

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Liedtke WB, Heller S, editors. TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades. Boca Raton (FL): CRC Press/Taylor & Francis; 2007.

TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades.
Show detailsINTRODUCTION
Proteolytic enzymes comprise approximately 2 percent of the human genome [1]. Given their abundance, it is not surprising that proteases have diverse biological functions, ranging from the degradation of proteins in lysosomes to the control of physiological processes such as the coagulation cascade. However, a subset of serine proteases (possessing serine residues within their catalytic sites), which may be soluble in the extracellular fluid or tethered to the plasma membrane, are signaling molecules that can specifically regulate cells by cleaving protease-activated receptors (PARs), a family of four G-protein-coupled receptors (GPCRs). These serine proteases include members of the coagulation cascade (e.g., thrombin, factor VIIa, and factor Xa), proteases from inflammatory cells (e.g., mast cell tryptase, neutrophil cathepsin G), and proteases from epithelial tissues and neurons (e.g., trypsins). They are often generated or released during injury and inflammation, and they cleave PARs on multiple cell types, including platelets, endothelial and epithelial cells, myocytes, fibroblasts, and cells of the nervous system. Activated PARs regulate many essential physiological processes, such as hemostasis, inflammation, pain, and healing. These proteases and their receptors have been implicated in human disease and are potentially important targets for therapy.
Proteases and PARs participate in regulating most organ systems and are the subject of several comprehensive reviews [2, 3]. Within the central and peripheral nervous systems, proteases and PARs can control neuronal and astrocyte survival, proliferation and morphology, release of neurotransmitters, and the function and activity of ion channels, topics that have also been comprehensively reviewed [4, 5]. This chapter specifically concerns the ability of PARs to regulate TRPV channels of sensory neurons and thereby affect neurogenic inflammation and pain transmission [6, 7].
PAR CLEAVAGE AND ACTIVATION
Molecular Mechanisms of PAR Activation
The four cloned PARs share the typical structural features of GPCRs, with seven transmembrane domains, three intracellular and extracellular loops, an extracellular amino terminus, and an intracellular carboxyl terminus. PAR1 was initially identified by expressing libraries of RNA from thrombin-responsive cells in Xenopus oocytes, and identifying clones that exhibited thrombin responsiveness [8,9]. The discovery of PAR2 was accidental and occurred while screening a mouse genomic library using degenerate primers to the bovine neurokinin 2 receptor [10]. The existence of at least one further PAR, PAR3, was inferred from the observation that platelets from PAR1 knockout mice still respond to thrombin [11]. Finally, PAR4 was identified by searching expressed sequence tag libraries for PAR-like sequences [12, 13]. All four PARs share a general common mechanism of activation by proteases, although there are some subtle differences in activation that depend on the receptor and protease in question (these differences will be addressed below). In general, activating proteases cleave receptors at specific sites within the extracellular amino terminus to reveal a new amino terminus that acts as a tethered ligand, binding to conserved regions of the second extracellular loop, thereby activating the receptor (Table 31.1, Figure 31.1). Support for this mechanism of activation comes from the observation that synthetic activating peptides (APs), as short as six amino acids, that correspond to the tethered ligand domain can directly bind to and activate PAR1, PAR2, and PAR4. Although APs generally have a very low potency compared to proteases, they are valuable reagents for investigating the physiological functions of PARs, avoiding the use of proteases that can have many other mechanisms of action. Moreover, APs have been used as the starting materials for developing specific and potent antagonists and agonists of certain PARs [14, 15]. However, the peptide corresponding to the tethered ligand for PAR3 is unable to activate this receptor, for unknown reasons. The activating proteases and peptides, intracellular signals, and in vivo locations of the four PARs are summarized in Table 31.1.
TABLE 31.1
A Summary of the Major Known PAR-Activating Proteases, Cleavage Sites, Sequences of Activating Peptides, Signaling Mechanisms, and the Sites of PAR Expression
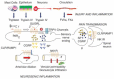
FIGURE 31.1
Protease cleavage and activation of PAR2 on primary spinal afferent neurons to cause neurogenic inflammation and pain. (1) Injury and inflammation trigger the generation and release of proteases from mast cells (tryptase), epithelial cells, and neurons (more...)
PAR Activating Proteases
The great diversity of proteases found within living organisms means that there are many different sources for potential activators of PARs. However, the presence of a particular protease within a system does not mean that it will necessarily be able to initiate a signal. Indeed, the capacity of proteases to activate PARs may depend on the presence of binding domains that tether proteases to receptors, of accessory proteins that tether proteases to the cell surface, and of protease inhibitors that modulate enzyme activity.
Certain proteases potently activate PARs only when they are concentrated at the cell surface, either through direct interaction with the receptor or by binding to other membrane proteins. As an example of receptor interaction, the anion-binding site of the coagulation factor thrombin binds to charged domains of PAR1 and PAR3, enabling the potent activation of these receptors [16, 17]. Higher concentrations of thrombin are required to activate PAR4, which lacks a thrombin-binding site [12, 13]. As an example of interaction with other membrane proteins, the coagulation factors VIIa and Xa interact with tissue factor, an integral membrane protein that is normally expressed by extravascular cells and expressed by endothelial cells and monocytes during inflammation, and thereby can potently activate PAR1 and PAR2 [18, 19]. Activated protein C, an anticoagulant protease that degrades coagulation factors Va and VIIa when released from the cell surface, can activate PAR1 when concentrated at the cell surface by the endothelial protein C receptor [20]. Interactions between PARs can also influence the capacity of proteases to activate receptors. The binding of an enzyme to one receptor can activate a different PAR, as in the case of PAR3 and PAR4, the thrombin receptors on murine platelets, which lack PAR1 [21]. Thrombin interacts with the charged domains of PAR3, which concentrates thrombin at the cell surface of murine platelets to promote thrombin cleavage and activation of PAR4. In addition, there is evidence for intermolecular signaling of certain PARs. The activating peptide sequence for PAR1 (SFLLRN) can activate PAR2, with reduced potency. When coexpressed in the same cell, the tethered ligand domain of PAR1 can interact with the binding site of PAR2, activating an uncleaved receptor [22].
Other proteases can potently activate PARs without being concentrated at the cell surface by interactions with receptors or other membrane proteins. For example, trypsins potently activate PAR2 and PAR4 without interacting with receptors or accessory membrane proteins. There are three trypsinogen genes in humans: protease serine (PRSS) I encodes trypsinogen I; PRSS2 encodes trypsinogen II; and PRSS3 encodes mesotrypsinogen [23, 24]. Trypsinogen IV is a splice variant of mesotrypsinogen [25]. Trypsinogen I and II are highly expressed in the pancreas and are secreted into the intestinal lumen, where they can cleave PAR2 on enterocytes [26]. These trypsins may also cleave PAR2 in the pancreas during inflammation, when trypsins are prematurely activated [27, 28]. Trypsinogen IV is expressed by brain neurons and epithelial cells of the intestine, airway, and prostate [25, 29]. Although the physiological functions of trypsinogen IV and mesotrypsinogen are unknown, they can activate PAR1, PAR2, and PAR4 [29] (Bunnett, unpublished). The observations that trypsinogen IV is resistant to polypeptide inhibitors, such as the pancreatic secretory trypsin inhibitor and soybean trypsin inhibitor, and also degrades such inhibitors [24, 29–31] implies that trypsinogen IV could show sustained activity in tissues, in contrast to trypsinogen I and II, which are susceptible to endogenous inhibitors and unable to degrade them.
Posttranslational modifications of PARs may also affect the ability of certain proteases to activate receptors. This appears to be the case for tryptase, an enzyme that may play an important role in activating PAR2 in the nervous system [32]. Tryptase is found in almost all subsets of human mast cells, where it forms up to 25 percent of total cellular protein [33, 34]. Tryptase is released from mast cells when they degranulate, and because mast cells are often in close proximity to sensory nerve fibers, both in normal and inflamed tissues [35], the activation of neuronal PARs by tryptase may be one mechanism by which mast cells regulate neuronal functions. Compared to trypsinogen, tryptase is a weak activator of PAR2. However, deglycosylation of PAR2 at a residue near the cleave site markedly increases the potency with which tryptase activates this receptor [36, 37]. The reason for this effect is unknown, although glycosylation may impede the interaction of the tryptase tetramer, with its active sites in the center of a doughnut shape, with the PAR2 cleavage site. Different forms of tryptase could also activate PAR2 with different potencies. In humans there are five different tryptase genes, and further splice variants are present in human mast cells. Recombinant forms of these tryptases have demonstrated mixed results in PAR2 activation assays, with α and βII tryptase activating PAR2 in a lymphoid cell line [38], while βI tryptase showed no PAR activation [39]. In addition to tryptase, cells of the immune system release several other PAR-activating proteases. Leukocytes, particularly neutrophils, release cathepsin G, elastase, and proteinase C, all of which can activate PARs.
MECHANISMS OF PAR SIGNAL TRANSDUCTION
PAR Coupling to G Proteins and Downstream Signaling Events
The unique nature of PAR activation, where a single protease molecule can activate multiple receptors, suggests that differences must exist between their signal transduction pathways and those for other GPCRs. Typical GPCR responses involve producing secondary messenger molecules in proportion to the number of receptors occupied by ligands at any one time. A single protease molecule could eventually cleave and activate every PAR on the surface of a cell, so the intensity of PAR responses is determined by the rate at which the receptors are cleaved, in proportion to the number of protease molecules present [40].
The identity of the G proteins through which PARs couple to mediate their cellular effects varies depending on the receptor in question and the cell type, allowing PARs to produce a wide range of physiological responses. The majority of studies of the mechanism of signal transduction by PARs have been carried out with PAR1, which can couple through Gαq/11, GαI, and Gα12/13. In platelets, thrombin activation of PAR1 activates Gαq/11, which in turn activates phospholipase C-β1. [41] This enzyme catalyzes the hydrolysis of the membrane phospholipid PIP2, releasing inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG), thus releasing Ca2+ from intracellular stores and activating protein kinase C (PKC). Activation of PAR1 on platelets also activates Gα12/13, coupling the receptor to the Rho family of guanine-nucleotide exchange factors that regulate Rho-kinase and myosin light chain kinase, mediating thrombin-induced changes in platelet shape [42]. In contrast, IP3 and DAG mediate the platelet aggregation and degranulation seen in response to thrombin [43]. The potential of PAR1 to also activate GαI, and the ability of the βγ subunits of the G proteins to themselves couple to other signaling pathways (such as phosphatidylinositol 3-kinase activation), further expands the range of intracellular processes mediated by PAR1.
Less is known about the signal transduction mechanisms of the other PARs. Activation of PAR2 induces generation of IP3 and a subsequent release of Ca2+ into the cytosol, in transfected cell lines [44], and in cells that endogenously express PAR2, such as neurons and enterocytes [45, 46], suggesting that PAR2 couples to Gαq/11. PAR2-mediated release of arachidonic acid and a subsequent generation of prostaglandins occur in enterocytes and transfected epithelial cells, indicating activation of phospholipase A2, although the coupling mechanism by which this occurs is not identified [26]. In addition, there is evidence of an interaction between PAR2 and TRPV channels requiring activation of protein kinase A (PKA), although it is not clear whether this is a result of Gαs activation and the resultant cAMP generation by adenylyl cyclase or whether it is a downstream component of a separate pathway (Bunnett, in press).
PAR agonists also couple to the activation of MAP kinases by a variety of mechanisms. One mechanism by which PAR2 activates ERK1/2 involves the β-arrestins, cytosolic proteins that interact with activated GPCRs at the plasma membrane. In addition to their role in receptor desensitization and endocytosis, β-arrestins are molecular scaffolds that form an ERK1/2 “signaling module” at the plasmamembrane or in endosomes. Thus, PAR2 agonists stimulate the assembly of a large molecular weight complex containing PAR2, β-arrestins, raf-1, and activated ERK1/2, which forms at the plasma membrane or in early endosomes [47]. The function of this module is to retain activated ERK1/2 in the cytosol, where they may regulate cytosolic or membrane targets, including ion channels.
Mechanisms of PAR Signal Arrest
Proteases activate PARs in an irreversible manner; once exposed, the tethered ligand is always available to interact with the cleaved receptor, which could lead to sustained signaling without the existence of efficient mechanisms to attenuate the response. Transient and permanent mechanisms dampen PAR signaling. The transient mechanism involves phosphorylation of PARs by members of the family of G-protein receptor kinases (GRKs), followed by interaction with β-arrestins, which uncouple PARs from heterotrimeric G proteins to desensitize G-protein signaling. GRK3 mediates agonist-induced phosphorylation of PAR1, because GRK3 overexpression enhances thrombin-stimulated PAR1 phosphorylation and suppresses thrombin signaling [48]. The overexpression of GRK3 in transgenic mice also attenuates thrombin-induced signaling in vivo [49]. In endothelial cells GRK5 mediates PAR1 desensitization; GRK5 expression suppresses thrombin signaling, and a dominant-negative GRK5 mutant prolongs thrombin signals [50]. The purpose of GRK-induced phosphorylation of a GPCR is to increase its affinity for β-arrestins to interact with GRK-phosphorylated GPCRs and disrupt their association with heterotrimeric G proteins to terminate the signal. PAR1 desensitization is diminished in fibroblasts lacking β-arrestins [51]. Similarly, a PAR2 mutant that is unable to interact with β-arrestins shows sustained signaling and diminished desensitization [47].
Receptor endocytosis followed by trafficking to lysosomes for degradation permanently arrests PAR signaling. Agonists promote endocytosis and lysosomal trafficking of PAR1 and PAR2 [52–54]. Both receptors internalize by a clathrin-mediated process involving the GTPase dynamin, which mediates detachment of clathrin-coated pits [55, 56]. β-arrestins also mediate endocytosis of many GPCRs by acting as adaptors that link receptors to clathrin. However, β-arrestins do not mediate agonist- induced endocytosis of PAR1, which proceeds normally in β-arrestin-deficient fibroblasts [51]. Endocytosis of PAR2 does requires β-arrestins. PAR2 agonists induce rapid translocation of β-arrestins from the cytosol to the plasma membrane, followed by a prolonged colocalization of PAR2 and β-arrestins in early endosomes [47, 54]. Compared to our understanding of endocytosis, less is known about the molecular mechanisms of post-endocytic sorting of GPCRs to lysosomes. Sorting nexin 1, a membrane-associated protein that interacts with the EGF receptor, is required for lysosomal trafficking of PAR1 [57]. A large amount of the E3 ligase c-Cbl is necessary to sort PAR2 from early endosomes to lysosomes [58].
PARs AND PROTEASE SIGNALING IN THE NERVOUS SYSTEM
Expression of PARs in the Nervous System
PARs1–4 are expressed in the brain, spinal cord, and associated ganglia, where they have been detected in both neurons and astrocytes. PAR1 mRNA is present in the rat neocortex, cingulate/retrosplenial cortex, subiculum, hypothalamic and thalamic nuclei, and in layers of the hippocampus, cerebellum, and olfactory bulb, where it is present in neurons and astroglia [59]. Immunoreactive PAR1 is found in the human cerebellum, temporal and frontal lobes, and the striatum, where it is prominently detected in astrocytes [60]. PAR1 is found in dopaminergic neurons in the substantia nigra, GABA-containing granule cells of the olfactory bulb, and glutaminergic neurons of the anterior thalamus. In rats, PAR1 is expressed in spinal cord motoneurons, preganglionic autonomic neurons, and dorsal root ganglia where it colocalizes with the neuropeptides substance P (SP) and calcitonin gene-related peptide (CGRP) [61]. PAR2 is also expressed by neurons and glial cells throughout the brain, including the hypothalamus [62, 63], and by primary spinal afferent neurons containing SP and CGRP [32]. Functional experiments confirm the widespread expression of PARs in astrocytes and neurons. Astrocytes express all four PARs [64], and thrombin increases [Ca2+]i in a glioma cell line and in astrocytes by activating both PAR1 and PAR4 [65]. Astrocytes also express PAR2 and respond to PAR2 agonists [66]. PAR1 and PAR2 agonists also signal to dorsal root ganglia neurons to increase [Ca2+]i, providing functional evidence for expression of these receptors [32, 46, 61].
PARs are also expressed in the peripheral nervous system, notably by enteric neurons. A large proportion of myenteric neurons respond to agonists of PAR1, PAR2, and PAR4 [46, 67, 68]. Agonists of these receptors depolarize myenteric neurons and increase their excitability [67, 68]. Approximately 50–60 percent of AH-type neurons (sensory neurons) and slightly fewer S neurons (inter and motor neurons) respond to PAR2 agonists. The PAR2-responsive S neurons also express nitric oxide (NO) synthase and project axons in an anal direction. These are inhibitory motor neurons, and activation would be expected to suppress motility. Submucosal neurons also express PARs, where activation may affect fluid and electrolyte secretion in the intestine. PAR2 is expressed in submucosal neurons of the guinea pig small intestine, some of which also express vasoactive intestinal polypeptides and are thus secretomotor neurons that play an important role in controlling fluid and electrolyte transport [69]. PAR2 agonists evoke transient depolarization of submucosal neurons followed by a prolonged hyperexcitability that can last for several hours. This long-term hyperexcitability mimics that observed after degranulation of mast cells, suggesting that mast cell proteases such as tryptase may induce long-term excitability of sub-mucosal neurons through PAR2 activation, which could result in enhanced fluid and electrolyte secretion from the mucosa [70, 71].
Proteases That Activate PARs in the Nervous System
Proteases from a variety of sources (including the circulatory system, inflammatory cells, and even the neurons themselves) have the potential to activate PARs in the central and peripheral nervous systems (Table 31.1). Although many of these enzymes activate PARs on neurons both in vitro and in vivo, in general the proteases that activate neuronal PARs under physiological and pathophysiological conditions have not been unequivocally identified.
Injury and inflammation generate several proteases of the coagulation cascade that can activate multiple PARs. Disruption of the integrity of the blood–brain barrier during pathological events such as head trauma and stroke may allow coagulation proteases to enter neural tissue, where they could activate PARs on neurons and astrocytes. During injury these proteases may also activate PARs in peripheral neurons, although this possibility has not been examined. Many inflammatory cells are recruited to sites of injury and inflammation, where they could release multiple proteases that activate neuronal PARs. In this regard, mast cells may be of particular importance because they are closely associated with sensory nerve fibers in peripheral tissues that contain SP and that are thus nociceptive neurons [35] (Figure 31.1). Indeed, tryptase released from mast cells can cleave and activate PAR2 on primary spinal afferent neurons to trigger the release of SP and CGRP in peripheral tissues, where they cause neurogenic inflammation, and within the dorsal horn of the spinal cord, where they cause pain transmission [6, 27, 28, 32, 72]. This mechanism could explain the poorly understood pain of irritable bowel syndrome, where there is a marked influx of tryptase-containing mast cells in close proximity to sensory nerve fibers in the colon, which is accompanied by hyperalgesia to distension of the bowel [73]. In addition, neuronal tissue itself expresses proteases that may activate PARs. In the olfactory bulb, mRNA coding for prothrombin and PAR1 colocalize in the same cell populations, suggesting that neuronally derived thrombin may activate neurons by cleaving PAR1 [59]. Moreover, several trypsinogens, including trypsinogen IV, are also expressed by neural tissues and, if released, could activate PARs [25, 29, 74]. However, nothing is known about the mechanisms that control the release of these neuronal proteases, thus their physiological role remains to be defined.
PAR Regulation of Neuronal Excitability
Accumulating evidence indicates that certain proteases can regulate the excitability of neurons, with important functional consequences. For example, trypsin and tryptase increase the excitability of primary spinal afferent neurons to induce hyperalgesia to thermal and mechanical stimuli [6, 72] and also to induce hyperexcitability of enteric neurons, with consequences for intestinal secretion and motility [67–69]. This increased activity of neurons depends on protease regulation of the function of ion channels.
Proteases could regulate ion channels by two general mechanisms: a direct mechanism by which proteases cleave the channel protein itself, and an indirect mechanism by which proteases cleave and activate PARs, which couple to signaling cascades that regulate the activity, localization, or level of expression of the channel. There is good evidence that certain proteases interact directly with ion channels to alter their activity, affecting both ionic transport and electrical excitability of target cells. Channel-activating proteases (CAPs) cleave certain epithelial sodium channels (ENaCs) to increase channel open probability sixfold, an activity mimicked by trypsin, although the precise target of enzymatic cleavage remains obscure [75]. Proteases can also regulate the acid-sensing ion channels (ASICs), members of the ENaC family that are found on neurons and that open in response to decreasing extracellular pH [76]. ASICs are expressed by nociceptive fibers and may transduce pain signals in response to local acidity [77]. ASIC1a and ASIC1b are cleaved by several serine proteases (e.g., trypsin, which only affects ASIC1a, chymotrypsin, and proteinase K), shifting the pH of channel opening and steady-state inactivation to a more acidic pH [78]. Proteases can also modify the activity of another neuronal ion channel, the NMDA receptor for the transmitter glutamate. Tissue-plasminogen activator, which is released by cortical neurons upon depolarization, interacts with NMDA receptors on the same neurons to increase their sensitivity to glutamate, leading to an increase in excitotoxic cell death [79]. The altered activity occurs as a result of tissue-plasminogen activator cleavage of the NR1 subunit of the NMDA receptor. However, although there is an increasing amount of evidence that proteases can regulate the activity of sensory ion channels, cleavage of TRPV channels by proteases has not been identified to date. The major known mechanism by which proteases regulate TRPV channels on sensory nerves is through the activation of PARs.
