Diagnosis
In its most characteristic form, Aicardi-Goutières syndrome (AGS) can be considered an early-onset encephalopathy associated with significant intellectual and physical disability.
Suggestive Findings
Aicardi-Goutières syndrome (AGS) should be suspected in individuals with the following clinical, neuroimaging, and supportive laboratory findings [Goutières et al 1998, Lanzi et al 2002, Rice et al 2007b, Livingston et al 2013].
Clinical features
Encephalopathy and/or significant intellectual disability
Acquired microcephaly during the first year of life
Dystonia and spasticity
Sterile pyrexias
Hepatosplenomegaly
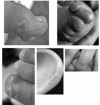
Chilblain lesions on the feet, hands, ears, and sometimes more generalized mottling of the skin. See .
Examples of chilblains seen in AGS Rice et al [2007b]; reprinted with permission of The American Journal of Human Genetics, University of Chicago Press.
Exclusion criteria include the following:
Evidence of prenatal/perinatal infection including, but not limited to, CMV, toxoplasmosis, rubella, herpes simplex, Zika, and HIV
Evidence of a known other metabolic disorder or neurodegenerative disorder
Neuroimaging
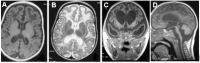
Calcification (best visualized on CT scan) of the basal ganglia, particularly the putamen, globus pallidus and thalamus but also extending into the white matter, sometimes in a para- (rather than true peri-) ventricular distribution [
Lanzi et al 2002,
Uggetti et al 2009,
Livingston et al 2013]. See .
Note: Intracranial calcification is not always recognized on MRI, the initial imaging modality employed in most units.
White matter changes, particularly affecting the frontotemporal regions with (in severe cases) temporal lobe cyst-like formation. See .
On MRI, appears on T2-weighted images as a hyperintense signal most commonly located around the horns of the ventricles ()
Cerebral atrophy, which may be progressive and involve the periventricular white matter and sulci
Bilateral striatal necrosis
Intracerebral vasculopathy including intracranial stenosis, moyamoya, and aneurysms
Examples of intracranial calcification on CT scan in individuals with AGS. Calcification is seen: A. In the basal ganglia;
The spectrum of brain changes seen on MRI in AGS A. Hypointensity on T1-weighted imaging of the white matter
Supportive Laboratory Findings
Peripheral blood
Elevated liver enzymes
Thrombocytopenia
Cerebrospinal fluid (CSF)
Chronic CSF leukocytosis, defined as more than five lymphocytes/mm3 CSF.
Increased interferon-alpha (IFN-α) activity in the CSF (normal: <2 IU/mL)
Levels are highest in the early stages of the disease and can normalize over time.
Levels of the neurotransmitter metabolites 5HIAA, HVA, and 5MTHF are normal.
Establishing the Diagnosis
The diagnosis of AGS is established in a proband with typical clinical findings and characteristic abnormalities on cranial CT (calcification of the basal ganglia and white matter) and MRI (leukodystrophic changes) and/or by the identification of biallelic pathogenic (or likely pathogenic) variants in ADAR, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, or TREX1; specific heterozygous autosomal dominant pathogenic (or likely pathogenic) variants in TREX1 and ADAR; or a variety of heterozygous autosomal dominant pathogenic (or likely pathogenic) variants in IFIH1.
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) The identification of variant(s) of uncertain significance cannot be used to confirm or rule out the diagnosis.
Molecular testing approaches can include serial single-gene testing, use of a multigene panel, and more comprehensive
genomic testing.
Serial single-gene testing can be pursued based on the individual's ethnicity and/or in the order in which pathogenic variants most commonly occur (see Table 1).
A multigene panel that includes ADAR, IFIH1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, TREX1, and other genes of interest (see Differential Diagnosis) may also be considered. Note: (1) The genes included and the sensitivity of multigene panels vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview; thus, clinicians need to determine which multigene panel is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
More comprehensive genomic testing (when available) including exome sequencing, mitochondrial sequencing, and genome sequencing may be considered if serial single-gene testing (and/or use of a multigene panel that includes ADAR, IFIH1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and TREX1) fails to confirm a diagnosis in an individual with features of AGS. Such testing may provide or suggest a diagnosis not previously considered (e.g., mutation of a different gene or genes that results in a similar clinical presentation). For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Aicardi-Goutières Syndrome
View in own window
| Gene 1 | Proportion of AGS Attributed to Pathogenic Variants in Gene | Proportion of Pathogenic Variants 2 Detectable by Method |
|---|
| Sequence analysis 3, 4 | Gene-targeted deletion/duplication analysis 5 |
|---|
|
ADAR
| 7% | 32/32 alleles 6 | Unknown 7 |
|
IFIH1
| 3% | 17/17 alleles 8 | Unknown 7 |
|
RNASEH2A
| 5% | 34/34 alleles 9 | Unknown 7 |
|
RNASEH2B
| 36% | ~99% 10 | Unknown 11 |
|
RNASEH2C
| 12% | ~99% 12, 13 | Unknown 7 |
|
SAMHD1
| 13% | Up to 95% 14 | Up to 30% 14, 15 |
|
TREX1
| 23% | ~99% 16 | Unknown 7 |
| Unknown | 1% | NA |
- 1.
- 2.
- 3.
- 4.
Sequence analysis of the coding regions and splice sites of ADAR, IFIH1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and TREX1 has identified pathogenic variants in approximately 90%-95% of individuals with clinical and MRI presentation of AGS [Rice et al 2007b, Rice et al 2009, Crow et al 2015].
- 5.
Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods used may include a range of techniques including quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications.
- 6.
- 7.
No deletions or duplications involving ADAR, IFIH1, RNASEH2A, RNASEH2C, or TREX1 have been reported to cause Aicardi-Goutières syndrome.
- 8.
- 9.
- 10.
- 11.
- 12.
- 13.
- 14.
- 15.
- 16.
Clinical Characteristics
Clinical Description
In its most characteristic form, Aicardi-Goutières syndrome (AGS) can be considered an early-onset encephalopathy associated with significant intellectual and physical disability.
Pregnancy, delivery, and the neonatal period are normal in approximately 80% of infants with Aicardi-Goutières syndrome (AGS) [Rice et al 2007b]. However, brain calcifications can be identified in utero [Le Garrec et al 2005] and 20% of cases, mainly those caused by biallelic pathogenic variants in TREX1, present at birth with abnormal neurologic findings, hepatosplenomegaly, elevated liver enzymes, and thrombocytopenia, a picture reminiscent of congenital infection.
All other affected infants present at variable times after the first few weeks of life, frequently after a period of apparently normal development. The majority of these later-presenting infants exhibit subacute onset of a severe encephalopathy characterized by extreme irritability, intermittent sterile pyrexias, loss of skills, and slowing of head growth. The encephalopathic phase usually lasts several months. The opinion of most pediatricians caring for such children is that the disease does not progress beyond the encephalopathic period; occasionally, however, affected individuals do appear to show progression and/or episodes of regression. Death is usually considered to be secondary to the neurologic damage incurred during the initial disease episode, not to further regression. Several affected individuals older than age 30 years show no obvious signs of disease progression.
Neurologic features. Typically, affected individuals have peripheral spasticity, dystonic posturing (particularly of the upper limbs), truncal hypotonia, and poor head control. Seizures are reported in up to half of affected children, but are usually relatively easily controlled [Goutières et al 1998, Rice et al 2007b]. A number of children demonstrate a marked startle reaction to sudden noise, and the differentiation from epilepsy can be difficult. Most affected individuals have severe intellectual and physical impairment. Variability in the severity of the neurologic outcome can be observed among sibs. Most affected children exhibit a severe acquired microcephaly; in children with preserved intellect head circumference can be normal.
Hearing is almost always normal.
Ophthalmology. Visual function varies from normal to cortical blindness. Ocular structures are almost invariably normal on examination. However, there is a risk of congenital glaucoma or later-onset glaucoma [Crow et al 2004a, Crow et al 2015].
Skin lesions. As many as 40% of affected individuals [Rice et al 2007b] have skin lesions with chilblains on the fingers and toes and sometimes the ears and other pressure points (e.g., elbows) [Tolmie et al 1995, Stephenson 2002] (see ). The cutaneous lesions may be complicated by periungual infection and necrosis.
