Clinical Description
In the premolecular era, the acronym CHARGE was proposed for the combination of the clinical features coloboma, heart defect, choanal atresia, retarded growth and development, genital hypoplasia, ear anomalies (including deafness) of unknown cause [Pagon et al 1981]. Clinical diagnostic criteria were refined for what became called CHARGE association [Blake et al 1998, Verloes 2005]. Following the discovery that heterozygous CHD7 variants and deletions cause CHARGE syndrome [Vissers et al 2004], molecular genetic testing of family members of probands with CHARGE syndrome expanded the phenotypic spectrum to include phenotypes that do not fulfill the previously proposed CHARGE syndrome clinical diagnostic criteria [Lalani et al 2006, Delahaye et al 2007, Jongmans et al 2009, Bergman et al 2011b, Hale et al 2016]. Thus, CHD7 disorder exhibits a high degree of clinical variability even among individuals in the same family and among individuals from different families with the same pathogenic variant [Jongmans et al 2008].
This section discusses only those reports in which a CHD7 pathogenic variant has been confirmed in affected individuals. To date reports of isolated manifestations of CHD7 disorder have been rare – many of which did not document a clinical workup sufficient to identify other features in the CHD7 disorder phenotypic spectrum. Thus, the percentages in Table 2 (based on persons with molecularly confirmed CHARGE syndrome [van Ravenswaaij-Arts & Martin 2017]) are likely to change over time as individuals with a CHD7 pathogenic variant ascertained through use of a multigene panel or genomic testing undergo a complete clinical evaluation (see Table 5).
Table 2.
Features of CHD7 Disorder in Individuals Ascertained for CHARGE Syndrome
View in own window
| Feature | % of Persons w/Feature | Comment |
|---|
Ocular coloboma
(ranging from small retinal coloboma to anophthalmia) | 80% | Light sensitivity, refractive error, loss of upper visual field/central visual field, blindness, ↑ risk of retinal detachment |
|
Choanal atresia/stenosis
| 45% |
Interferes w/breathing & feeding May require several surgeries to remain patent Unilateral stenosis may be easily missed.
|
Cranial nerve dysfunction/
anomaly
| I: hyposmia or anosmia | 90% | ↓ or absent sense of smell predicts hypogonadotropic hypogonadism. |
| VI: facial palsy | 40% |
Asymmetric face or lack of facial expression Facial nerve often has an aberrant course, which correlates w/SNHL & can be damaged during cochlear implant surgery.
|
| VIII: SNHL &/or vestibular dysfunction | >95% |
|
| IX/X: suck & swallow, abnormal GI motility | 60%-80% |
Lack of coordination of suck & swallow, aspiration, &/or gastroesophageal reflux Oral defensiveness Digestive & constipation issues
|
|
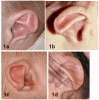
Ear malformations
| Abnormal auricle | 90% |
|
| Ossicular malformations | 80% |
Conductive hearing loss, which may fluctuate w/middle ear disease Complex mixed hearing loss may present as a wedge-shaped audiogram.
|
| Mondini defect | 90% | SNHL, esp high frequency |
| Semicircular canal defect | 94% | Affects balance & visual processing, → delayed motor development |
|
Cleft lip and/or palate
| 25%-50% | |
|
Endocrine 1
| Hypogonadotropic hypogonadism | 50%-70% |
Micropenis, cryptorchidism Small labia, uterine abnormality Delayed or absent puberty & infertility Often in combination w/anosmia
|
| Growth deficiency | 70% | May be due to growth hormone deficiency (in ~10%) |
| Hypothyroidism | 15%-20% | |
|
Developmental delay / Intellectual disability
| >90% / 60% | DD due to sensory deficits (hearing, vision, balance), illness, & hospitalizations |
|
Cardiovascular malformation
| 74% |
|
|
Tracheoesophageal anomalies
| 20% | Esophageal atresia w/or w/o fistula, laryngotracheomalacia, & gastroesophageal reflux, → feeding & breathing difficulties, aspiration (pneumonia), & sinusitis |
|
Brain
| Clivus hypoplasia
Hypoplasia/J-shaped sella | 95% | |
| Other | 50% | Microcephaly, ventriculomegaly, Dandy-Walker malformation, hypoplastic corpus callosum (30%), hypoplasia of cerebellar vermis (50%), brain stem, &/or frontal lobe |
|
Seizures
| 30% | Onset at any age, mostly general tonic-clonic convulsions as well as absence epilepsy |
|
Renal anomalies
| 30% |
Missing, hypoplastic, horseshoe, ectopic, or cystic kidney Vesicoureteral reflux & hydronephrosis
|
ASD = atrial septal defect; DD = developmental delay; PDA = patent ductus arteriosus; PFO = patent foramen ovale; SNHL = sensorineural hearing loss; VSD = ventricular septal defect
Based on individuals with molecularly confirmed typical or partial CHARGE syndrome [van Ravenswaaij-Arts & Martin 2017]. Note: percentages in this table are highly ascertainment dependent (i.e., the reason for molecular genetic testing). With the increasing use of multigene panels and genomic testing, it is likely that more individuals with presentations atypical for classic CHARGE syndrome will be diagnosed with CHD7 disorder.