PAR Sensitization of TRPV Channels
Proteases that derive from mast cells (e.g., tryptase) and epithelial tissues (e.g., trypsins) can cleave PAR2 on the fibers of primary spinal afferent neurons to trigger the release of SP and CGRP in peripheral tissues and in the dorsal horn of the spinal cord [6, 27, 28, 32, 72]. In a similar manner, thrombin activates PAR1 on sensory nerves in peripheral tissues to cause neurogenic inflammation [61]. However, in contrast to PAR2 agonists, thrombin causes analgesia by mechanisms that are not fully understood [80]. The important roles played by TRPV1 and TRPV4 in detecting noxious thermal and mechanical stimuli, respectively, suggests that PAR2-induced thermal and mechanical hyperalgesia may be mediated by sensitization of these ion channels. This sensitization could occur through increased channel activity, decreased channel inhibition, insertion of additional channels into the cell membrane, or a combination of the three. Indeed, agonists of both GPCRs and receptor tyrosine kinases can sensitize TRPV1 by these mechanisms and thereby induce thermal hyperalgesia and inflammatory pain.
Exposure of sensory neurons to other inflammatory mediators (e.g., ATP, bradykinin, NGF), which are released in response to tissue damage or other stresses, induces thermal nociception by sensitizing TRPV1. The mechanisms of this sensitization have been thoroughly investigated. ATP and bradykinin, acting via metabotropic P2Y1 receptors and B2 receptors, respectively, activate PLCβ and consequently PKCɛ [81, 82]. Activated PKCɛ translocates to the plasma membrane and phosphorylates TRPV1 at S502 and S800 to enhance its open probability and inhibit its desensitization [83, 84]. Binding of the membrane phospholipid PIP2 to TRPV1 has been suggested to inhibit channel gating [85], and exposure to NGF sensitizes TRPV1 by removing this inhibition [86]. NGF is thought to bind to its tyrosine kinase receptor TrkA to activate PLCγ, which hydrolyzes PIP2 and removes its inhibitory effects on TRPV1. However, a recent study disputes this mechanism of NGF sensitization of TRPV1, suggesting instead that the most significant effect occurs through activated TrkA receptors that insert TRPV1-containing vesicles into the plasma membrane [87]. Activated TrkA phosphorylates phosphatidylinositol 3-kinase, with a downstream activation of Src kinase leading to phosphorylation of vesicular TRPV1 molecules on Y200, targeting the vesicles to the membrane.
Several observations suggest that PAR2 may regulate TRPV1 channels in sensory neurons to induce hyperalgesia to thermal stimuli (Figures 31.1 and 31.2). PAR2 is coexpressed in primary sensory neurons, alongside CGRP, SP, and TRPV1, which are all important components of nociceptive pathways [6, 32]. Activation of PARs induces neurogenic inflammation and nociception and, at subinflammatory doses, both mechanical and thermal hyperalgesia [6, 32, 72]. Furthermore, other inflammatory agents, such as bradykinin, ATP, and NGF, acting through protein kinases, can sensitize TRPV1 [81, 82, 86]. The first report of an interaction between PAR2 and TRPV1 was that PAR2 agonists enhance both capsaicin-induced release of CGRP from dorsal root ganglia and fos expression in spinal neurons [27]. Capsazepine, a TRPV1 antagonist, also inhibits the hyperalgesia [88] and gastric mucus secretion [89] induced by PAR2 activation in vivo, providing further evidence for TRPV1’s role in PAR2 regulation of neuronal function. PAR2 agonists also sensitize TRPV1-mediated calcium mobilization and TRPV1 currents in HEK cells expressing TRPV1 and in primary spinal afferent neurons [6, 7]. PAR2-induced thermal hyperalgesia, measured as a decrease in the paw withdrawal latency to a radiant heat, is abolished in mice lacking TRPV1 [6, 7]. Furthermore, nonalgesic doses of PAR2 agonists and capsaicin act synergistically to produce hyperalgesia [6]. Together, these results suggest that proteases released during trauma or inflammation can potentially activate PAR2 expressed in primary sensory neurons to sensitize TRPV1, resulting in exacerbated responses to TRPV1 activators, exemplified by thermal hyperalgesia. However, there are certain gaps in our understanding of this process.
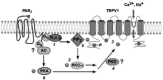
FIGURE 31.2
Potential mechanisms by which PAR2 sensitizes TRPV1. (1) Activated PAR2 couples to Gαq11 and activates PLCβ. (2) This pathway leads to the activation of PKCɛ, which translocates to the plasma membrane. (3) PKCɛ can phosphorylate (more...)
One deficiency is that the signal transduction mechanism by which PAR2 sensitizes TRPV1 is not fully understood. PAR2 couples to Gαq/11 and activates PLCβ, releasing IP3 and DAG. PAR2-induced activation of PLCβ could enhance TRPV1 by reducing the levels of its substrate, the membrane phospholipid PIP2, consequently freeing TRPV1 from PIP2-mediated inhibition [85, 86] (Figure 31.2). In addition, PLCβ-induced release of IP3 and DAG could activate PKC, which phosphorylates TRPV1 to increase its open probability and decrease desensitization [83, 84]. Indeed, PAR2-mediated potentiation of TRPV1 responses is diminished by PLC inhibitors and also by the nonselective inhibition of PKC inhibitors, supporting both hypotheses [6]. The identity of the PKC isozyme responsible for PAR2 sensitization of TRPV1 remains to be determined. One particularly promising candidate is PKCɛ, which plays a major role in transmitting mechanical and thermal hyperalgesia [90]. Also, PKCɛ phosphorylates TRPV1 [82] to mediate bradykinin-induced sensitization of TRPV1 currents [81]. Indeed, selective antagonism of PKCɛ suppresses PAR2-induced sensitization of TRPV1 currents in nociceptive neurons [7]. However, the contribution of other PKC isoforms cannot be excluded, because Gö6976, an inhibitor of conventional PKC isoforms (α, β1, β2, and γ), but not ɛ, also inhibits PAR2-mediated potentiation of TRPV1 signals [6]. This implicates protein kinase D1 (PKD1), as Gö6976 is also a PKD1 inhibitor. Initially thought to be an atypical member of the PKC family (PKCμ), PKD1 is now recognized as a member of a new family of three PKD isozymes that resemble the CAM kinase family [91]. Certain novel PKC isoforms activate PKD (e.g., thrombin activation of PKD in platelets involves PKCδ [92]), which may mediate some of the effects of PKC activation. This possibility is particularly relevant in light of the finding that PKD1 binds and phosphorylates TRPV1 on S116, enhancing TRPV1-mediated responses, while anti-sense oligonucleotides directed against PKD1 decreased TRPV1 responses [93]. There is also evidence that sensitization of TRPV1 may also occur through activation of PKA. PKA is required for injury-induced hyperalgesia [94], and it phosphorylates TRPV1 on S116 to regulate its desensitization [95, 96], thus sensitizing TRPV1 currents in nociceptive neurons [97, 98].
Another gap in our understanding of the role of PARs in pain transmission is the mechanism of PAR2-induced mechanical hyperalgesia, which is evident after both somatic and visceral mechanical stimuli [72, 99]. Although the channel that mediates responses to mechanical stimuli is not unequivocally identified, TRPV4 is a strong candidate. TRPV4 is the mammalian homologue of the C. elegans gene osm-9, which encodes a protein conferring sensitivity to osmotic pressure, touch, and odorants [100]. Originally called VR-OAC, this channel responds to small reductions in tonicity; in rats it is expressed in circumventricular organs, which detect changes in systemic osmolarity, and in neurosensory structures such as Merkel cells and sensory neurons [101]. Based on its structure, ionic selectivity (preference for Ca2+), and thermal sensitivity (>27°C), VR-OAC was placed in the same TRPV family as TRPV4 [102, 103]. The phorbol ester 4α-phorbol 12,13-didecanoate (4αPDD) is a synthetic TRPV4 agonist [104], and the cytochrome P450 product of arachidonic acid, 5’,6’-epoxyeicosatrienoic acid (5’,6’-EET), is a potential endogenous TRPV4 agonist [105] (hypoosmotic stimuli leads to cell swelling, phospholipase A2 activation, and release of arachidonic acid [106]). The expression of TRPV4 in sensory neurons and its activation by changes in cell volume suggest that it detects osmotic and mechanical stimuli. TRPV4−/− mice show abnormal osmotic regulation and decreased acid and pressure nociception (not impaired touch or heat sensation) [107, 108]. Although injection of hypotonic saline into rat hindpaws does not induce nociception, hypertonic saline causes pain, and pretreatment with an inflammatory mediator, PGE2, sensitizes responses to both stimuli [101, 109]. These effects require TRPV4, because they are prevented by TRPV4 deletion or downregulation. In a similar manner to PGE2, PAR2 agonists also increase the magnitude of the Ca2+ mobilization in response to hypotonic saline and 4αPDD in both human bronchial epithelial cells, which constitutively express both TRPV4 and PAR2, and in HEK cells heterologously expressing TRPV4 (Bunnett, unpublished). In slices of rat spinal cord, exposure to 4αPDD or hypotonic saline stimulates the release of SP and CGRP, which is potentiated by preexposure to PAR2 agonist. Hypotonic saline injected into mice paws does not produce any nociceptive behavior, but the combination of PAR2–AP and hypotonic saline increases the sensitivity to mechanical pain to a greater degree than PAR2–AP alone, indicating a sensitization of TRPV4 by PAR2 activation. TRPV4−/− mice do not show any increased sensitivity with either injection, indicating that both the mechanical hyperalgesia induced by PAR2 activation and the pain induced by hypotonic saline are mediated by TRPV4 (Bunnett, unpublished). Together, these findings suggest that PAR2 agonists can sensitize TRPV4 to induce hyperalgesia to mechanical stimuli, possibly by a mechanism that resembles those that mediate PAR2-induced sensitization of TRPV1.
CONCLUSIONS AND FUTURE PERSPECTIVES
PARs are widely expressed in the central and peripheral nervous systems, and a variety of protease sources can signal to neurons by cleaving PARs to regulate fundamentally important processes, which include pain transmission and neurogenic inflammation as well as neuronal survival, degeneration, neurite outgrowth, and release of neurotransmitters. Because PAR-activating proteases are generated or released during injury and inflammation, these mechanisms may be of considerable interest in treating diseases, and protease inhibitors and PAR antagonists could be useful therapeutic agents.
However, there are major gaps in our understanding of the role of proteases and PARs in the nervous system. First, although several proteases have been characterized with the potential to activate neuronal PARs, the endogenous proteases that activate PARs in neuronal tissues under physiological and pathophysiological conditions have not been unequivocally identified. Second, the cellular mechanisms that transduce and regulate PAR signaling in neurons have not been defined. Most information about PAR signaling and signal arrest derives from studies of cell lines, often transfected to express the receptor of interest, and it remains to be determined whether similar processes occur in neurons. Third, the mechanisms by which PARs regulate excitability of sensory nerves are not fully understood. Although it is well established that PAR2 agonists can sensitize TRPV1 and thereby induce thermal hyperalgesia, the signaling pathways responsible for this sensitization are not completely defined. Moreover, the mechanisms of PAR2-induced mechanical hyperalgesia are not well characterized, and the mechanisms of the analgesic actions of PAR1 agonists are unknown. Fourth, although the roles of PAR1 and PAR2 have been studied in neurons, very little is known about the functions of PAR3 and PAR4, which are also present in the nervous system. Finally, the roles of proteases and PARs in experimental models of pain and neurogenic inflammation have not been fully explored, in part due to the lack of specific protease inhibitors and PAR antagonists. The use of such tools and of genetically modified animals will allow investigation of the contributions of proteases and PARs in experimental models of human diseases.
ACKNOWLEDGMENTS
Research in the authors’ laboratory in this area is supported by NIH grants DK43207, DK57489, and DK39957, and by the Wellcome Trust.
REFERENCES
- 1.
- Southan C. A genomic perspective on human proteases. FEBS Lett. 2001;498:214. [PubMed: 11412860]
- 2.
- Macfarlane SR, et al. Proteinase-activated receptors. Pharmacol Rev. 2001;53:245. [PubMed: 11356985]
- 3.
- Ossovskaya VS, Bunnett NW. Protease-activated receptors: contribution to physiology and disease. Physiol Rev. 2004;84:579. [PubMed: 15044683]
- 4.
- Vergnolle N, et al. Proteinase-activated receptors: novel signals for peripheral nerves. Trends Neurosci. 2003;26:496. [PubMed: 12948661]
- 5.
- Saito T, Bunnett NW. Protease-activated receptors: regulation of neuronal function. Neuromolecular Med. 2005;7:79. [PubMed: 16052040]
- 6.
- Amadesi S, et al. Protease-activated receptor 2 sensitizes the capsaicin receptor transient receptor potential vanilloid receptor 1 to induce hyperalgesia. J Neurosci. 2004;24:4300. [PMC free article: PMC6729438] [PubMed: 15128844]
- 7.
- Dai Y, et al. Proteinase-activated receptor 2–mediated potentiation of transient receptor potential vanilloid subfamily 1 activity reveals a mechanism for proteinase-induced inflammatory pain. J Neurosci. 2004;24:4293. [PMC free article: PMC6729433] [PubMed: 15128843]
- 8.
- Rasmussen UB, et al. cDNA cloning and expression of a hamster alpha-thrombin receptor coupled to Ca2+ mobilization. FEBS Lett. 1991;288:123. [PubMed: 1652467]
- 9.
- Vu TK, et al. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991;64:1057. [PubMed: 1672265]
- 10.
- Nystedt S, et al. Molecular cloning of a potential proteinase activated receptor. Proc Natl Acad Sci USA. 1994;91:9208. [PMC free article: PMC44781] [PubMed: 7937743]
- 11.
- Connolly AJ, et al. Role of the thrombin receptor in development and evidence for a second receptor. Nature. 1996;381:516. [PubMed: 8632823]
- 12.
- Xu WF, et al. Cloning and characterization of human protease-activated receptor 4. Proc Natl Acad Sci USA. 1998;95:6642. [PMC free article: PMC22580] [PubMed: 9618465]
- 13.
- Kahn ML, et al. A dual thrombin receptor system for platelet activation. Nature. 1998;394:690. [PubMed: 9716134]
- 14.
- Andrade-Gordon P, et al. Design, synthesis, and biological characterization of a peptide-mimetic antagonist for a tethered-ligand receptor. Proc Natl Acad Sci USA. 1999;96:12257. [PMC free article: PMC22903] [PubMed: 10535908]
- 15.
- Ferrell WR, et al. Essential role for proteinase-activated receptor-2 in arthritis. J Clin Invest. 2003;111:35. [PMC free article: PMC151840] [PubMed: 12511586]
- 16.
- Vu TK, et al. Domains specifying thrombin-receptor interaction. Nature. 1991;353:674. [PubMed: 1717851]
- 17.
- Ishihara H, et al. Protease-activated receptor 3 is a second thrombin receptor in humans. Nature. 1997;386:502. [PubMed: 9087410]
- 18.