Other
Additional infrequently observed features of AGS are summarized in Table 2.
Table 2.
Infrequent Features Seen in a Cohort of 123 Individuals with Molecularly Confirmed AGS
View in own window
| Feature | Number of Affected Individuals by Gene |
|---|
|
RNASEH2A
|
RNASEH2B
|
RNASEH2C
|
TREX1
|
|---|
| Scoliosis | 0 | 9 | 0 | 0 |
| Cardiomegaly | 1 | 0 | 1 | 4 |
| Abnormal antibody profile | 0 | 3 | 1 | 2 |
| Preserved language | 0 | 6 | 0 | 0 |
| Demyelinating peripheral neuropathy | 0 | 2 | 1 | 1 |
| Congenital glaucoma | 0 | 0 | 1 | 2 |
| Micropenis | 0 | 0 | 1 | 1 |
| Hypothyroidism | 0 | 1 | 0 | 1 |
| Insulin-dependent diabetes mellitus | 0 | 1 | 0 | 1 |
| Transitory deficiency of antidiuretic hormone | 0 | 0 | 0 | 1 |
Rice et al [2007b]. Note: This paper was published before it was determined that pathogenic variants in ADAR, IFIH1, and SAMHD1 can cause AGS.
Neuroimaging/neuropathologic findings
Phenotype Correlations by Gene
In general, the early-onset neonatal form of AGS is most frequently seen in association with biallelic pathogenic variants in RNASEH2A, RNASEH2C, or TREX1. The later-onset presentation (sometimes occurring after several months of normal development and occasionally associated with remarkably preserved neurologic function) is most frequently seen in association with biallelic pathogenic variants in RNASEH2B, SAMHD1, or ADAR but may also be seen in individuals who have an autosomal dominant heterozygous pathogenic variant in ADAR or IFIH1 [Crow et al 2015].
Mortality is correlated with genotype: 34% of individuals with RNASEH2A, RNASEH2C, and TREX1 pathogenic variants were known to have died compared to 8% with RNASEH2B pathogenic variants (p=0.001) [Rice et al 2007b]. The mortality associated with other genotypes is less clear.
ADAR. Pathogenic variants in ADAR have been associated with a clinical presentation of acute bilateral striatal necrosis.
A subgroup of individuals with ADAR pathogenic variants can present with symmetric signal changes in the caudate and putamen, often associated with swelling and later shrinkage in the context of an acute or subacute onset of refractory four-limb dystonia. Cases have been identified with onset as late as age four years on a background of completely normal development [La Piana et al 2014, Livingston et al 2014a].
ADAR-related disease should be considered in the differential diagnosis of apparently nonsyndromic bilateral striatal necrosis (BSN) with severe dystonia of varying evolution. The finding of an interferon signature provides a useful screening test for the presence of ADAR pathogenic variants in this context.
RNASEH2B. Some individuals with biallelic pathogenic variants in RNASEH2B have relatively preserved intellectual function, with a few having a completely normal IQ and head circumference [McEntagart et al 1998].
RNASEH2C. A family with a recurrent Asian founder variant in RNASEH2C and striking intrafamilial variability has been described [Crow et al 2015].
SAMHD1
Intracerebral vasculopathy, including intracranial stenosis and aneurysms, is observed more frequently in individuals who have
biallelic pathogenic variants in
SAMHD1.
Dale et al [2010] reported two sibs who had
biallelic null variants in
SAMHD1. The older girl showed mild intellectual disability with microcephaly. Her younger brother had significant spastic paraparesis with normal intellect and head size. Both children had an unclassified chronic inflammatory skin condition with chilblains and recurrent mouth ulcers. One of the sibs had a chronic progressive deforming arthropathy of the small and large joints with secondary contractures. Similar joint involvement was described by
Ramantani et al [2010] (see also
Crow et al [2015]).
Biallelic pathogenic variants in
SAMHD1 are most frequently associated with chilblains and glaucoma [
Crow et al 2015].
