Clinical Description
Beta-propeller protein-associated neurodegeneration (BPAN) typically presents with seizures, infantile-onset developmental delay, intellectual disability, absent-to-limited expressive language, and abnormal behaviors with Rett syndrome-like features. During adolescence and early adulthood (mean age 25 years; range 15-37 years) some of the hallmark childhood features (e.g., seizures) resolve or become less prominent, while cognitive decline and progressive parkinsonism and dystonia emerge as characteristic findings [Hayflick et al 2013, Nishioka et al 2015].
The majority of individuals with BPAN are female; however, some males have been reported. Significant phenotypic variability is observed in both males and females.
With increasing use of genomic testing, individuals with BPAN now tend to be identified at younger ages than those originally described by Haack et al [2012].
Core Clinical Features
Seizures are common during childhood, often starting as febrile seizures [Nishioka et al 2015]. Seizures are frequently multiform and include focal seizures with impaired consciousness, epileptic spasms [Hogarth, personal communication], and generalized seizures, including absence, atonic, myoclonic, and tonic-clonic [Hayflick et al 2013].
One young boy included in a cohort of children with early-onset epileptic encephalopathy had a de novo deletion of WDR45 and two contiguous genes (CCDC120 and PRAF2) and clinical findings consistent with BPAN (brain iron accumulation, dystonia, and spastic tetraparesis) [Abidi et al 2016].
Although seizures often require pharmacologic treatment and may be intractable, they often resolve or become less prominent with age.
Developmental delay / intellectual disability. Individuals with BPAN typically have developmental delay as children. The majority of children with BPAN have no expressive language; some develop limited language. Rarely, girls with BPAN can be high functioning with normal or nearly normal language development and less severe learning disabilities [Long et al 2015].
Onset of progressive dementia occurs between adolescence and early to middle adulthood (mean age 25 years; range 15-37 years) [Hayflick et al 2013]. Similar neurologic decline was observed in seven affected Japanese women between ages 29 to 39 years [Nishioka et al 2015].
In some instances, parents of children with BPAN have reported regression during early childhood; however, it is not clear whether this is due to the underlying disease, unrecognized seizures, or both [Hogarth, personal communication]. The aggressive epilepsy profile eventually seen in some children with BPAN may itself contribute to cognitive dysfunction, as in other epileptic encephalopathies.
Motor dysfunction. Children are typically clumsy and have a broad-based or ataxic gait. Some never learn to walk; others who achieve walking eventually become non-ambulatory. Some have mild spasticity that generally does not require treatment.
Fine motor skills are also impaired. Some also have limited purposeful hand use (reminiscent of Rett syndrome) that can contribute to functional impairments such as difficulty with dressing and use of utensils.
The neurologic deterioration that occurs during adolescence and early to middle adulthood also includes onset of movement disorders, dystonia, and parkinsonism [Hayflick et al 2013, Nishioka et al 2015].
Dystonia often starts in the upper extremities.
Parkinsonism is characterized by prominent bradykinesia, rigidity, freezing of gait, and postural instability. Tremor is not as common as in other forms of parkinsonism.
Abnormal behaviors that overlap with Rett syndrome include lack of purposeful hand movements, stereotypic hand movements such as repetitive midline hand-wringing, bruxism when awake, abnormal sleep patterns, features of autism spectrum disorder, and diminished response to pain [Haack et al 2012, Hayflick et al 2013, Ohba et al 2014, Khalifa & Naffaa 2015]. Some young girls have been noted to have episodes of deep breathing during waking hours [Authors, personal observation]. Several individuals with BPAN have been diagnosed with autism spectrum disorder – due in part to their limited language and social skills [Verhoeven et al 2014].
Other Features
Disordered sleep. Many families report that because their young children with BPAN have difficulty falling asleep and staying asleep, they sleep for only short periods of time. When performed, sleep studies revealed shortened mean sleep latency and abnormal REM sleep, hypersomnolence, hyposomnolence, and "dance-like" movements of the extremities with sleep onset [Hayflick et al 2013]. Unrecognized nocturnal seizure activity may contribute to abnormal sleep patterns.
Some adults with BPAN continue to have sleep difficulties or develop new manifestations such as waking and vocalizing during the night. Two of seven Japanese women had sleep problems as adults [Nishioka et al 2015].
Ophthalmologic findings including bilateral partial retinal colobomas, high myopia, astigmatism with myopia, spontaneous retinal detachment, and patchy loss of the pupillary ruff were observed in seven of 23 individuals [Hayflick et al 2013].
Bilateral optic atrophy has been described in one individual who also had bilateral sensorineural hearing loss [Rathore et al 2014].
Feeding and nutritional issues. Some infants and young children have feeding difficulties, most commonly texture sensitivity, oropharyngeal dysfunction, and aspiration. In some instances, GE reflux in young children has also required medical management. The neurologic deterioration that occurs in adolescence to early or middle adulthood frequently involves progressive feeding difficulty related to cognitive decline, dystonia, and parkinsonism and often results in significant weight loss.
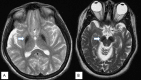
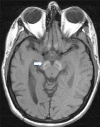
Neuropathologic features. Findings in a woman who died from pneumonia at age 27 years included mild cerebellar atrophy, thinned cerebral peduncles, and dark gray-brown appearance of the substantia nigra and (to a lesser extent) the globus pallidus. The substantia nigra and globus pallidus stained strongly for iron and demonstrated numerous axonal spheroids – both findings consistent with the pathology of other types of neurodegeneration with brain iron accumulation. Numerous tau-positive neurofibrillary tangles were seen in several regions; no beta-amyloid plaques or Lewy bodies were observed [Hayflick et al 2013].
In a second, more advanced case the findings were similar, but with more extensive neuronal loss and tau pathology [Paudel et al 2015]. Recent, unpublished findings by the authors suggest a more complex pathology.
BPAN in Affected Males and Females
Although WDR45 is X-linked and in females is subject to X-chromosome inactivation, the clinical features of BPAN follow a pattern that is somewhat atypical for an X-linked disorder: while there are far fewer affected males than females, the phenotype is similar in the two sexes.
In the original report of 20 individuals, the three affected males had pathogenic variants predicted to render the protein nonfunctional (all frameshifts leading to premature stop codons). One of these males had evidence suggestive of somatic mosaicism [Haack et al 2012]. A hypothesis of somatic mosaicism of a WDR45 pathogenic variant can explain both the viability of these males and the similarity of their phenotype to that of affected heterozygous females who are functionally mosaic due to X-chromosome inactivation [Haack et al 2012, Saitsu et al 2013].
Males with germline pathogenic variants in WDR45 were originally predicted to be non-viable, but five such males from three families have since been reported [Dufke et al 2014, Nakashima et al 2016, Zarate et al 2016]. Two families had missense variants while the other had a 3-bp in-frame deletion.
The overall paucity of affected males relative to females suggests that germline pathogenic variants are rare in males and that affected male conceptuses are less likely to survive than female conceptuses. In two sets of male-female sibs with inherited WDR45 pathogenic variants, the males were more severely affected than the females [Nakashima et al 2016, Zarate et al 2016]; of note, one female showed strongly skewed X-chromosome inactivation with the abnormal allele being preferentially active [Zarate et al 2016].
In summary, the following factors are all proposed to contribute to the variability in phenotype and to the predominance of affected females relative to males: