

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Liedtke WB, Heller S, editors. TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades. Boca Raton (FL): CRC Press/Taylor & Francis; 2007.

TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades.
Show detailsREQUIREMENT OF CALCIUM ENTRY FOR CELL-CYCLE PROGRESSION
Actions of growth factors are essential for mammalian cells to proliferate. For example, fibroblasts continue to grow in the presence of growth factors in serum, and the removal of serum attenuates proliferation. Serum-deprived cells eventually leave the cell cycle, fall into the G0 state, and become quiescent. Quiescent cells reenter the cell cycle when exposed to serum and progress toward the S phase. Two classes of growth factors exist in serum: the competence factor and the progression factor [1]. The competence factor activates the quiescent cells, forces them to enter the cell cycle again, and renders them competent to progress through the G1 phase. Then the progression factor acts and brings the competent cells toward the S phase. Hence, it is the progression factor that promotes the cells to progress through the G1 phase to the S phase. A major competence factor in serum is platelet-derived growth factor (PDGF), while a major progression factor in serum is insulin-like growth factor-I (IGF-I) [2]. The competence factor exerts its action by acting transiently, whereas the progression factor should act continuously: when the progression factor is removed during the G1 phase, the cell-cycle progression is blocked immediately. When the progression factor is restored within three hours, cells again progress to the S phase upon readdition of the factor. In contrast, when cells are deprived of the factor for more than three hours, they eventually return to the quiescent state [3]. An interesting aspect of the action of the progression factor is that extracellular calcium is absolutely necessary to promote cell-cycle progression [3]. When extracellular calcium concentration is reduced to less than 0.3 mM, IGF-I is not able to exert its action as a progression factor [3]. An inorganic calcium channel blocker—for example, cobalt or nickel—also blocks the action of IGF-I on cell-cycle progression. Interestingly, when calcium entry is blocked for more than three hours during the G1 phase, cells return to the quiescent state even in the presence of IGF-I. Attenuation of calcium entry is equivalent to the removal of the growth factor. These observations suggest that IGF-I stimulates calcium entry, which is the prerequisite for cell-cycle progression. In accordance with this notion, IGF-I increases the calcium influx rate in competent fibroblasts, and this effect lasts as long as IGF-I is present [4]. It is well known that the IGF-I receptor resembles the insulin receptor and has an intrinsic tyrosine kinase activity. Binding of IGF-I to the receptor leads to phosphorylation of many substrates including insulin receptor substrates (IRSs). Phosphorylated IRSs act as docking proteins and eventually activate the Ras and phosphatidylinositol (PI) 3-kinase pathways [5]. In addition to the activation of the tyrosine phosphorylation cascade, the IGF-I receptor also continuously activates the calcium entry pathway. Tyrosine phosphorylation of IRSs and subsequent activation of the Ras and PI 3-kinase pathway are not affected by the removal of extracellular calcium or an addition of cobalt or nickel. In this regard, activation of PI 3-kinase and the Ras pathway is independent of the calcium influx pathway. Transfection of the dominant negative Ras does not affect the IGF-induced calcium entry, whereas inhibitors of PI 3-kinase inhibit calcium entry.
PROPERTY OF THE IGF-REGULATED CHANNEL
An electrophysiological study reveals that IGF-I activates a calcium-permeable channel in fibroblasts. The IGF-regulated channel is a nonselective calcium-permeable cation channel, the activity of which is regulated by the IGF-I receptor [4, 6]. Interestingly, activation of the channel by IGF-I is not immediate and requires several minutes for full activation. Once activated, however, the calcium-permeable channel remains activated as long as the ligand binds to the receptor, which is gradually inactivated soon after removal of the ligand [6]. These properties of the IGF-regulated channel are suitable for regulation of long-term action (i.e., cell growth). We screened the compound that blocks the IGF-regulated cation channel and found that tranilast, an anti-allergic compound known to inhibit calcium entry in mast cells, blocks the IGF-regulated calcium-permeable channel. Indeed, this compound effectively blocks the growth-promoting action of IGF-I in fibroblasts without affecting either PI 3-kinase or the Ras activity. Tranilast is also effective in other types of normal cells as well as in cancer cells [7]. This raises the possibility that the IGF-regulated calcium-permeable channel can be a molecular target to treat certain types of cancer.
The IGF-regulated channel is a ligand-operated voltage-independent cation channel [6]. Of interest is the fact that pretreatment of the cells with pertussis toxin completely blocks IGF-induced calcium entry [4]. Pertussis toxin-sensitive trimetric G protein may be involved in regulating the IGF-regulated calcium-permeable channel.
MOLECULAR IDENTIFICATION OF THE IGF-REGULATED CHANNEL
We were interested in the molecular nature of the IGF-regulated channel and identified it to be TRPV2 [8]. It is structurally related to the vanilloid receptor channel VR1 [9] and is a mouse homologue of the VR1-like channel, VRL-1 [10]. Mouse TRPV2 is composed of 756 amino acids with a relative molecular weight of 86,000 dalton. Like other members of the TRP family channels, TRPV2 has six hydrophobic putative transmembrane domains and an additional short hydrophobic stretch between the fifth and sixth hydrophobic domains. The amino terminal segment contains ankyrin-repeat domains. TRPV2 has 40 percent overall amino acid identity with rat VR1 and 11–12 percent with other TRP family channels. There is a potential N-linked glycosylation site between the fifth and sixth transmembrane domains. There are multiple potential phosphorylation sites for protein kinase A (residues 98 and 326), protein kinase C (residues 97, 111, 322, and 734), and tyrosine kinase (residues 106, 223, and 330). When the expression of TRPV2 is examined by Northern blotting, the transcript is abundantly found in the brain, lungs, and liver. RT-PCR analysis reveals that TRPV2 is also expressed in various other tissues and organs, including the gastrointestinal tract, pancreas, kidney, heart, blood vessels, skeletal muscle, and fat tissues. Histologically, the expression of TRPV2 is abundant in neurons, including Purkinje cells, neuroendocrine cells in the intestine and pancreas, and macrophages in the lung and spleen [11].
REGULATION OF TRPV2 BY IGF-I
As with other members of the TRP family channels, TRPV2 functions as a calcium-permeable cation channel. Unlike other members of the TRPV family, however, TRPV2 has an intriguing property in which localization of the channel protein is regulated by ligands. Indeed, TRPV2 translocates from an intracellular compartment to the plasma membrane in response to IGF-I [8]. This is the first example of a channel whose trafficking is regulated by extracellular signals. To directly monitor the localization of the channel in living cells, green fluorescent protein (GFP)–tagged TRPV2 (TRPV2-GFP) is expressed in fibroblasts. In a quiescent condition, TRPV2-GFP is barely detected in the plasma membrane (Figure 7.1A). Instead, TRPV2-GFP is distributed diffusely in cytoplasm. Regarding the intracellular localization, the TRPV2-GFP signal colocalizes with the marker of the endoplasmic reticulum but not with that of mitochondria, the trans-Golgi network, endosomes, or lysosomes. Immunoelectron microscopy reveals that TRPV2 is located in the endoplasmic reticulum in unstimulated cells. In cells stimulated with IGF-I for 15 minutes, the TRPV2 signal found in the intracellular compartment is reduced, and some of the TRPV2 becomes detectable in the plasma membrane (Figure 7.1B). Because localization of a membrane-spanning TRPV2 protein is changed by IGF-I action, it seems likely that TRPV2 moves from the endoplasmic reticulum to the plasma membrane carried on the vesicles. However, the vesicles containing TRPV2 have not been detected to date by immunoelectron microscopy. An alternate possibility is that a portion of the endoplasmic reticulum is directly connected to the plasma membrane and transfers the TRPV2 to the plasma membrane. At present, it is not certain whether or not translocation of the TRPV2 channel involves vesicle trafficking. We were also able to monitor translocation of TRPV2 by measuring the current through TRPV2. Figure 7.2A depicts changes in the whole-cell Cs+ current in fibroblasts stimulated with IGF-I. In fibroblasts, the expression of other members of the TRPV family is negligible by RT-PCR. Since the Cs+ current observed in fibroblasts is inhibited by ruthenium red, the Cs+ current is mostly through the TRPV2 channel. Adding IGF-I induces a gradual increase in the Cs+ current, which reaches the plateau level within twenty minutes. Upon removal of IGF-I, the Cs+ current decreases gradually and returns to the basal level after 60 minutes. Although we cannot rule out the possibility that IGF-I directly modifies the gating of TRPV2, augmentation of the Cs+ current is slow and perhaps largely due to translocation of TRPV2 to the plasma membrane [8].
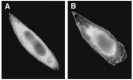
FIGURE 7.1
Effect of IGF-I on the distribution of TRPV2. CHO cells expressing TRPV2-GFP were incubated for fifteen minutes with or without 1 nM IGF-I and the GFP fluorescence was monitored. A: none, B: IGF-I.