Nomenclature
The microcephaly-intracranial calcification syndrome (MICS; also known as pseudo-TORCH syndrome or Baraitser-Reardon syndrome) was previously differentiated from AGS on the basis of congenital microcephaly and the presence of non-neurologic abnormalities including elevation of liver enzymes, hepatomegaly, and thrombocytopenia at birth [Reardon et al 1994]. However, recent studies have shown that these same features can be seen in persons with AGS in whom pathogenic variants in one of the associated genes have been identified [Rice et al 2007b]. Of note, in the majority of MICS cases reported no information is available on CSF cell count and IFN-α concentration; thus it is probable that most cases of MICS are in fact AGS.
"Familial systemic lupus erythematosus."
Dale et al [2000] described two children of consanguineous parents with early-onset encephalopathy, intracranial calcifications, chilblain skin lesions, and the progressive production of high levels of autoantibodies. CSF was not analyzed. These cases most likely represent AGS [Aicardi & Goutières 2000]. Additional individuals with AGS who have signs and symptoms similar to systemic lupus erythematosus have also been described.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Autosomal Recessive Inheritance – Risk to Family Members
Parents of a proband
The parents of an affected child are obligate heterozygotes (i.e., carriers of one AGS-related
pathogenic variant).
Sibs of a proband
At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic
carrier, and a 25% chance of being unaffected and not a carrier.
Heterozygotes (carriers) are not at risk of developing AGS; however, heterozygotes may be at increased risk of developing later-onset systemic lupus erythematosus (SLE) or retinal vasculopathy with cerebral leukodystrophy (RVCL), depending on the specific
gene and
pathogenic variant involved (see
Genetically Related Disorders).
Offspring of a proband. Most individuals with AGS do not reproduce.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of an AGS-related pathogenic variant.
Carrier (heterozygote) detection. Carrier testing for at-risk relatives requires prior identification of the AGS-related pathogenic variants in the family.
Autosomal Dominant Inheritance – Risk to Family Members
Parents of a proband
If the
pathogenic variant found in the
proband cannot be detected in leukocyte DNA of either parent, the proband most likely has a
de novo pathogenic variant. Another possible explanation is
germline mosaicism in a parent (though theoretically possible, no instances of germline mosaicism have been reported).
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's parents:
If a parent of the
proband is affected, the risk to the sibs is 50%.
If the
TREX1,
ADAR, or
IFIH1 pathogenic variant found in the
proband cannot be detected in the leukocyte DNA of either parent, the risk to sibs is presumed to be slightly greater than that of the general population (though still <1%) because of the theoretic possibility of parental
germline mosaicism.
Offspring of a proband. Each child of an individual with autosomal dominant AGS has a 50% chance of inheriting the AGS-related pathogenic variant; however, most individuals with AGS do not reproduce.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent is affected, the parent's family members may be at risk.
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Aicardi-Goutieres Syndrome: Genes and Databases
View in own window
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Table B.
View in own window
|
146920 | ADENOSINE DEAMINASE, RNA-SPECIFIC; ADAR |
|
225750 | AICARDI-GOUTIERES SYNDROME 1; AGS1 |
|
606034 | RIBONUCLEASE H2, SUBUNIT A; RNASEH2A |
|
606609 | 3-PRIME @REPAIR EXONUCLEASE 1; TREX1 |
|
606754 | SAM DOMAIN- AND HD DOMAIN-CONTAINING PROTEIN 1; SAMHD1 |
|
606951 | INTERFERON-INDUCED HELICASE C DOMAIN-CONTAINING PROTEIN 1; IFIH1 |
|
610181 | AICARDI-GOUTIERES SYNDROME 2; AGS2 |
|
610326 | RIBONUCLEASE H2, SUBUNIT B; RNASEH2B |
|
610329 | AICARDI-GOUTIERES SYNDROME 3; AGS3 |
|
610330 | RIBONUCLEASE H2, SUBUNIT C; RNASEH2C |
|
610333 | AICARDI-GOUTIERES SYNDROME 4; AGS4 |
|
612952 | AICARDI-GOUTIERES SYNDROME 5; AGS5 |
|
615010 | AICARDI-GOUTIERES SYNDROME 6; AGS6 |
|
615846 | AICARDI-GOUTIERES SYNDROME 7; AGS7 |
ADAR
Gene structure.
