Clinical Description
To date, nine individuals from nine families have been reported with a pathogenic variant in ODC1 [Bupp et al 2018, Rodan et al 2018, Rajasekaran et al 2021, VanSickle et al 2021]. The following description of the phenotypic features associated with this condition is based on these reports.
Table 2.
Bachmann-Bupp Syndrome: Frequency of Select Features
View in own window
| Feature | % of Persons
w/Feature | Comment |
|---|
| Alopecia | 9/9 (100%) | |
| Nonspecific dysmorphic features | 9/9 (100%) | No specific pattern identified |
| Developmental delay 1 | 8/8 (100%) | |
| Hypotonia 1 | 8/8 (100%) | |
| Macrocephaly | 6/9 (66%) | |
| Pregnancy notable for polyhydramnios | 5/9 (55.5%) | |
| Skin findings 1 | 4/8 (50%) | Keratosis pilaris and follicular cysts |
| Constipation 1 | 3/8 (37.5%) | |
| Macrosomia | 2/5 (40%) | Measured in infancy; growth parameters tend to normalize w/age. |
| Seizures 1 | 1/8 (12.5%) | |
- 1.
One reported case was of a late-term stillbirth; thus, some features pertaining to this person are unknown [Rodan et al 2018].
Alopecia appears to be the most distinctive feature of BABS, but it does have some variability.
Hair is typically present at birth but is sometimes sparse and sometimes has atypical color (darker or lighter than anticipated).
Loss of hair, if present, begins in the first few weeks of life with hair falling out in large clumps.
Absent or sparse eyebrows and eyelashes are typically
congenital.
Some affected individuals undergo regrowth of scalp hair that usually remains sparse, although one affected individual had full, thick hair with no reported loss of hair postnatally but absent eyebrows and eyelashes.
Nonspecific dysmorphic features. Dysmorphic features have been identified in most affected individuals, but not with any discernible pattern or consistency.
Developmental delay / intellectual disability. Developmental delay is evident early in life with both motor and speech delays. Walking was achieved between age 17 months and four years, although two affected individuals still had not walked when reported at ages three and five years. First words were said between ages three and six years; three affected individuals were nonverbal when reported at ages ten months, 32 months, and 16 years.
Hypotonia is uniform and likely contributes to motor developmental delay.
Behavior. Attention-deficit/hyperactivity disorder (ADHD), autism, and aggression have all been reported in five of the nine published individuals.
Growth. Larger head circumference for age and sex is often seen. One affected individual with a normal head circumference had sagittal craniosynostosis, which has not been seen in any other reported individuals.
Macrosomia at birth has been reported; in four of five affected individuals measured, length was >95% percentile, and in two of five weight was >95% percentile. However, growth parameters tend to normalize with age: at later childhood examination, only two of eight had height >95% percentile and none had weight >95% percentile.
Gastrointestinal and feeding. Ability to feed varies with developmental level, with some affected individuals requiring gastrostomy tube. Others can take food by mouth. Constipation has been seen and may be related to hypotonia.
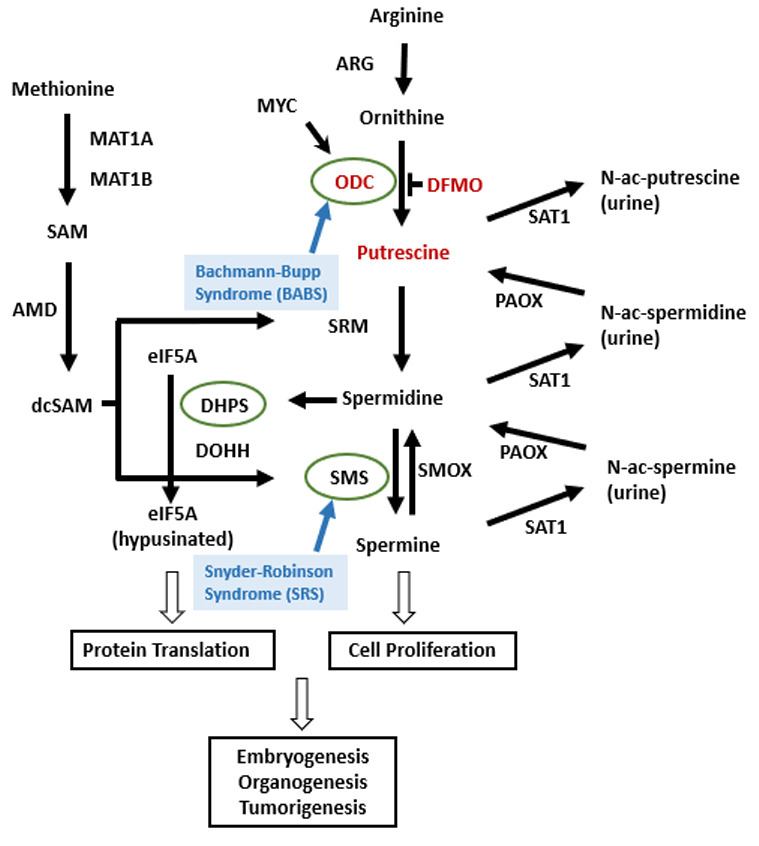
Skin findings including keratosis pilaris, recurrent follicular cysts (particularly on the back, axilla, and posterior of head), and dry skin have been observed in childhood. Cysts were noted in the first few years of life and resolved with DFMO treatment (see Therapies Under Investigation).
Seizures are seen in one individual, at age 23 years the oldest known with BABS. His seizures emerged at age 14 years, and multiple seizure types have been observed including atypical absence, atonic, and generalized tonic-clonic. Treatment has been challenging as no controlling medication has been identified [VanSickle et al 2021].
Brain MRI findings. Abnormalities identified on brain MRI vary quite broadly without a unifying pattern, although cystic lesions have been reported, and nonspecific white matter changes (loss, hyperintensity, signal abnormality) have been observed in six of nine affected individuals.
Other. Because this is a newly recognized condition, it is unclear if the following findings are associated with BABS or represent rare co-occurrences, as the findings have been found in a small number of affected individuals.
Hearing loss. One individual had unilateral
congenital sensorineural hearing loss. Three individuals have required myringotomy.
Congenital heart disease. Mild pulmonary stenosis has been reported in one affected individual and ventricular septal defect (self-resolved) was reported in another.
Nail anomalies. Two affected individuals had brittle nails and another had hypoplastic toenails.
Ophthalmologic findings. One reported individual had esotropia, pseudostrabismus, and bilateral myopic astigmatism.
Prognosis. The availability of experimental targeted treatment options (see Table 7) may change the natural history of this condition. It is unclear if diagnosis prior to symptom onset, coupled with targeted therapies, will decrease the morbidity and mortality of this condition in the future.