- Camerer E, et al. Tissue factor– and factor X–dependent activation of protease-activated receptor 2 by factor VIIa. Proc Natl Acad Sci USA. 2000;97:5255. [PMC free article: PMC25815] [PubMed: 10805786]
- 19.
- Riewald M, Ruf W. Mechanistic coupling of protease signaling and initiation of coagulation by tissue factor. Proc Natl Acad Sci USA. 2001;98:7742. [PMC free article: PMC35412] [PubMed: 11438726]
- 20.
- Riewald M, et al. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science. 2002;296:1880. [PubMed: 12052963]
- 21.
- Nakanishi-Matsui M, et al. PAR3 is a cofactor for PAR4 activation by thrombin. Nature. 2000;404:609. [PubMed: 10766244]
- 22.
- O’Brien PJ, et al. Thrombin responses in human endothelial cells. Contributions from receptors other than PAR1 include the transactivation of PAR2 by thrombin-cleaved PAR1. J Biol Chem. 2000;275:13502. [PubMed: 10788464]
- 23.
- Emi M, et al. Cloning, characterization, and nucleotide sequences of two cDNAs encoding human pancreatic trypsinogens. Gene. 1986;41:305. [PubMed: 3011602]
- 24.
- Nyaruhucha CN, et al. Identification and expression of the cDNA-encoding human mesotrypsin(ogen), an isoform of trypsin with inhibitor resistance. J Biol Chem. 1997;272:10573. [PubMed: 9099703]
- 25.
- Wiegand U, et al. Cloning of the cDNA encoding human brain trypsinogen and characterization of its product. Gene. 1993;136:167. [PubMed: 8294000]
- 26.
- Kong W, et al. Luminal trypsin may regulate enterocytes through proteinase-activated receptor 2. Proc Natl Acad Sci USA. 1997;94:8884. [PMC free article: PMC23180] [PubMed: 9238072]
- 27.
- Hoogerwerf WA, et al. The proteinase-activated receptor 2 is involved in nociception. J Neurosci. 2001;21:9036. [PMC free article: PMC6762290] [PubMed: 11698614]
- 28.
- Hoogerwerf WA, et al. Trypsin mediates nociception via the proteinase-activated receptor 2: a potentially novel role in pancreatic pain. Gastroenterology. 2004;127:883. [PubMed: 15362043]
- 29.
- Cottrell GS, et al. Trypsin IV, a novel agonist of protease-activated receptors 2 and 4. J Biol Chem. 2004;279:13532. [PubMed: 14726524]
- 30.
- Szmola R, et al. Human mesotrypsin is a unique digestive protease specialized for the degradation of trypsin inhibitors. J Biol Chem. 2003;278:48580. [PMC free article: PMC1393292] [PubMed: 14507909]
- 31.
- Sahin-Toth M. Human mesotrypsin defies natural trypsin inhibitors: from passive resistance to active destruction. Protein Pept Lett. 2005;12:457. [PMC free article: PMC1488880] [PubMed: 16029158]
- 32.
- Steinhoff M, et al. Agonists of proteinase-activated receptor 2 induce inflammation by a neurogenic mechanism. Nat Med. 2000;6:151. [PubMed: 10655102]
- 33.
- Caughey GH. Mast cell chymases and tryptases: phylogeny, family relations, and biogenesis. In: Caughey GH, editor. Mast cell proteases in immunology and biology. Marcel Dekker; New York: 1995. p. 305.
- 34.
- Fiorucci L, Ascoli F. Mast cell tryptase, a still enigmatic enzyme. Cell Mol Life Sci. 2004;61:1278. [PubMed: 15170507]
- 35.
- Stead RH, et al. Intestinal mucosal mast cells in normal and nematode-infected rat intestines are in intimate contact with peptidergic nerves. Proc Natl Acad Sci USA. 1987;84:2975. [PMC free article: PMC304783] [PubMed: 2437589]
- 36.
- Compton SJ, et al. Glycosylation and the activation of proteinase-activated receptor 2 (PAR2) by human mast cell tryptase. Br J Pharmacol. 2001;134:705. [PMC free article: PMC1572998] [PubMed: 11606310]
- 37.
- Compton SJ, et al. Glycosylation of human proteinase-activated receptor-2 (hPAR2): role in cell surface expression and signalling. Biochem J. 2002;368:495. [PMC free article: PMC1222997] [PubMed: 12171601]
- 38.
- Mirza H, et al. Mitogenic responses mediated through the proteinase-activated receptor-2 are induced by expressed forms of mast cell alpha- or beta-tryptases. Blood. 1997;90:3914. [PubMed: 9354658]
- 39.
- Huang C, et al. Evaluation of the substrate specificity of human mast cell tryptase beta I and demonstration of its importance in bacterial infections of the lung. J Biol Chem. 2001;276:26276. [PubMed: 11335723]
- 40.
- Ishii K, et al. Kinetics of thrombin receptor cleavage on intact cells. Relation to signaling. J Biol Chem. 1993;268:9780. [PubMed: 7683662]
- 41.
- Hung DT, et al. The cloned platelet thrombin receptor couples to at least two distinct effectors to stimulate phosphoinositide hydrolysis and inhibit adenylyl cyclase. J Biol Chem. 1992;267:20831. [PubMed: 1328213]
- 42.
- Klages B, et al. Activation of G12/G13 results in shape change and Rho/Rho-kinase–mediated myosin light chain phosphorylation in mouse platelets. J Cell Biol. 1999;144:745. [PMC free article: PMC2132941] [PubMed: 10037795]
- 43.
- Offermanns S, et al. Defective platelet activation in G alpha(q)-deficient mice. Nature. 1997;389:183. [PubMed: 9296496]
- 44.
- Bohm SK, et al. Molecular cloning, expression, and potential functions of the human proteinase-activated receptor-2. Biochem J. 1996;314:1009. [PMC free article: PMC1217107] [PubMed: 8615752]
- 45.
- Corvera CU, et al. Mast cell tryptase regulates rat colonic myocytes through proteinase-activated receptor 2. J Clin Invest. 1997;100:1383. [PMC free article: PMC508316] [PubMed: 9294103]
- 46.
- Corvera CU, et al. Thrombin and mast cell tryptase regulate guinea-pig myenteric neurons through proteinase-activated receptors-1 and -2. J Physiol. 1999;517:741. [PMC free article: PMC2269379] [PubMed: 10358115]
- 47.
- DeFea KA, et al. beta-arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J Cell Biol. 2000;148:1267. [PMC free article: PMC2174299] [PubMed: 10725339]
- 48.
- Ishii K, et al. Inhibition of thrombin receptor signaling by a G-protein-coupled receptor kinase. Functional specificity among G-protein-coupled receptor kinases. J Biol Chem. 1994;269:1125. [PubMed: 8288570]
- 49.
- Iaccarino G, et al. Myocardial overexpression of GRK3 in transgenic mice: evidence for in vivo selectivity of GRKs. Am J Physiol. 1998;275:H1298. [PubMed: 9746479]
- 50.
- Tiruppathi C, et al. G-protein-coupled receptor kinase-5 regulates thrombin-activated signaling in endothelial cells. Proc Natl Acad Sci USA. 2000;97:7440. [PMC free article: PMC16564] [PubMed: 10861009]
- 51.
- Paing MM, et al. beta-arrestins regulate protease-activated receptor-1 desensitization but not internalization or down-regulation. J Biol Chem. 2002;277:1292. [PubMed: 11694535]
- 52.