ADAR is a single-copy 16-exon gene that encodes two main isoforms constitutively expressed in mammalian cells: a truncated protein (p110 NP_001020278.1) encoded by a transcript variant of 15 exons (NM_001025107.2) and an IFN inducible full-length protein (p150 NP_001102.2) induced isoform encoded by transcript variant NM_001111.4. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. The majority of pathogenic variants in ADAR result in autosomal recessive disease. Missense, nonsense, frameshift, and splice site pathogenic variants have been reported. One missense variant, p.Pro193Ala, is common in individuals of European origin. One dominant variant, p.Gly1007Arg, has been reported [Rice et al 2012, Livingston et al 2014a].
Table 4.
Selected ADAR Pathogenic Variants
View in own window
| DNA Nucleotide Change | Predicted Protein Change | Reference Sequences |
|---|
| c.577C>G | p.Pro193Ala |
NM_001111.4
NP_001102.2
|
| c.3019G>A | p.Gly1007Arg |
Variants listed in the table have been provided by the author. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
Normal gene product. DRADA, double-stranded RNA-specific adenosine deaminase, is a modular protein with a C-terminal deaminase catalytic domain, three centrally located dsRNA-binding domains (dsRBDs) and one or two N-terminal Z-DNA–binding domains; compared with p110, the p150 isoform of human DRADA possesses an additional 295 N-terminal amino acids containing a nuclear export signal and an extra Z-DNA/Z-RNA-binding domain.
Abnormal gene product. Pathogenic variants in ADAR are believed to cause AGS as a result of loss of protein activity; the precise mechanism leading to disease is unclear.
IFIH1
Gene structure.
IFIH1 has 16 exons. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. All IFIH1 pathogenic variants associated with AGS to date have been missense variants that cause dominant disease.
Normal gene product.
IFIH1 encodes the interferon-induced helicase C domain-containing protein 1, a cytosolic double-stranded RNA receptor.
Abnormal gene product. Pathogenic variants in IHIF1 associated with AGS encode missense variants that cause up-regulation of type I interferon signaling [Rice et al 2014].
RNASEH2A
Gene structure.
RNASEH2A has eight exons. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. The majority of pathogenic variants in RNASEH2A are missense; splicing and frameshift pathogenic variants have also been reported [Crow et al 2006b, Rice et al 2007b] (see Table 5).
Table 5.
Selected RNASEH2A Pathogenic Variants
View in own window
| DNA Nucleotide Change 1 | Predicted Protein Change | Reference Sequences |
|---|
| c.69G>A 2 | p.Val23= |
NM_006397.2
NP_006388.2
|
| c.75C>T 2 | p.Arg25= |
| c.109G>A | p.Gly37Ser |
| c.207_208insG | p.Thr69AspfsTer50 |
| c.322C>T | p.Arg108Trp |
| c.556C>T | p.Arg186Trp |
| c.690C>A | p.Phe231Leu |
| c.704G>A | p.Arg236Gln |
| c.716_717dupGC | p.Thr239AlafsTer77 |
| c.719C>T | p.Thr241Met |
| c.872G>A | p.Arg292His |
Variants listed in the table have been provided by the author. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
- 1.
Frequency of each allele is less than 1%.
- 2.
Two synonymous variants (c.69G>A and c.75C>T) in RNASEH2A are pathogenic due to an alteration in RNA splicing [Rice et al 2013b].
Normal gene product.
RNASEH2A encodes the ribonuclease H2 subunit A, which comprises 299 amino acids. Ribonuclease H (RNASEH) enzymes endonucleolytically cleave ribonucleotides from RNA:DNA duplexes. RNASEH2 has been proposed to function in the removal of lagging strand Okazaki fragment RNA primers during DNA replication, as well as in the excision of single ribonucleotides from DNA:DNA duplexes. However, the precise biologic function of the human RNASEH2 complex in the context of AGS is uncertain.
Abnormal gene product. See RNASEH2B,
Abnormal gene product.