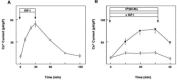
FIGURE 7.2
Time course of the effect of IGF-I on the Cs+ current. (A) CHO cells expressing TRPV2 were stimulated for 30 minutes with 1 nM IGF-I, and the changes in the whole-cell Cs+ current were monitored by the perforate mode patch clump. Values are the mean ± (more...)
REGULATION OF TRANSLOCATION OF TRPV2
Glucose transporter 4 (GLUT4) is an insulin-regulated glucose transporter, which translocates from an intracellular pool to the plasma membrane in adipocytes and skeletal muscle cells. GLUT4 translocates from the intracellular storage pool to the plasma membrane by moving on microvesicles [12]. Under basal conditions, some small portion of the GLUT4-containing vesicles moves to the plasma membrane. Simultaneously, some of the GLUT4 undergoes endocytosis and eventually returns to the intracellular pool. Therefore, even in an unstimulated condition, GLUT4 in the plasma membrane is in a dynamic equilibrium of exocytosis and endocytosis. When stimulated with insulin, a large amount of GLUT4-containing vesicles leaves the storage pool and moves toward the plasma membrane. This results in a large increase in the amount of GLUT4 expressed in the plasma membrane. Concomitantly, GLUT4 undergoes endocytosis and returns to the intracellular pool. Collectively, a large portion of GLUT4 recycles in insulin-stimulated conditions.
We examined whether trafficking of TRPV2 resembles that of GLUT4 by monitoring the Cs+ current in fibroblasts. Under basal conditions, the Cs+ current is low. We first addressed whether or not TRPV2 shuttles under basal conditions. If so, it would be expected that blocking internalization of TRPV2 would lead to an increase in the amount of TRPV2 in the plasma membrane and increase the Cs+ current. To examine this experimentally, we needed to block internalization of TRPV2 from the plasma membrane. To this end, we used Dk(62–85), a synthetic peptide derived from the α1 domain of the murine major histocompatibility complex class I antigen known to inhibit endocytosis of GLUT4 and transferrin [13]. When Dk(62–85) is added to an unstimulated fibroblast, the Cs+ current increases slightly (Figure 7.2B). Upon removal of the Dk(62–85), the Cs+ current decreases and returns to the basal value. Therefore, a small fraction of TRPV2 shuttles even under basal conditions. Presumably, TRPV2 undergoes endocytosis by a mechanism similar to those of GLUT4 and transferrin. In fact, transfection of the dominantly negative mutant of dynamin, a GTP-binding protein involved in endocytosis of various membrane proteins, including GLUT4 [14] blocks endocytosis of TRPV2. TRPV2 undergoes endocytosis by a mechanism involving dynamin GTPase. The second issue is whether or not the major site of action of IGF-I stimulates exocytotic recruitment of TRPV2 to the plasma membrane. To examine this, we again used Dk(62–85) to block endocytosis. Using Dk(62–85), we were able to assess the unidirectional exocytotic recruitment of TRPV2. Indeed, an addition of IGF-I in Dk(62–85)-treated fibroblasts resulted in a large increase in the Cs+ current (Figure 7.2B). Since endocytosis of TRPV2 is blocked in these cells, the results demonstrate that the major site of action of IGF-I on TRPV2 trafficking is the stimulation of the exocytotic step of TRPV2 (Figure 7.2B). Collectively, most of the TRPV2 is located in the endoplasmic reticulum in unstimulated fibroblasts. Some small portion of TRPV2 moves to the plasma membrane, which is balanced by the endocytosis of TRPV2 (Figure 7.3). When cells are stimulated with IGF-I, a relatively large amount of TRPV2 is recruited from the intracellular pool and translocates to the plasma membrane. As a result, the expression of TRPV2 in the plasma membrane increases considerably and calcium entry is augmented. Some portion of TRPV2 also is internalized by endocytosis. When the action of IGF-I is removed, the supply of TRPV2 from the intracellular pool is terminated. In addition, TRPV2 expressed in the plasma membrane undergoes endocytosis and returns to the endoplasmic reticulum. Consequently, the amount of TRPV2 expressed in the plasma membrane is reduced gradually (Figure 7.3). Thus, the major site of action of IGF-I is the recruitment of TRPV2 to the plasma membrane.
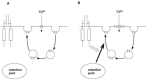
FIGURE 7.3
Regulation of TRPV2 by IGF-I. (A) In unstimulated conditions, most of TRPV2 locates in the storage pool (endoplasmic reticulum), and a small fraction of TRPV2 is recycling. (B) When IGF-I is added, TRPV2 is recruited from the storage pool and translocates (more...)