- Hoxie JA, et al. Internalization and recycling of activated thrombin receptors. J Biol Chem. 1993;268:13756. [PubMed: 8390469]
- 53.
- Bohm SK, et al. Mechanisms of desensitization and resensitization of proteinase-activated receptor-2. J Biol Chem. 2003;2712:1996. [PubMed: 8703006]
- 54.
- Dery O, et al. Trafficking of proteinase-activated receptor-2 and beta-arrestin-1 tagged with green fluorescent protein. Beta-arrestin-dependent endocytosis of a proteinase receptor. J Biol Chem. 1999;274:18524. [PubMed: 10373461]
- 55.
- Trejo J, et al. Protease-activated receptor-1 down-regulation: a mutant HeLa cell line suggests novel requirements for PAR1 phosphorylation and recruitment to clathrin-coated pits. J Biol Chem. 2000;275:31255. [PubMed: 10893235]
- 56.
- Roosterman D, et al. Rab5a and rab11a mediate agonist-induced trafficking of protease-activated receptor 2. Am J Physiol Cell Physiol. 2003;284:C1319. [PubMed: 12540381]
- 57.
- Wang Y, et al. Down-regulation of protease-activated receptor-1 is regulated by sorting nexin 1. Mol Biol Cell. 1965;13:2002. [PMC free article: PMC117618] [PubMed: 12058063]
- 58.
- Jacob C, et al. c-Cbl mediates ubiquitination, degradation, and down-regulation of human protease-activated receptor 2. J Biol Chem. 2005;280:16076. [PubMed: 15708858]
- 59.
- Weinstein JR, et al. Cellular localization of thrombin receptor mRNA in rat brain: expression by mesencephalic dopaminergic neurons and codistribution with pro-thrombin mRNA. J Neurosci. 1995;15:2906. [PMC free article: PMC6577761] [PubMed: 7722637]
- 60.
- Junge CE, et al. Protease-activated receptor-1 in human brain: localization and functional expression in astrocytes. Exp Neurol. 2004;188:94. [PubMed: 15191806]
- 61.
- de Garavilla L, et al. Agonists of proteinase-activated receptor 1 induce plasma extravasation by a neurogenic mechanism. Br J Pharmacol. 2001;133:975. [PMC free article: PMC1572861] [PubMed: 11487506]
- 62.
- Smith-Swintosky VL, et al. Protease-activated receptor-2 (PAR-2) is present in the rat hippocampus and is associated with neurodegeneration. J Neurochem. 1997;69:1890. [PubMed: 9349532]
- 63.
- D’Andrea MR, et al. Characterization of protease-activated receptor-2 immunore-activity in normal human tissues. J Histochem Cytochem. 1998;46:157. [PubMed: 9446822]
- 64.
- Wang H, et al. Four subtypes of protease-activated receptors, co-expressed in rat astrocytes, evoke different physiological signaling. Glia. 2002;37:53. [PubMed: 11746783]
- 65.
- Kaufmann R, et al. The two-receptor system PAR-1/PAR-4 mediates alpha-thrombin-induced [Ca(2+)](i) mobilization in human astrocytoma cells. J Cancer Res Clin Oncol. 2000;126:91. [PubMed: 10664248]
- 66.
- Ubl JJ, et al. Co-existence of two types of [Ca2+]i-inducing protease-activated receptors (PAR-1 and PAR-2) in rat astrocytes and C6 glioma cells. Neuroscience. 1998;86:597. [PubMed: 9881872]
- 67.
- Linden DR, et al. Agonists of proteinase-activated receptor 2 excite guinea pig ileal myenteric neurons. Eur J Pharmacol. 2001;431:311. [PubMed: 11730723]
- 68.
- Gao C, et al. Serine proteases excite myenteric neurons through protease-activated receptors in guinea pig small intestine. Gastroenterology. 2002;123:1554. [PubMed: 12404230]
- 69.
- Reed DE, et al. Mast cell tryptase and proteinase-activated receptor 2 induce hyperexcitability of guinea-pig submucosal neurons. J Physiol. 2003;547:531. [PMC free article: PMC2342663] [PubMed: 12562962]
- 70.
- Green BT, et al. Intestinal type 2 proteinase-activated receptors: expression in opoid-sensitive secretomotor neural circuits that mediate epithelial ion transport. J Pharmacol Exp Ther. 2000;295:410. [PubMed: 10992008]
- 71.
- Cuffe JE, et al. Basolateral PAR-2 receptors mediate KCl secretion and inhibition of Na+ absorption in the mouse distal colon. J Physiol. 2002;539:209. [PMC free article: PMC2290120] [PubMed: 11850514]
- 72.
- Vergnolle N, et al. Proteinase-activated receptor-2 and hyperalgesia: a novel pain pathway. Nat Med. 2001;7:821. [PubMed: 11433347]
- 73.
- Barbara G, et al. Activated mast cells in proximity to colonic nerves correlate with abdominal pain in irritable bowel syndrome. Gastroenterology. 2004;126:693. [PubMed: 14988823]
- 74.
- Minn A, et al. Enhanced GFAP expression in astrocytes of transgenic mice expressing the human brain-specific trypsinogen IV. Glia. 1998;22:338. [PubMed: 9517566]
- 75.
- Chraibi A, et al. Protease modulation of the activity of the epithelial sodium channel expressed in Xenopus oocytes. J Gen Physiol. 1998;111:127. [PMC free article: PMC1887769] [PubMed: 9417140]
- 76.
- Krishtal O. The ASICs: signaling molecules? modulators? Trends Neurosci. 2003;26:477. [PubMed: 12948658]
- 77.
- Sutherland SP, et al. Acid-sensing ion channel 3 matches the acid-gated current in cardiac ischemia-sensing neurons. Proc Natl Acad Sci USA. 2001;98:711. [PMC free article: PMC14653] [PubMed: 11120882]
- 78.
- Poirot O, et al. Selective regulation of acid-sensing ion channel 1 by serine proteases. J Biol Chem. 2004;279:38448. [PubMed: 15247234]
- 79.
- Nicole O, et al. The proteolytic activity of tissue-plasminogen activator enhances NMDA receptor-mediated signaling. Nat Med. 2001;7:59. [PubMed: 11135617]
- 80.
- Asfaha S, et al. Proteinase-activated receptor-1 agonists attenuate nociception in response to noxious stimuli. Br J Pharmacol. 2002;135:1101. [PMC free article: PMC1573232] [PubMed: 11877315]
- 81.
- Cesare P, et al. Ion channels gated by heat. Proc Natl Acad Sci USA. 1999;96:7658. [PMC free article: PMC33597] [PubMed: 10393876]
- 82.
- Numazaki M, et al. Direct phosphorylation of capsaicin receptor VR1 by protein kinase C epsilon and identification of two target serine residues. J Biol Chem. 2002;277:13375. [PubMed: 11884385]
- 83.
- Vellani V, et al. Protein kinase C activation potentiates gating of the vanilloid receptor VR1 by capsaicin, protons, heat, and anandamide. J Physiol. 2001;534:813. [PMC free article: PMC2278732] [PubMed: 11483711]
- 84.
- Mandadi S, et al. Activation of protein kinase C reverses capsaicin-induced calcium-dependent desensitization of TRPV1 ion channels. Cell Calcium. 2004;35:471. [PubMed: 15003856]
- 85.
- Prescott ED, Julius D. A modular PIP2 binding site as a determinant of capsaicin receptor sensitivity. Science. 2003;300:1284. [PubMed: 12764195]
- 86.