RNASEH2B
Gene structure.
RNASEH2B has 11 exons and codes for a 308-amino acid protein. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. Almost all pathogenic variants so far identified in RNASEH2B are missense [Crow et al 2006b, Rice et al 2007b] (see Table 6).
The frequency of alleles identified in affected individuals is:
p.Ala177Thr (62%)
p.Thr163Ile (7%)
p.Val185Gly (7%)
c.136+1delG (4%)
Table 6.
Selected RNASEH2B Pathogenic Variants
View in own window
| DNA Nucleotide Change | Predicted Protein Change | Reference Sequences |
|---|
| c.64+1G>A | -- |
NM_024570.1
|
| c.136+1delG | -- |
| c.244+1G>T | -- |
| c.436+1G>T | -- |
| c.510+1G>A | -- |
| c.128C>A | p.Pro43His |
NM_024570.1
NP_078846.1
|
| c.132T>A | p.Cys44Ter |
| c.172C>T | p.Gln58Ter |
| c.179T>G | p.Leu60Arg |
| c.218G>T | p.Trp73Leu |
| c.247G>A | p.Gly83Ser |
| c.257A>G | p.His86Arg |
| c.412C>T | p.Leu138Phe |
| c.476G>T | p.Ser159Ile |
| c.485A>C | p.Lys162Thr |
| c.488C>T | p.Thr163Ile |
| c.529G>A | p.Ala177Thr |
| c.547C>A | p.Val183Met |
| c.554T>G | p.Val185Gly |
| c.655T>C | p.Tyr219His |
Variants listed in the table have been provided by the author. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
Normal gene product. The precise function of the ribonuclease H2 subunit B protein within the human RNASEH2 complex is unknown.
Abnormal gene product. Pathogenic variants in genes encoding any of the three subunits of the ribonuclease H2 complex are thought to cause AGS resulting from a loss of enzymatic function.
RNASEH2C
Gene structure.
RNASEH2C is a four-exon gene encoding a 164-amino acid protein. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. All pathogenic variants so far identified in RNASEH2C are missense [Crow et al 2006b, Rice et al 2007b] (see Table 7).
The frequency of alleles identified in affected individuals is:
Table 7.
Selected RNASEH2C Pathogenic Variants
View in own window
| DNA Nucleotide Change | Predicted Protein Change | Reference Sequences |
|---|
| c.38G>A | p.Arg13His |
NM_032193.3
NP_115569.2
|
| c.205C>T | p.Arg69Trp |
| c.227C>T | p.Pro76Leu |
| c.412C>T | p.Pro138Leu |
| c.428A>T | p.Lys143Ile |
| c.451C>T | p.Pro151Ser |
Variants listed in the table have been provided by the author. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
Normal gene product. The function of ribonuclease H2 subunit C (RNASEH2C) within the RNASEH2 complex is unknown.
Abnormal gene product. See RNASEH2B,
Abnormal gene product.
SAMHD1
Gene structure.
SAMHD1 has 16 exons. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. The majority of pathogenic variants are missense, splice site, nonsense, or frameshift variants [Rice et al 2009].
Three large deletions plus a small deletion have also been identified. A large deletion of exon 1 is common among individuals of Ashkenazi Jewish descent [Crow et al 2015].
Table 8.
Selected SAMHD1 Pathogenic Variants
View in own window
DNA Nucleotide Change
(Alias 1) | Predicted Protein Change | Reference Sequences |
|---|
| c.359_370del12 | p.Asp120_His123del |
NM_015474.3
NP_056289.2
|
| c.368A>C | p.His123Pro |
| c.427C>T | p.Arg143Cys |
| c.428G>A | p.Arg143His |
| c.433C>T | p.Arg145Ter |
| c.434G>A | p.Arg145Gln |
| c.445C>T | p.Gln149Ter |
| c.602T>A | p.Ile201Asn |
| c.625G>A | p.Gly209Ser |
| c.649_650insG | p.Phe217CysTer2 |
| c.760A>G | p.Met254Val |
| c.1106T>C | p.Leu369Ser |
| c.1153A>G | p.Met385Val |
| c.1324C>T | p.Arg442Ter |
| c.1411-2A>G | (splice acceptor) |
| c.1503+1G>T | (splice donor) |
| c.1642C>T | p.Gln548Ter |
| c.1609-1G>C | (splice acceptor) |
| (exons 12-16del) | -- |
NM_015474.3
|
| (8984bp promoter+ex1del) | -- |
| (exons 1-13del) | -- |
Variants listed in the table have been provided by the author. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
- 1.