IGF-I-induced translocation of TRPV2 is blocked by inhibitors of PI 3-kinase, LY294002, and wortmannin. Translocation is also blocked by transfection of the dominant-negative mutant of the p85 subunit of the PI 3-kinase. Although activation of PI 3-kinase is required for IGF-I-induced translocation of TRPV2, it is not a sufficient signal. Thus, activation of PI 3-kinase by adding phosphatidylinositol 3,4,5-trisphosphate does not induce translocation. Obviously, an additional signal is necessary to mobilize TRPV2 from the intracellular compartment. At present, little is known about the molecular mechanism downstream of the PI 3-kinase. In this regard, an interesting machinery regulated by PI 3-kinase is the cytoskeletal proteins involved in cell motility. When actin filaments are disrupted by latrunculin A or cytochalasin D, distribution of TRPV2 is altered considerably. Under basal conditions, most of the TRPV2-GFP localizes in the endoplasmic reticulum. However, TRPV2 does not translocate to the plasma membrane after the stimulation with IGF-I. Similarly, the Cs+ current is only slightly higher in the basal condition but does not respond to IGF-I when the actin filament is disrupted by latrunculin A or cytochalasin D (Figure 7.4). Disruption of the actin filament does not affect the decrease in the TRPV2 current, suggesting that the actin filament is not necessary for endocytosis of TRPV2. These results indicate that the actin filament plays a critical role in recruitment of TRPV2 to the plasma membrane from the endoplasmic reticulum. In contrast, disruption of microtubules by nocodazole does not affect the translocation of TRPV2 induced by IGF-I. Among various cytoskeletal proteins, reorganization of the actin filament may be involved in IGF-I–mediated translocation of TRPV2.
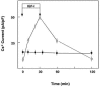
FIGURE 7.4
Effect of latrunculin A on IGF-I-induced Cs+ current. Cells were preincubated with or without 50μM latrunculin A and then 1 nM IGF-I was added. Changes in the Cs+ current were monitored. ○: without latrunculin A, ●: with latrunculin (more...)
Many issues still remain unsolved. First, the molecular mechanism regulating translocation is largely elusive. For example, it is not totally certain whether or not vesicle trafficking is involved in translocating TRPV2. Suppose TRPV2 is carried on vesicles, the formation, trafficking, and exocytosis of the vesicles should be regulated by the IGF-I signal. These regulatory mechanisms need to be identified. Second, it is not clear at present that translocation is the sole mechanism by which IGF-I regulates TRPV2. In other words, it is possible that IGF-I also modulates TRPV2 gating. If this is the case, it is necessary to identify the regulatory mechanism. Third, TRPV2 is located in the endoplasmic reticulum under basal conditions. It is not certain that TRPV2 functions as a calcium-permeable channel in this organella. If so, trafficking of TRPV2 may alter the calcium handling in the endoplasmic reticulum. Fourth, translocation of TRPV2 is regulated by IGF-I in fibroblasts and neuroendocrine cells [8], but other ligands may regulate TRPV2 in other types of cells. In fact, translocation of TRPV2 is induced by ligands that bind to G-protein-coupled receptors in other types of cells [15]. Regulation of the TRPV2 translocation by those ligands may be different. It is also shown that membrane stretch induces translocation of TRPV2 in myocytes [16]. Translocation may be a major regulatory mechanism for TRPV2 activation. Further works are clearly required to elucidate the mechanism and the role of TRPV2 translocation.
REFERENCES
- 1.
- Scher CD, et al. Platelet-derived growth factor and the regulation of the mammalian fibroblast cell cycle. Biochem Biophys Acta. 1979;560:217. [PubMed: 380651]
- 2.
- Stiles CD, et al. Dual control of cell growth by somatomedins and platelet-derived growth factor. Proc Natl Acad Sci USA. 1979;76:1279. [PMC free article: PMC383234] [PubMed: 312500]
- 3.
- Kojima I, et al. Role of calcium entry and protein kinase C in the progression activity of insulin-like growth factor-I in Balb/c 3T3 cells. J Biol Chem. 1993;268:10003. [PubMed: 8486672]
- 4.
- Kojima I, et al. Calcium influx: an intracellular message of the mitogenic action of insulin-like growth factor-I. J Biol Chem. 1988;263:16561. [PubMed: 2460452]
- 5.
- Nakae J, Kido Y, Accili D. Distinct and overlapping function of insulin and IGF-I receptors. Endocrine Rev. 2001;22:818. [PubMed: 11739335]
- 6.
- Matsunaga H, et al. Activation of a calcium-permeable cation channel by insulin-like growth factor-Ii in Balb/c 3T3 cells. Am J Physiol. 1988;255:C442. [PubMed: 2845795]
- 7.