- Chuang HH, et al. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature. 2001;411:957. [PubMed: 11418861]
- 87.
- Zhang X, et al. NGF rapidly increases membrane expression of TRPV1 heat-gated ion channels. Embo J. 2005;24:4211. [PMC free article: PMC1356334] [PubMed: 16319926]
- 88.
- Kawao N, et al. Capsazepine inhibits thermal hyperalgesia but not nociception triggered by protease-activated receptor-2 in rats. Jpn J Pharmacol. 2002;89:184. [PubMed: 12120762]
- 89.
- Kawabata A, et al. Capsazepine partially inhibits neurally mediated gastric mucus secretion following activation of protease-activated receptor 2. Clin Exp Pharmacol Physiol. 2002;29:360. [PubMed: 11985550]
- 90.
- Khasar SG, et al. A novel nociceptor signaling pathway revealed in protein kinase C epsilon mutant mice. Neuron. 1999;24:253. [PubMed: 10677042]
- 91.
- Rozengurt E, et al. Protein kinase D signaling. J Biol Chem. 2005;280:13205. [PubMed: 15701647]
- 92.
- Tan M, et al. Thrombin rapidly induces protein kinase D phosphorylation, and protein kinase C delta mediates the activation. J Biol Chem. 2003;278:2824. [PubMed: 12431976]
- 93.
- Wang Y, et al. Interaction between protein kinase Cmu and the vanilloid receptor type 1. J Biol Chem. 2004;279:53674. [PubMed: 15471852]
- 94.
- Malmberg AB, et al. Diminished inflammation and nociceptive pain with preservation of neuropathic pain in mice with a targeted mutation of the type I regulatory subunit of cAMP-dependent protein kinase. J Neurosci. 1997;17:7462. [PMC free article: PMC6573437] [PubMed: 9295392]
- 95.
- Bhave G, et al. cAMP-dependent protein kinase regulates desensitization of the capsaicin receptor (VR1) by direct phosphorylation. Neuron. 2002;35:721. [PubMed: 12194871]
- 96.
- Mohapatra DP, Nau C. Desensitization of capsaicin-activated currents in the vanilloid receptor TRPV1 is decreased by the cyclic AMP-dependent protein kinase pathway. J Biol Chem. 2003;278:50080. [PubMed: 14506258]
- 97.
- Lopshire JC, Nicol GD. The cAMP transduction cascade mediates the pros-taglandin E2 enhancement of the capsaicin-elicited current in rat sensory neurons: whole-cell and single-channel studies. J Neurosci. 1998;18:6081. [PMC free article: PMC6793178] [PubMed: 9698303]
- 98.
- Rathee PK, et al. PKA/AKAP/VR-1 module: a common link of Gs-mediated signaling to thermal hyperalgesia. J Neurosci. 2002;22:4740. [PMC free article: PMC6758778] [PubMed: 12040081]
- 99.
- Coelho AM, et al. Proteinases and proteinase-activated receptor 2: a possible role to promote visceral hyperalgesia in rats. Gastroenterology. 2002;122:1035. [PubMed: 11910355]
- 100.
- Liedtke W, et al. Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell. 2000;103:525. [PMC free article: PMC2211528] [PubMed: 11081638]
- 101.
- Alessandri-Haber N, et al. Hypotonicity induces TRPV4-mediated nociception in rat. Neuron. 2003;39:497. [PubMed: 12895423]
- 102.
- Guler AD, et al. Heat-evoked activation of the ion channel, TRPV4. J Neurosci. 2002;22:6408. [PMC free article: PMC6758176] [PubMed: 12151520]
- 103.
- Clapham DE, et al. International Union of Pharmacology. XLIII. Compendium of voltage-gated ion channels: transient receptor potential channels. Pharmacol Rev. 2003;55:591. [PubMed: 14657417]
- 104.
- Watanabe H, et al. Activation of TRPV4 channels (hVRL-2/mTRP12) by phorbol derivatives. J Biol Chem. 2002;277:13569. [PubMed: 11827975]
- 105.
- Watanabe H, et al. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature. 2003;424:434. [PubMed: 12879072]
- 106.
- Pedersen S, et al. Hypotonic cell swelling induces translocation of the alpha isoform of cytosolic phospholipase A2 but not the gamma isoform in Ehrlich ascites tumor cells. Eur J Biochem. 2000;267:5531. [PubMed: 10951212]
- 107.
- Liedtke W, Friedman JM. Abnormal osmotic regulation in trpv4−/− mice. Proc Natl Acad Sci USA. 2003;100:13698. [PMC free article: PMC263876] [PubMed: 14581612]
- 108.
- Suzuki M, et al. Impaired pressure sensation in mice lacking TRPV4. J Biol Chem. 2003;278:22664. [PubMed: 12692122]
- 109.
- Alessandri-Haber N, et al. TRPV4 mediates pain-related behavior induced by mild hypertonic stimuli in the presence of inflammatory mediator. Pain. 2005 [PubMed: 16213085]
- Review Protease-activated receptors: regulation of neuronal function.[Neuromolecular Med. 2005]Review Protease-activated receptors: regulation of neuronal function.Saito T, Bunnett NW. Neuromolecular Med. 2005; 7(1-2):79-99.
- Review Protease-activated receptors: how proteases signal to cells to cause inflammation and pain.[Semin Thromb Hemost. 2006]Review Protease-activated receptors: how proteases signal to cells to cause inflammation and pain.Bunnett NW. Semin Thromb Hemost. 2006 Apr; 32 Suppl 1:39-48.
- Review Protease-activated receptors: contribution to physiology and disease.[Physiol Rev. 2004]Review Protease-activated receptors: contribution to physiology and disease.Ossovskaya VS, Bunnett NW. Physiol Rev. 2004 Apr; 84(2):579-621.
- Review Role of PAR-2 in Neuroimmune Communication and Itch.[Itch: Mechanisms and Treatment...]Review Role of PAR-2 in Neuroimmune Communication and Itch.Kempkes C, Buddenkotte J, Cevikbas F, Buhl T, Steinhoff M. Itch: Mechanisms and Treatment. 2014
- Review Pathophysiological actions of protease activated receptors (PARs).[Pharmazie. 2007]Review Pathophysiological actions of protease activated receptors (PARs).Pandya NM, Jain SM, Santani DD. Pharmazie. 2007 Mar; 62(3):163-9.
- Protease-Activated Receptors: Mechanisms by Which Proteases Sensitize TRPV Chann...Protease-Activated Receptors: Mechanisms by Which Proteases Sensitize TRPV Channels to Induce Neurogenic Inflammation and Pain - TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades
- ATPase family gene 2 protein homolog A isoform X8 [Homo sapiens]ATPase family gene 2 protein homolog A isoform X8 [Homo sapiens]gi|2462595243|ref|XP_054205053.1|Protein
- Lantaneae pentatricopeptide repeat-containing protein 97 gene, partial cds.Lantaneae pentatricopeptide repeat-containing protein 97 gene, partial cds.PopSet: 2085706842PopSet
- TRPV4: A Multifunctional Nonselective Cation Channel with Complex Regulation - T...TRPV4: A Multifunctional Nonselective Cation Channel with Complex Regulation - TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades
- Molecular Mechanisms of TRPV4 Gating - TRP Ion Channel Function in Sensory Trans...Molecular Mechanisms of TRPV4 Gating - TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades
Your browsing activity is empty.
Activity recording is turned off.
See more...