Variant designation that does not conform to current naming conventions
Normal gene product. SAMHD1 is a 626-amino acid protein that consists of a SAM domain and an HD domain; the protein acts as a dNTP triphosphohydrolase [Goldstone et al 2011].
Abnormal gene product. Pathogenic variants in SAMHD1 are believed to cause AGS as a result of mislocalization of the protein or loss of protein function.
TREX1 (AGS1)
Gene structure.
TREX1 has one exon. For a detailed summary of gene and protein information, see Table A, Gene.
Note: A great deal of confusion regarding TREX1 and the overlapping gene ATRIP exists in the databases. TREX1 and ATRIP are distinct genes that encode distinct proteins; they are not known to be relevant to one another [Yang et al 2007].
Pathogenic variants. Stop variants, deletions, and insertions are common in TREX1, but the most prevalent pathogenic variant is a missense variant (p.Arg114His) that affects the dimerization of the TREX1 protein (3' repair exonuclease 1) and is likely to be a functional null allele. The pathogenic variant p.Arg114His is particularly common in people from northern Europe.
Affected individuals are almost always homozygotes or compound heterozygotes for pathogenic variants within the same gene. However, children with clinically typical AGS had a de novo heterozygous pathogenic variant in TREX1 [Rice et al 2007a, Haaxma et al 2010, Abe et al 2014] (see Table 9).
The frequency of alleles identified in affected individuals is [Rice et al 2007b]:
p.Arg114His (50%)
c.58_59insG (1%)
Remaining alleles (<1%)
Table 9.
Selected TREX1 Pathogenic Variants
View in own window
| DNA Nucleotide Change | Predicted Protein Change | Reference Sequences |
|---|
| c.52G>A | p.Asp18Asn |
AAK07616.1
AF319569.1
|
| c.58_59insG | p.Glu20GlyfsTer81 |
| c.212_213dupTG | p.Ala72Trpfster16 |
| c.341G>A | p.Arg114His |
| c.365T>C | p.Val122Ala |
| c.366_368dupGGC | p.Ala123dup |
| c.397delC | p.Leu133CysfsTer26 |
| c.393_408dup16 | p.Glu137ProfsTer23 |
| c.490C>T | p.Arg164Ter |
| c.500delG | p.Ser166ThrfsTer12 |
| c.598G>T | p.Asp200Asn |
| c.600_601insGAT | p.Asp200dup |
| c.602T>A | p.Val201Asp |
| c.609_662dup54 | p.Leu204_Ala221dup |
| c.625_628dupCAGT | p.Trp210SerfsTer31 |
| c.868_885del18 | p.Pro289_Ala294del |
| c.907A>C | p.Thr303Pro |
Variants listed in the table have been provided by the author. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
Normal gene product.
TREX1 is a single-exon gene encoding a 314-amino acid residue protein.
TREX1 protein represents the major 3'→5' DNA exonuclease activity measured in mammalian cells. The protein has three conserved sequence motifs known as Exo I, II, and III. These motifs contain four conserved acidic residues that participate in coordination of divalent metal ions required for catalysis. In addition, the protein contains a C-terminal domain of about 75 amino acids, which is probably involved in subcellular localization of the protein, and a polyproline motif that may be involved in the interaction with other proteins. TREX1 appears to play a role in the disposal of single-stranded DNA possibly produced as a normal replication intermediate during S phase [Yang et al 2007], or derived from retro-elements [Stetson et al 2008].
Abnormal gene product. The most prevalent pathogenic variant in TREX1 is a missense change (p.Arg114His) that affects the dimerization of the TREX1 protein (3' repair exonuclease 1) and is likely to be a functional null allele, as are other reported frameshift variants.