- Nie L, et al. Inhibition of proliferation of MCF-7 breast cancer cells by a blocker of Ca2+-permeable channel. Cell Calcium. 1997;22:75. [PubMed: 9292225]
- 8.
- Kanzaki M, et al. Translocation of a calcium-permeable channel induced by insulin-like growth factor-I. Nature Cell Biol. 1999;1:165. [PubMed: 10559903]
- 9.
- Caterina MJ, et al. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816. [PubMed: 9349813]
- 10.
- Caterina MJ, et al. A capsaicin receptor homologue with a high threshold for noxious heat. Nature. 1999;398:436. [PubMed: 10201375]
- 11.
- Kowase T, et al. Immunohistochemical localization of growth factor–regulated channel (GRC) in human tissues. Endocrine J. 2002;49:349. [PubMed: 12201220]
- 12.
- Haney PM, et al. Intracellular targeting of the insulin-regulatable glucose transporter (GLUT4) is isoform specific and independent of cell type. J Cell Biol. 1991;114:689. [PMC free article: PMC2289882] [PubMed: 1651337]
- 13.
- Shibata H, et al. Dissection of GLUT4 recycling pathway into exocytosis and endocytosis in rat adipocytes. J Biol Chem. 1995;270:11489. [PubMed: 7744788]
- 14.
- Omata W, et al. Subcellular distribution of GLUT4 in CHO cells overexpressing mutant dynamin. Biochem Biophys Res Commun. 1997;241:401. [PubMed: 9425283]
- 15.
- Boels K, et al. The neuropeptide head activator induces activation and translocation of the growth factor–regulated Ca2+-permeable channel GRC. J Cell Sci. 2001;114:3599. [PubMed: 11707512]
- 16.
- Iwata Y, et al. A novel mechanism of myocyte degeneration involving the Ca2+-permeable growth factor–regulated channel. J Cell Biol. 2003;161:957. [PMC free article: PMC2172975] [PubMed: 12796481]
- Prolonged vs transient roles for early cell cycle signaling components.[Oncogene. 1998]Prolonged vs transient roles for early cell cycle signaling components.Rose DW, Xiao S, Pillay TS, Kolch W, Olefsky JM. Oncogene. 1998 Aug 20; 17(7):889-99.
- Expression of calcium-permeable cation channel CD20 accelerates progression through the G1 phase in Balb/c 3T3 cells.[J Biol Chem. 1995]Expression of calcium-permeable cation channel CD20 accelerates progression through the G1 phase in Balb/c 3T3 cells.Kanzaki M, Shibata H, Mogami H, Kojima I. J Biol Chem. 1995 Jun 2; 270(22):13099-104.
- Role of calcium entry and protein kinase C in the progression activity of insulin-like growth factor-I in Balb/c 3T3 cells.[J Biol Chem. 1993]Role of calcium entry and protein kinase C in the progression activity of insulin-like growth factor-I in Balb/c 3T3 cells.Kojima I, Mogami H, Shibata H, Ogata E. J Biol Chem. 1993 May 15; 268(14):10003-6.
- Review [Calcium influx: an intracellular message of the mitogenic action of insulin-like growth factors].[Gan To Kagaku Ryoho. 1989]Review [Calcium influx: an intracellular message of the mitogenic action of insulin-like growth factors].Kojima I. Gan To Kagaku Ryoho. 1989 Mar; 16(3 Pt 2):489-98.
- Review Growth-stimulatory actions of insulin in vitro and in vivo.[Endocr Rev. 1984]Review Growth-stimulatory actions of insulin in vitro and in vivo.Straus DS. Endocr Rev. 1984 Spring; 5(2):356-69.
- TRPV2: A Calcium-Permeable Cation Channel Regulated by Insulin-Like Growth Facto...TRPV2: A Calcium-Permeable Cation Channel Regulated by Insulin-Like Growth Factors - TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades
- zinc transporter ZIP13 isoform X4 [Homo sapiens]zinc transporter ZIP13 isoform X4 [Homo sapiens]gi|767966821|ref|XP_011518771.1|Protein
- UI-H-BI4-aom-h-08-0-UI.s1 NCI_CGAP_Sub8 Homo sapiens cDNA clone IMAGE:3085670 3'...UI-H-BI4-aom-h-08-0-UI.s1 NCI_CGAP_Sub8 Homo sapiens cDNA clone IMAGE:3085670 3', mRNA sequencegi|11595040|gnl|dbEST|7039107|gb|BF 2.1|Nucleotide
Your browsing activity is empty.
Activity recording is turned off.
See more...