamic stimuli, the endothelium produces in principle three types of vasodilating factors: nitric oxide (NO), prostacyclin (PGI2), and the endothelium-derived hyperpolarizing factor (EDHF) (Figure 27.1). In contrast to nitric oxide and PGI2, the EDHF is not considered a chemical vasodilator [3,4] but rather an electrical phenomenon in which an initial endothelial hyperpolarization spreads to the vascular smooth muscle via a myoendothelial gap junction, closing L-type voltage-gated Ca2+ channels and thus vasorelaxation.
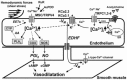
FIGURE 27.1
Schematic illustration of the hypothetical role of TRPC and mechanosensitive TRPV4 channels in endothelial function. 4αPDD, 4α-phorbol-12,13-didecanoate; AA, arachidonic acid; EETs, epoxyeicosatrienoic acids; COX, cyclooxygenase-1; CYP, (more...)
The synthesis of all three vasodilators is Ca2+ dependent. The endothelial nitric oxide synthase is activated in a Ca2+/calmodulin-dependent fashion [5], and also PGI2 synthesis by cyclooxgenase-1 requires the Ca2+-dependent release of arachidonic acid by phospholipase-A2. The generation of the EDHF signaling is Ca2+ dependent because the initial endothelial hyperpolarization is caused by opening endothelial Ca2+-activated K+ channels [4,6–10].
Upon endothelial stimulation by binding of classical agonists to their G-protein-coupled receptors, the intracellular Ca2+ concentration ([Ca2+]i) increases rapidly within seconds due to an IP3-mediated Ca2+ release from internal stores. Following the initial Ca2+ peak, [Ca2+]i remains elevated for up to several minutes, due to Ca2+ influx from the extracellular space. Ca2+-permeable cation channels located in the plasma membrane play an important role in this plateau phase of elevated [Ca2+]i as they provide the Ca2+-influx pathway [11]. Besides the well-documented Ca2+ mobilization following receptor stimulation, stimulation of the endothelium by hemodynamic stimuli (i.e., shear stress) also results in an increase in [Ca2+]i. Such a shear stress–induced Ca2+ mobilization has been observed following stimulation in endothelial cells in vitro [9,12] as well as in the endothelium in intact vessel preparations [13]. This flow/shear stress–induced increase in [Ca2+]i is thought to involve Ca2+ influx through mechanosensitive Ca2+-permeable cation channels (MSC), which are believed to act as mechanosensors in recognizing alterations of hemodynamic forces, as well as the Ca2+ release from internal IP3- [14] and ryanodine-sensitive stores [9]. The Ca2+ influx through MSC- and Ca2+-release events result in [Ca2+]i oscillations with a complex spatial and temporal pattern [9,12]. Frequency and amplitude modulation of such [Ca2+]i fluctuations is thought to fine-tune the synthesis of endothelial vasodilators depending on the degree of mechanical stimulation [9]. At the cell membrane level, MSC has been identified in EC in vitro [11,15] and in situ [16,17] as mostly stretch-activated channels (SAC) based on their activation by applying negative pressure to the patch pipette in single-channel patch-clamp recordings. However, molecular identity (i.e., the MSC/SAC encoding gene[s] as well as the intrinsic molecular determinants of mechanosensitivity) is still elusive. However, the recent identification of cation channels of the TRP gene family with mechanosensitive properties may bring some progress in this field [18,19].
TRPS IN THE ENDOTHELIUM
In recent years, the mammalian genes encoding for Ca2+-permeable cation channels were identified as mammalian homologues of the “transient receptor potential” trp gene expressed in photoreceptors of Drosophila [20]; meanwhile, expressions of several members of the TRP superfamily of cation channels have been shown in endothelial cells (EC) of humans and other species (for review, see reference 21). Within the diverse subfamilies of TRP cation channels, EC express several members of the canonical TRP subfamily (TRPC): TRPC1, TRPC3, TRPC4, and TRPC6 with expression patterns varying within different species. Homomeric or heteromeric TRPC channel complexes are principally thought to serve as Ca2+ influx channels after receptor activation or store depletion (Figure 27.1) [21,22]. Of the endothelial TRPCs, particularly TRPC4 has been shown to contribute to endothelial-dependent vasodilatation as concluded from an altered vasorelaxation response in TRPC4 knockout mice [23]. Concerning the other TRP subfamilies, the melastatin (TRPM) and vanilloid (TRPV) subfamilies (e.g., TRPM4 and TRPV4) have also been reported to be endothelial TRPs [21,24].
The functional role of TRPM4 in the endothelium is unclear. In addition to its regulation by ATP- and PKC-mediated phosphorylation, this channel is itself activated by increases in intracellular Ca2+ in a Ca2+/calmodulin-dependent fashion, but it is not Ca2+ permeable [21]. The depolarizing Na+ current through this channel leads to membrane depolarization. Possibly, this channel is needed (in terms of a negative feedback mechanism) to counteract prolonged membrane hyperpolarization, which is caused by open Ca2+-activated K+ channels.
TRPV4 was first identified in the endothelium of mouse aorta by Bernd Nilius’s group [24]. Thus far, it appears that this channel is the only member of the TRPV subfamily expressed in the endothelium. TRPV4 was also identified by us in the endothelium of the carotid artery of the rat. The precise role of this TRPV subtype in endothelial function is not elucidated so far. However, in this chapter we provide new insight into how TRPV4 contributes to endothelial function and endothelium-dependent vasodilatation.
TRPV4 IN THE ENDOTHELIUM
Within this diverse expression pattern of TRP channels in the endothelium, TRPV4 is especially interesting because of its moderately high Ca2+ permeability [24,25], which would offer a significant Ca2+-influx pathway (Figure 27.1). Unlike TRPC channels, TRPV4 is not considered a classical receptor/second-messenger–activated channel or store-operated channel [20], but it exhibits extremely diverse gating behavior upon physical and chemical stimuli and importantly mechanical and osmotic challenges [26]. Due to this mechano- and osmosensitivity, the new term mechano- and osmo- TRP was assigned to this TRPV4 [19]. Moreover, the physiological importance of this channel and of OSM-9, the TRPV homologue in invertebrate C. elegans, was highlighted by the finding of a disturbed mechanosensation and osmoregulation in TRPV4 knockout mice [27] and the impaired mechanosensation, olfaction, and osmoregulation in C. elegans with a mutant osm-9-gene [28]. The detailed biophysical properties of this channel and its molecular mechanism of activation were outlined in other chapters of this book. After a brief summary of general functional features of endothelial TRPV4, we focus in this chapter on the potential physiological roles of TRPV4 in the endothelium and in the mechanism of endothelial control of vascular tone.
Similar to TRPV4 in heterologous expression systems, TRPV4 in aortic endothelial cells in mice [24,29] and in the EC of the rat carotid artery is activated by moderate warmth (>27°C), cell swelling in response and hypotonic stress (HTS), and pharmacologically by the non-PKC-activating phorbol ester, 4α-phorbol-12,13-didecanoate (4αPDD), leading to a considerable increase in endothelial [Ca2+]i (Figure 27.2A–C). Single-cell-RT-PCR analysis of the TRPV mRNA expression pattern in the in situ EC of carotid arteries revealed TRPV4 expression, but none of the other closely related TRPV1–3. Within the vascular wall of the rat carotid artery, TRPV4 expression seems to be limited to the endothelium because mRNA expression was not detectable in the vascular smooth muscle cells in the carotid artery. However, it should be noted that TRPV4 expression and channel functions were recently detected in the smooth muscle of cerebral arteries of rats [30], in which this channel forms a functional complex with the large-conductance Ca2+-activated K+ channel (BKCa), which modulates vascular smooth muscle membrane potential. This also indicates that TRPV4 expression may be heterogeneous within different vascular beds.
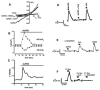
FIGURE 27.2
Electrophysiological properties of TRPV4 currents in RCAEC of rat carotid artery in situ. (A) Representative whole-cell recording of activation of cation currents by 4α-phorbol-12,13-didecanoate (4αPDD, 1 μM). Nonpermeable NMDG (more...)
With respect to mechanism of TRPV4 activation in the endothelium, arachidonic acid (AA) has been shown to mediate HTS-induced TRPV4 activation similar to findings in other cell types and in heterologous expression systems [31,32]. A more recent study in aortic ECs from mice suggests that arachidonic acid itself might not be the endogenous activator of the channel. But conversion of arachidonic acid to 5,6 epoxyeicosatrienoic acid (5,6 EET) and 8,9 epoxyeicosatrienoic acid (8,9 EET) mediated by cytochrome P450 epoxygenases leads to channel activation [33]. Thus, the authors proposed that EETs serve as the endogenous activators of TRPV4. This finding is highly relevant and offers some exciting insight into the putative role of TRPV4 in the endothelial function because EETs have been suggested to be molecular candidates for the EDHF (the third major vasodilating factor) in some vessels or to facilitate EDHF signaling by intra-endothelial mechanisms [4,33,34]. Therefore, EET activation of TRPV4 and subsequent Ca2+ entry may play a role in the generation or amplification of EDHF signaling after receptor stimulation.
