Clinical Description
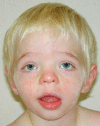
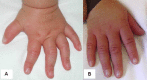
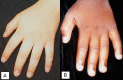
At the more severe end of the continuum of RPS6KA3-related intellectual disability (RPS6KA3-ID), clinically described Coffin-Lowry syndrome commonly involves developmental delay, intellectual disability, neurologic manifestations (hypotonia, stimulus-induced drop attacks, spastic paraparesis, and seizures), musculoskeletal manifestations (kyphoscoliosis and pectus deformity), and characteristic craniofacial and hand findings. The milder end of the continuum primarily manifests with neurodevelopmental features and variable but less pronounced multisystem involvement. Some heterozygous females exhibit clinical manifestations that are typically less severe than those seen in males. However, females can have recognizable craniofacial and hand findings that suggest the diagnosis of RPS6KA3-ID.
To date, more than 200 individuals have been identified with a pathogenic variant in RPS6KA3. A precise number is not known due to literature reports of individuals who were diagnosed based on clinical features prior to gene identification. The following description of the phenotypic features associated with RPS6KA3-ID is derived from an admixture of reported individuals whose diagnoses were molecularly confirmed as well as reported individuals who were diagnosed based on clinical findings, radiographic findings, and/or family history without molecular confirmation.
Affected Males
Developmental delay (DD) and intellectual disability (ID). Early milestones are variably delayed, with speech typically more severely affected than motor development. Coffin-Lowry syndrome (CLS) is typically characterized by severe-to-profound ID in males, although those with mild disability have been reported [Hanauer & Young 2002, Hunter 2002, Pereira et al 2010]. These individuals may now be better classified as having RPS6KA3-ID. Early developmental assessments may overestimate the ultimate developmental prognosis [Hunter 2002]. Manouvrier-Hanu et al [1999] reported two sibs with an unusually mild presentation associated with a missense variant.
Neurobehavioral/psychiatric manifestations. Males with CLS are often described as generally happy and easygoing, although behavioral issues can occur. These include attention-deficit/hyperactivity disorder [Matsumoto et al 2013], aggressive behavior [Hunter 2002], self-injury, and manifestations of autism spectrum disorder.
Neurologic. Detailed neurologic assessment may be hampered by the presence of severe ID. Findings reported include the following:
Musculoskeletal
Progressive kyphoscoliosis is one of the most difficult aspects of the long-term care of individuals with CLS. At least 40% of affected males have been reported to have progressive kyphoscoliosis [
Hunter 2002]. The rates were higher in a series reported from an orthopedic referral clinic [
Herrera-Soto et al 2007]. When other types of abnormalities such as thoracic lordosis and degenerative disc disease are included, the rate of spinal abnormalities in persons with CLS approaches 80% [
Welborn et al 2018]. Calcifications and/or hypertrophy of the ligamentum flavum have been noted in several reports, and other findings may include irregular vertebral end plates, anterior wedging, and narrowing of the intervertebral spaces [
Welborn et al 2018].
The severity of the spinal deformity often worsens significantly over time and can lead to severe complications:
Pectus carinatum and/or excavatum are frequently seen.
Other minor skeletal changes that may be seen on radiographs are of no clinical consequence.
Cardiovascular. Approximately 14% of affected males have cardiovascular disorders [Hunter 2002, Martinez et al 2011, Yoshida et al 2015, Wakami et al 2022]. This percentage may be an underestimate, as many individuals with CLS have not had thorough initial or ongoing cardiac assessment. Reports have included abnormalities of the mitral, tricuspid, and aortic valves, short chordae, cardiomyopathy (with endocardial fibroelastosis in one individual), unexplained congestive heart failure, and dilatation of the aorta and of the pulmonary artery. Facher et al [2004] reported a 14-year-old male with restrictive cardiomyopathy. Martinez et al [2011] reported a male with CLS who had left ventricular non-compaction cardiomyopathy with a restrictive pattern. Corrective surgery for mitral and triscupid insufficiency has been described in two males at ages 14 and 18 years, respectively [Yoshida et al 2015, Wakami et al 2022]. Cardiac anomalies may contribute to premature death. There has not been a systematic review of cardiovascular disorders in individuals with CLS.
Growth. Prenatal growth is usually normal; reduced growth usually occurs early in the postnatal period [Touraine et al 2002]. Males generally fall below the third centile in height but are expected to track a curve. Height in adult males has been reported to range between 115 and 158 cm, with an average of 143 cm [Hanauer & Young 2002]. Kyphoscoliosis may exacerbate the reduction in stature [Touraine et al 2002]. The safety and efficacy of growth hormone for the treatment of short stature in persons with CLS have not been studied, and it has been suggested that this could aggravate skeletal deformities and calcifications of the ligamentum flavum [Lv et al 2019].
While microcephaly is common, many individuals with CLS have a normal head circumference. Short stature, hypotonia, and decreased activity may lead to increased risk for obesity.
Dental. Dental anomalies commonly include small teeth, malpositioning, open bite, hypodontia of secondary teeth, advanced or delayed eruption of primary teeth, and premature loss [Hunter 2002, Igari et al 2006, Norderyd & Aronsson 2012]. The palate is high. With age, the retrognathia in the younger child tends to be replaced by prognathism.