- 1.
Because the majority of individuals with a pathogenic CHD7 variant have a typical CHARGE syndrome or CHARGE syndrome-like phenotype, the clinical features described below are relevant for most individuals with CHD7 disorder. In contrast, isolated hypogonadotropic hypogonadism with or without anosmia due to a pathogenic CHD7 missense variant appears to be rare [Xu et al 2018].
Development
Motor delay is invariably present due to vestibular anomalies and presents as poor head control, five-point crawl, delayed motor milestones, and reduced fine motor skills.
Language delay is caused by hearing loss, vision loss, vestibular anomalies, hospitalizations and illness, and/or cognitive impairment.
Assessment of cognitive abilities is difficult because of the multiple sensory deficits (vision, hearing, balance, smell), and much of the delay observed in motor and speech/language abilities is secondary to these deficits. Nonetheless, intellectual outcome is within the normal range in 50% of the individuals with clinical features consistent with CHARGE syndrome [Vesseur et al 2016b].
Children with better walking skills and fewer medical problems exhibit better adaptive behavior than children with less mobility and more medical problems [Salem-Hartshorne & Jacob 2005].
Behavioral features often reported are attention-deficit/hyperactivity disorder, repetitive behavior, and obsessive-compulsive behaviors. Self-abuse is occasionally seen. An increased pain threshold may predispose children to behaviors that are incorrectly interpreted by others as aggressive [Hartshorne et al 2005].
Many adults with clinical features consistent with CHARGE syndrome live independently, including many who have college or even advanced degrees. However, the level of independence comprises a broad spectrum [Blake et al 2005, Hartshorne et al 2016], depending, for each individual, on the combination of clinical features, educational program designed to address specific needs, and resources available.
Other Features
Gastrointestinal problems are frequently seen, mainly GI-related motility issues such as gastroesophageal reflux disease, constipation, and abdominal pain. Feeding challenges often result in tube feeding and problems with aspiration.
Late-onset issues can include malrotation of intestines, intussusception, and choking due to mouth overstuffing [Hudson et al 2015, Blake & Hudson 2017].
Immunodeficiency due to absent thymus (rarely) or decreased number or function of T-cells may occur [Wong et al 2015b]. Recurrent upper airway infections are common.
Skeletal involvement can include craniosynostosis, vertebral anomalies, scoliosis (in the majority of affected individuals) [Doyle & Blake 2005], extra or missing ribs, absent long bones (rare), ectrodactyly, polydactyly, finger-like thumb, and (more commonly) brachydactyly [Van de Laar et al 2007].
Hypermobility and contractures can be part of the syndrome.
Neuromuscular problems are common in CHARGE syndrome, mostly hypotonia (often resulting in scoliosis) and abnormal shoulder girdle muscles [O'Grady et al 2016]. Proprioception is diminished and, when in combination with balance problems, often results in a preference for pressure-building postures (upside-down position, legs twisted around one another) [Brown 2005].
Dental problems may include overbite, hypodontia, and poor mineralization of teeth [Chetty et al 2020].
Life expectancy highly depends on the severity of manifestations, since the phenotypic spectrum of CHD7 disorder is substantial. Mortality can be high in the first few years, when severe birth defects (particularly complex heart defects) are present, and are often complicated by airway and feeding issues. Feeding difficulties are usually due to cranial nerve abnormalities and improve gradually.
Multiple complex surgeries, along with the breathing problems or difficulty with anesthesia reported in CHARGE syndrome [Blake et al 2009], increase the risks associated with procedures.
After the first two or three years, mortality (and certainly morbidity and medical fragility) remains increased, with parents reporting frequent illnesses, infections, and hospitalizations [Bergman et al 2010].
In childhood, adolescence, and adulthood, increased mortality is likely related to a combination of residual heart defects, infections, aspiration or choking [Corsten-Janssen et al 2016], respiratory issues including obstructive and central apnea, and possibly seizures.
A number of families have reported serious (and in some instances lethal) intestinal issues such as volvulus [Lai & Feng 2006] and intussusception.
Despite these complications, the life span for many individuals can be normal. Individuals with clinical features consistent with CHARGE syndrome in their 60s who are in good health have been observed.