TRPV4 AND ENDOTHELIUM-DEPENDENT VASODILATATION
The mechanosensitivity of TRPV4 may point to a role of the channel as an endothelial mechanosensor and thus in the mechanisms of flow- or shear stress–induced vasodilatation. It is noteworthy that a shear stress–induced TRPV4-mediated Ca2+ entry has also been shown in heterologous expression systems and renal tubular epithelial cells [35,36]. As mentioned before, such flow- and shear stress–induced Ca2+ signals have also been observed in the EC in vitro [9,12] and in the endothelium in intact vessel preparations [13]. The inhibition of MSC by the rather nonselective lanthanide gadolinium (Gd3+) abolishes this flow- and shear stress–induced Ca2+ mobilization [9]. As already stated, the molecular identity of this shear stress–activated Ca2+-permeable MSC is still unknown. But in keeping with the mechanosensitivity of TRPV4, this channel might be a good molecular candidate for the shear stress–activated Ca2+-entry channel in the endothelium. In our own studies, we characterized the functional role of TRPV4 channels in the endothelium of rat carotid arteries by pressure myography, and we determined vasodilatory responses after pharmacological activation of the channel by 4αPDD and after increasing shear stress.
Our myograph experiments revealed that pharmacological opening of TRPV4 by 4αPDD caused a robust vasodilatation in the carotid artery (Figure 27.2D). This 4αPDD-induced vasodilatation was almost as potent as that achieved by physiologically relevant concentrations of acetylcholine (10 nM–1 μM; Figure 27.2D). The 4αPDD-induced vasodilatation required a functionally intact endothelium, and endothelial inactivation by mechanical damage prevented vasodilatation to 4αPDD. This indicates that the 4αPDD-induced vasodilatation is strictly endothelium dependent. This was further supported by the observation that 4αPDD just caused vasodilatation when applied to the luminal and thus endothelium-covered face of the artery. Neither vasodilatation nor vasoconstriction occurred when 4αPDD was directly applied to the extravasal side (i.e., to the smooth muscle), thus indicating that TRPV4 is most likely not expressed in the vascular smooth muscle of the carotid artery or is not of considerable importance in smooth muscle cell functions. This is also in line with the lack of TRPV4–mRNA expression as determined by single-cell RT-PCR analysis in freshly isolated smooth muscle cells.
That 4αPDD-induced vasodilatation is indeed caused by opening of endothelial TRPV4 is further supported by the observation that 4αPDD elicited vasodilatation with a KD of 0.3 μM, which is in fact similar to the KD reported for TRPV4 activation [24]. 4αPDD-induced vasodilatation was prevented by buffering intracellular [Ca2+]i with BAPTA-AM in the endothelium and by the TRPV4 channel blocker ruthenium red (1 μM; Figure 27.2D), indicating that 4αPDD exerts its vasodilating effect by inducing Ca2+ influx and subsequently synthesis of endothelial vasodilators. Regarding the classic acetylcholine-induced vasodilatation, we found that ruthenium red, if intraluminally applied, also reduced this endothelium-dependent vasodilatation, although only modestly. The sole intraluminal application of ruthenium red was without effect on basal vessel diameter. These observations suggest that TRPV4 does not contribute substantially to either agonist-induced Ca2+ signaling and thus vasodilatation or to basal control of vascular tone. This also suggests that ruthenium red does not exert gross unspecific effects or other effects caused by blocking ryanodine-sensitive Ca2+-release channels in smooth muscle.
TRPV4-MEDIATED VASODILATATION IS NITRIC OXIDE DEPENDENT IN CAROTID ARTERIES
Inhibition of nitric oxide synthase alone or in combination with the blockade of prostacyclin synthesis almost completely suppressed the 4αPDD-induced vasodilatation (Figure 27.2E), suggesting that this type of vasodilatation largely relies on the synthesis and action of nitric oxide (NO), whereas the other two major vasodilator systems (i.e., the prostacyclin system or the EDHF system) do not seem to make significant contributions to the vasodilating effect of 4αPDD in this conduit artery. NO synthesis following TRPV4 activation is most likely related to the Ca2+ influx through TRPV4 channels and subsequent stimulation of Ca2+-dependent eNOS (Figure 27.1). Regarding EDHF-mediated vasodilatation, Ca2+-dependent activation of endothelial KCa channels of the KCa3.1 and KCa2.3 types and subsequent endothelial hyperpolarization have been considered prerequisites for generating the EDHF signal (Figure 27.1); selective inhibition of these KCa channels abolishes EDHF-type vasodilatation in many vessels [4] and species including the rat carotid artery [6]. Moreover, EDHF-type vasodilatations have been shown to become more important when vessel size decreases [37]. In the large-conduit carotid artery, in which the EDHF system is apparently less important than the NO system, 4αPDD-induced TRPV4 activation and subsequent Ca2+ entry did not cause major EDHF-mediated vasodilatation (Figure 27.2E). In contrast, 4αPDD was able to produce EDHF-mediated vasodilatation in small-sized arteries (A. gracilis 200 μM in diameter). Therefore, pharmacological opening of TRPV4 appears to be sufficient to induce EDHF-type vasodilatation in small-sized arteries, in which EDHF plays a significant role.
A FUNCTIONAL ROLE OF TRPV4 IN ENDOTHELIAL MECHANOTRANSDUCTION?
In keeping with the proposed mechanosensitivity of TRPV4 [21,25,31,36], we speculated that TRPV4 activation and Ca2+ entry may occur by mechanical stimulation of the endothelium by increased fluid viscosity and thus shear stress. As stated in previous paragraphs, shear stress– or flow-induced elevations of endothelial [Ca2+]i are due to both Ca2+ influx and Ca2+ release from internal stores [9,12]. Moreover, such a shear stress–induced increase in [Ca2+]i is prevented by strongly buffering extracellular Ca2+ or by the MSC and TRP blocker Gd3+ [9], indicating that an increase in shear stress activates a “directly” or “indirectly” mechanosensitive Ca2+-entry channel.
In pressure myography of small vessels, shear stress can be experimentally increased by increasing the viscosity of the perfusion medium by adding dextran. The advantage of this procedure is that pressure gradients are not changed during the experiments, which could lead to additional myogenic effects. By adding 5 percent dextran to the perfusion buffer, the viscosity of the buffer increases from 0.7 to 2.9 mPa*s, which leads to an increase of shear stress from 1 to 3 dyn/cm2 in carotid arteries and from 8 to 19 dyn/cm2 in small-sized arteries, according to the law of Hagen-Poiseuille: τ = 4Q/r3; τ = shear stress; η = viscosity; Q = flow; and r = radius. These experiments revealed that such an increase in shear stress caused substantial vasodilatation of the rat carotid artery (Figure 27.2F), which was strictly NO dependent as an inhibitor of NO-synthesis NG-nitro-L-arginine (L-NNA) completely abolished this vasodilatation. This NO dependency of shear stress–induced vasodilatation is also in agreement with findings in arteries of humans [38] and other species [39], whereas in mice both prostaglandins as well as NO mediate this type of vasodilatation [40]. Similar to sensitivity of 4αPDD-induced vasodilatation to the pharmacological TRPV4 inhibition, shear stress–induced vasodilatation in the rat carotid artery was greatly blocked by ruthenium red, suggesting TRPV4’s involvement in this response. In addition, the buffering of endothelial [Ca2+]i with BAPTA-AM resulted in complete suppression of shear stress–induced vasodilatation, which clearly demonstrates that this is a Ca2+-dependent event and that it mostly depends on Ca2+-dependent NO generation.
Inhibition of protein kinase C and of tyrosine kinases was without effect, suggesting that protein phosphorylation in general or a potential TRPV4 phosphorylation by one of these kinases does not seem to play a major role in shear stress–induced vasodilatation. Importantly, shear stress–induced vasodilatation was prevented by inhibition of PLA2 and thus the release of arachidonic acid in rat carotid arteries. Release of AA and production of its metabolites in response to flow is well documented in cultured endothelial cells [41], and a role of AA metabolites in flow-induced vasodilatation has been proposed previously [40]. With respect to TRPV4, exogenously applied AA has been shown to activate rat TRPV4 as shown here and cloned TRPV4 previously [33] and endogenously produced AA mediates mechanical activation (i.e., by cell swelling) [32]. These roles of AA in both shear stress–induced vasodilatation and TRPV4 activation tempt us to speculate that PLA2-mediated release of AA following shear stress stimulation mediates TRPV4 activation. This interpretation also implies that TRPV4 is unlikely to be the mechanosensor per se. Nonetheless, AA-dependent TRPV4 activation might be an essential component in the signal transduction mechanism of endothelial mechanotransduction.
Collectively, these observations suggest that Ca2+ entry through endothelial TRPV4 channels triggers NO-dependent vasodilatation in the endothelium of the rat CA (conduit artery) and NO- and EDHF-dependent vasodilatation in small-sized A. gracilis (a more resistance artery-like vessel). Moreover, it is tempting to speculate that endothelial TRPV4 channels are involved in endothelial mechanosensing of shear stress–induced vasodilatation. Thus among the numerous TRP channels expressed in the endothelium, TRPV4 might be specifically assigned to endothelial mechanotransduction.
ENDOTHELIAL TRP CHANNELS AND CARDIOVASCULAR DISEASE
Endothelial dysfunction has contributed to several cardiovascular disease states and especially to the defective regulation of vascular tone in hypertension. Moreover, altered functions of endothelial cation channels have been proposed to contribute to this endothelial dysfunction and increased blood pressure. For instance, in rat models of experimental renal insufficiency [42], alterations in endothelial cation channel functions have been described for Ca2+-activated K+ channels, which play a crucial role in EDHF-mediated vasodilatation. Accordingly, impaired EDHF signaling and thus endothelial dysfunction was found in this animal model of chronic renal failure. Moreover, genetic manipulation of expression of endothelial Ca2+-activated K+ channels (i.e., the KCa2.3) increases myogenic responsiveness and leads to increased systemic blood pressure. Regarding endothelial Ca2+-permeable cation channels, alterations in mechanosensitive cation channels (MSC) have reported in rat models of genetic hypertension (spontaneously hypertensive rats) and of salt-sensitive hypertension [16,17,43]. Also it is not sufficiently investigated whether alterations in endothelial TRP channels also contribute to endothelial dysfunction and increased blood pressure. However, emerging and already existing TRP knockout mice may further elucidate the functional role of specific TRP in endothelium-dependent control of vascular tone and thus blood pressure control. For instance, the abnormal endothelium-dependent relaxation in TRPC4 knockout mice shows that at least this channel of the canonical TRPC subfamily is important for appropriate endothelial function [23]. Moreover, a recent report on increased myogenic tone and hypertension in TRPC6 knockout mice [44] indicates that also alterations in TRP function in vascular smooth muscle contribute to defective regulation of vascular tone. Whether endothelial functions are disturbed in these mice is currently under investigation. With respect to TRPV4, cardiovascular phenotyping of these mice is superficial so far and thus incomplete [45], and further studies are needed to determine whether endothelial dysfunction and abnormal regulation of vascular tone are present in these mice. Although conventional or even conditional single or double knockout strategies are helpful in elucidating the functional role of the respective TRP channel, one should also consider that early compensation during embryonic development and even rapid compensation in adult animals may occur, thus masking specific roles of a single channel.
DO ENDOTHELIAL CA2+-PERMEABLE CATION CHANNELS LIKE TRPV4 REPRESENT NOVEL PHARMACOTHERAPEUTIC TARGETS FOR ANTI-HYPERTENSIVE THERAPY?
Endothelial TRP channels and especially TRPV4 can be considered important regulators of vascular tone by modulating intracellular Ca2+ signaling and thus adequate synthesis of vasodilating factors. The functional importance of these ion channels may therefore suggest that they may represent novel pharmacotherapeutic targets in addition to the well-known voltage-gated calcium channels in vascular smooth muscles.
From a more general point of view, small molecule openers of TRP channels are more promising than blockers regarding a potential antihypertensive efficacy or therapeutic utility. This based on the assumption that a pharmacological activation of TRP channels and thus an increased Ca2+-influx would improve endothelial function by supporting the synthesis of endothelial vasodilators which is in fact Ca2+-dependent. Moreover, it should be considered that a specific channel is not similarly expressed in vascular smooth muscle since pharmacological activation of this channel would have counteracting vasocontracting effects, thus precluding the usefulness of such a compound. Nonetheless, within the different TRP expressed in the vascular wall, TRPV4 fulfills some of these criteria, and a fairly selective-opener (i.e., 4αPDD) is available to test this idea. As described here, there are some indications that 4αPDD might have blood pressure lowering properties: First, by opening of endothelial TRPV4, 4αPDD would stimulate Ca2+-influx and increase [Ca2+]i, which in turn stimulates the synthesis of vasodilator NO in larger vessel as well as NO- and EDHF-mediated vasodilatation in small resistance-sized arteries and arterioles. Second, 4αPDD exerts vasodilating effects already at submicromolar concentrations and is a completely synthetic compound. This may indicate moderate selectivity. Third, 4αPDD does not induce vasoconstriction in carotid arteries or small-sized arteries as shown here, which is most likely explained by the fact that TRPV4 is not expressed in smooth muscle of these arteries.
However, what about potential side effects? It should be considered that TRPV4 is also expressed in epithelia, i.e., airway and kidney epithelia, in the autonomic nervous system, and especially in vessel-innervating sympathetic nerves, and importantly in the ear and in the brain, e.g., in neurosensory cells of the circumventricular organs which are responsive to systemic osmotic pressure. Therefore, it remains to be determined whether TRPV4 openers—as new kind of antihypertensive drugs—might be of clinical usefulness.
REFERENCES
- 1.
- Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–76. [PubMed: 6253831]
- 2.
- Pohl U, et al. Crucial role of endothelium in the vasodilator response to increased flow in vivo. Hypertension. 1986;8:37–44. [PubMed: 3080370]
- 3.
- Griffith TM. Endothelium-dependent smooth muscle hyperpolarization: do gap junctions provide a unifying hypothesis. Br J Pharmacol. 2004;141:881–903. [PMC free article: PMC1574270] [PubMed: 15028638]
- 4.
- Busse R, et al. EDHF: bringing the concepts together. Trends Pharmacol Sci. 2002;23:374–380. [PubMed: 12377579]
- 5.
- Bredt DS, Snyder SH. Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc Natl Acad Sci USA. 1990;87:682–685. [PMC free article: PMC53329] [PubMed: 1689048]
- 6.
- Eichler I, et al. Selective blockade of endothelial Ca2+-activated small- and intermediate-conductance K+-channels suppresses EDHF-mediated vasodilation. Br J Pharmacol. 2003;138:594–601. [PMC free article: PMC1573692] [PubMed: 12598413]
- 7.
- Kohler R, et al. Expression and function of endothelial Ca2+-activated K+ channels in human mesenteric artery: a single-cell reverse transcriptase-polymerase chain reaction and electrophysiological study in situ. Circ Res. 2000;87:496–503. [PubMed: 10988242]
- 8.
- Kohler M, et al. Small-conductance, calcium-activated potassium channels from mammalian brain. Science. 1996;273:1709–1714. [PubMed: 8781233]
- 9.
- Hoyer J, Kohler R, Distler A. Mechanosensitive Ca2+ oscillations and STOC activation in endothelial cells. Faseb J. 1998;12:359–366. [PubMed: 9506480]
- 10.
- Ishii TM, et al. A human intermediate conductance calcium-activated potassium channel. Proc Natl Acad Sci USA. 1997;94:11651–11656. [PMC free article: PMC23567] [PubMed: 9326665]
- 11.
- Nilius B, Droogmans G. Ion channels and their functional role in vascular endothelium. Physiol Rev. 2001;81:1415–1459. [PubMed: 11581493]
- 12.
- Helmlinger G, Berk BC, Nerem RM. Pulsatile and steady flow-induced calcium oscillations in single cultured endothelial cells. J Vasc Res. 1996;33:360–369. [PubMed: 8862141]
- 13.
- Falcone JC, Kuo L, Meininger GA. Endothelial cell calcium increases during flow-induced dilation in isolated arterioles. Am J Physiol. 1993;26:4, H653–659. [PubMed: 8447477]
- 14.
- Nollert MU, Eskin SG, McIntire LV. Shear stress increases inositol tris-phosphate levels in human endothelial cells. Biochem Biophys Res Commun. 1990;170:281–287. [PubMed: 2372294]
- 15.
- Lansman JB, Hallam TJ, Rink TJ. Single stretch-activated ion channels in vascular endothelial cells as mechanotransducers. Nature. 1987;325:811–813. [PubMed: 2434860]
- 16.
- Hoyer J, Kohler R, Distler A. Mechanosensitive cation channels in aortic endothelium of normotensive and hypertensive rats. Hypertension. 1997;30:112–119. [PubMed: 9231830]
- 17.
- Hoyer J, et al. Up-regulation of pressure-activated Ca2+-permeable cation channel in intact vascular endothelium of hypertensive rats. Proc Natl Acad Sci USA. 1996;93:11253–11258. [PMC free article: PMC38316] [PubMed: 8855342]
- 18.
- Maroto R, et al. TRPC1 forms the stretch-activated cation channel in vertebrate cells. Nat Cell Biol. 2005;7:179–185. [PubMed: 15665854]
- 19.
- Liedtke W, Kim C. Functionality of the TRPV subfamily of TRP ion channels: add mechano-TRP and osmo-TRP to the lexicon! Cell Mol Life Sci. 2005;62:2985–3001. [PubMed: 16314934]
- 20.
- Clapham DE, et al. International Union of Pharmacology. XLIII. Compendium of voltage-gated ion channels: transient receptor potential channels. Pharmacol Rev. 2003;55:591–596. [PubMed: 14657417]
- 21.
- Nilius B, Droogmans G, Wondergem R. Transient receptor potential channels in endothelium: solving the calcium entry puzzle. Endothelium. 2003;10:5–15. [PubMed: 12699072]
- 22.
- Hofmann T, et al. Subunit composition of mammalian transient receptor potential channels in living cells. Proc Natl Acad Sci USA. 2002;99:7461–7466. [PMC free article: PMC124253] [PubMed: 12032305]
- 23.
- Freichel M, et al. Lack of an endothelial store-operated Ca2+ current impairs agonist-dependent vasorelaxation in TRP4−/− mice. Nat Cell Biol. 2001;3:121–127. [PubMed: 11175743]
- 24.
- Watanabe H, et al. Activation of TRPV4 channels (hVRL-2/mTRP12) by phorbol derivatives. J Biol Chem. 2002;277:13569–13577. [PubMed: 11827975]
- 25.
- Strotmann R, et al. OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nat Cell Biol. 2000;2:695–702. [PubMed: 11025659]
- 26.
- Nilius B, et al. TRPV4 calcium entry channel: a paradigm for gating diversity. Am J Physiol Cell Physiol. 2004;28:6, C195–205. [PubMed: 14707014]
- 27.
- Liedtke W, Friedman JM. Abnormal osmotic regulation in trpv4−/− mice. Proc Natl Acad Sci USA. 2003;100:13698–13703. [PMC free article: PMC263876] [PubMed: 14581612]
- 28.
- Colbert HA, Smith TL, Bargmann CI. OSM-9, a novel protein with structural similarity to channels, is required for olfaction, mechanosensation, and olfactory adaptation in Caenorhabditis elegans. J Neurosci. 1997;17:8259–8269. [PMC free article: PMC6573730] [PubMed: 9334401]
- 29.
- Watanabe H, et al. Heat-evoked activation of TRPV4 channels in a HEK293 cell expression system and in native mouse aorta endothelial cells. J Biol Chem. 2002;277:47044–47051. [PubMed: 12354759]
- 30.
- Earley S, et al. TRPV4 forms a novel Ca2+ signaling complex with ryanodine receptors and BKCa channels. Circ Res. 2005;97:1270–1279. [PubMed: 16269659]
- 31.
- Andrade YN, et al. TRPV4 channel is involved in the coupling of fluid viscosity changes to epithelial ciliary activity. J Cell Biol. 2005;168:869–874. [PMC free article: PMC2171792] [PubMed: 15753126]
- 32.
- Vriens J, et al. Cell swelling, heat, and chemical agonists use distinct pathways for the activation of the cation channel TRPV4. Proc Natl Acad Sci USA. 2004;101:396–401. [PMC free article: PMC314196] [PubMed: 14691263]
- 33.
- Watanabe H, et al. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature. 2003;424:434–438. [PubMed: 12879072]
- 34.
- Vriens J, et al. Modulation of the Ca2+ permeable cation channel TRPV4 by cytochrome P450 epoxygenases in vascular endothelium. Circ Res. 2005;97:908–915. [PubMed: 16179585]
- 35.
- Gao X, Wu L, O'Neil RG. Temperature-modulated diversity of TRPV4 channel gating: activation by physical stresses and phorbol ester derivatives through protein kinase C–dependent and –independent pathways. J Biol Chem. 2003;278:27129–27137. [PubMed: 12738791]
- 36.
- O'Neil RG, Heller S. The mechanosensitive nature of TRPV channels. Pflügers Arch. 2005;451:193–203. [PubMed: 15909178]
- 37.
- Shimokawa H, et al. The importance of the hyperpolarizing mechanism increases as the vessel size decreases in endothelium-dependent relaxations in rat mesenteric circulation. J Cardiovasc Pharmacol. 1996;28:703–711. [PubMed: 8945685]
- 38.
- Joannides R, et al. Nitric oxide is responsible for flow-dependent dilatation of human peripheral conduit arteries in vivo. Circulation. 1995;91:1314–1319. [PubMed: 7867167]
- 39.
- Holtz J, et al. Flow-dependent, endothelium-mediated dilation of epicardial coronary arteries in conscious dogs: effects of cyclooxygenase inhibition. J Cardiovasc Pharmacol. 1984;6:1161–1169. [PubMed: 6084775]
- 40.
- Sun D, et al. Enhanced release of prostaglandins contributes to flow-induced arteriolar dilation in eNOS knockout mice. Circ Res. 1999;85:288–93. [PubMed: 10436172]
- 41.
- Frangos JA, et al. Flow effects on prostacyclin production by cultured human endothelial cells. Science. 1985;227:1477–1479. [PubMed: 3883488]
- 42.
- Kohler R, et al. Impaired EDHF-mediated vasodilation and function of endothelial Ca2+-activated K+ channels in uremic rats. Kidney Int. 2005;67:2280–2287. [PubMed: 15882269]
- 43.
- Kohler R, et al. Impaired function of endothelial pressure-activated cation channel in salt-sensitive genetic hypertension. J Am Soc Nephrol. 2001;12:1624–1629. [PubMed: 11461934]
- 44.
- Dietrich A, et al. Increased vascular smooth muscle contractility in TRPC6−/− mice. Mol Cell Biol. 2005;25:6980–6989. [PMC free article: PMC1190236] [PubMed: 16055711]
- 45.
- Suzuki M, et al. Impaired pressure sensation in mice lacking TRPV4. J Biol Chem. 2003;278:22664–22668. [PubMed: 12692122]
- CA2+ AND ENDOTHELIAL FUNCTION
- TRPS IN THE ENDOTHELIUM
- TRPV4 IN THE ENDOTHELIUM
- TRPV4 AND ENDOTHELIUM-DEPENDENT VASODILATATION
- TRPV4-MEDIATED VASODILATATION IS NITRIC OXIDE DEPENDENT IN CAROTID ARTERIES
- A FUNCTIONAL ROLE OF TRPV4 IN ENDOTHELIAL MECHANOTRANSDUCTION?
- ENDOTHELIAL TRP CHANNELS AND CARDIOVASCULAR DISEASE
- DO ENDOTHELIAL CA2+-PERMEABLE CATION CHANNELS LIKE TRPV4 REPRESENT NOVEL PHARMACOTHERAPEUTIC TARGETS FOR ANTI-HYPERTENSIVE THERAPY?
- REFERENCES
- Arterial response to shear stress critically depends on endothelial TRPV4 expression.[PLoS One. 2007]Arterial response to shear stress critically depends on endothelial TRPV4 expression.Hartmannsgruber V, Heyken WT, Kacik M, Kaistha A, Grgic I, Harteneck C, Liedtke W, Hoyer J, Köhler R. PLoS One. 2007 Sep 5; 2(9):e827. Epub 2007 Sep 5.
- Review [Role of endothelial nitric oxide in the regulation of the vasomotor system].[Pathol Biol (Paris). 1998]Review [Role of endothelial nitric oxide in the regulation of the vasomotor system].Michel JB. Pathol Biol (Paris). 1998 Mar; 46(3):181-9.
- Review [The role of endothelium and nitric oxide in the regulation of vascular tone].[Cesk Fysiol. 2008]Review [The role of endothelium and nitric oxide in the regulation of vascular tone].Púzserová A, Kopincová J, Bernátová I. Cesk Fysiol. 2008; 57(2-3):53-60.
- Evidence for a functional role of endothelial transient receptor potential V4 in shear stress-induced vasodilatation.[Arterioscler Thromb Vasc Biol....]Evidence for a functional role of endothelial transient receptor potential V4 in shear stress-induced vasodilatation.Köhler R, Heyken WT, Heinau P, Schubert R, Si H, Kacik M, Busch C, Grgic I, Maier T, Hoyer J. Arterioscler Thromb Vasc Biol. 2006 Jul; 26(7):1495-502. Epub 2006 May 4.
- Review Secondary endothelial dysfunction: hypertension and heart failure.[J Mol Cell Cardiol. 1999]Review Secondary endothelial dysfunction: hypertension and heart failure.Boulanger CM. J Mol Cell Cardiol. 1999 Jan; 31(1):39-49.
- Role of TRPV4 in the Mechanotransduction of Shear Stress in Endothelial Cells - ...Role of TRPV4 in the Mechanotransduction of Shear Stress in Endothelial Cells - TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades
- The TRPC Family of Ion Channels: Relation to the TRP Superfamily and Role in Rec...The TRPC Family of Ion Channels: Relation to the TRP Superfamily and Role in Receptor- and Store-Operated Calcium Entry - TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades
Your browsing activity is empty.
Activity recording is turned off.
See more...