Hearing loss is reported in as many as 30% of individuals [Pereira et al 2010]. Hunter [2002] reported hearing loss in 14 of 89 affected males.
Vision. Significant visual issues appear to be uncommon, although cataract, retinal pigment atrophy, and optic atrophy have been reported, and the incidence of chronic eyelid irritation (blepharitis) may be increased [Hunter 2002].
Respiratory. Obstructive sleep apnea may occur. Tracheostomy was reported to improve both obstructive sleep apnea and SIDAs in an individual reported by Imataka et al [2016].
Persons with CLS may have issues with intubation and/or ventilation that require careful consideration of the strategy for airway management during anesthesia [Hirakawa et al 2017].
Restrictive lung disease may occur due to kyphoscoliosis; this can be severe and go unrecognized [Venter et al 2019].
Other. Findings reported in single individuals include rectal prolapse, jejunal diverticuli, colonic diverticuli with reduced ganglion cells, popliteal ganglion, pyloric stenosis, unilateral renal agenesis, anteriorly placed anus, increased facial pigment, and enlarged trachea [Hunter 2002]. A family was reported in which multiple individuals developed type II diabetes [Touma Boulos et al 2021].
Prognosis. A number of adult males with CLS have been described. One recently reported individual is alive at age 48 years [Di Stazio et al 2021]. Since many adults with disabilities have not undergone advanced genetic testing, it is likely that adults with RPS6KA3-ID are underrecognized and underreported.
Life span is reduced in some males with CLS. Of individuals reported in the literature, death occurred in 13.5% of males at a mean age of 20.5 years (range: 13-34) [Hunter 2002].
Complicating factors have included cardiac anomalies, panacinar emphysema, respiratory complications, progressive kyphoscoliosis, and seizure-associated aspiration.
Coffin [2003] reported that one of his original patients died at age 18.8 years of pneumonia superimposed on chronic lung and heart disease, and a second individual died at age 18 years of acute food aspiration.
The authors are aware of an individual with CLS who had life-threatening central and obstructive sleep apnea, and of another male who had a history of chronic obstructive and central sleep apnea who died from respiratory complications after surgery for jaw advancement.
One affected male died of Hodgkin disease [
Hunter 2002].
Heterozygous Females
Heterozygous females often manifest markedly variable clinical manifestations of RPS6KA3-ID, such as mild facial coarsening, tapered fingers, short stature, and varied degrees of ID. Females with facial, hand, and skeletal findings typical of those seen in hemizygous males have been reported [Fryssira et al 2002, Hunter 2002, Jurkiewicz et al 2010, Rojnueangnit et al 2014]. Some heterozygous females have typical development and intellectual ability and lack other systemic findings associated with RPS6KA3-ID.
DD and ID. Affected females tend to have ID in the mild-to-moderate range.
Neurobehavioral/psychiatric manifestations. The rate of psychiatric illness may be higher than that in the general population. Six (8.8%) of 68 women (22 females with RPS6KA3-ID, 38 unaffected heterozygotes, and 8 "affected" sisters) have had psychiatric diagnoses, including schizophrenia, bipolar disease, and "psychosis" (reviewed in Hunter [2002]). One of two women reported by Micheli et al [2007] had a "psychosis," and one of two affected sisters was reported by Wang et al [2006] to have schizophrenia. Pervasive developmental disorder has been described [Matsumoto et al 2013]. Compulsive eyebrow-pulling behavior was reported in one female [Gürsoy et al 2022].
Neurologic. Females with typical SIDAs have been reported [Jurkiewicz et al 2010, Arslan et al 2014, Rojnueangnit et al 2014].
Musculoskeletal. At least 32% of females have been reported to have progressive kyphoscoliosis [Hunter 2002]. Rojnueangnit et al [2014] reported a female who was diagnosed with both scoliosis and spondylolisthesis at age 10 years and required surgical fusion from L4 to S1 by age 11 years due to the severity and progressive nature of her spinal issues.
Cardiovascular. Approximately 5% of affected females have cardiovascular disorders [Hunter 2002]. Cong et al [2022] described a family in which three females with a pathogenic variant in RPS6KA3 all had mild mitral and tricuspid regurgitation. However, it should be noted that two of these individuals also had a distal chromosome 22q11 deletion, which may have contributed to their phenotypes.
Growth. Stature may be reduced or fall within the typical range.
Dental. Females may have dental manifestations, including hypodontia [Jurkiewicz et al 2010, Yamoto et al 2020, Song et al 2022].
Hearing loss.
Hunter [2002] reported hearing loss in one of 22 affected females.
Vision. Specific data are not available regarding the frequency or types of vision issues observed in females with RPS6KA3-ID.
Respiratory. Sleep apnea may occur.
Other. Idiopathic hypercalcemia requiring bisphosphonate therapy during the second year of life was described in a female with RPS6KA3-ID [Tise et al 2022]. Genitourinary tract anomalies including uterine prolapse, bicornuate uterus, and duplicationof the renal collecting system have also been reported [Hunter 2002, Tise et al 2022]. Central precocious puberty with advanced bone age was described in an affected female [Song et al 2022].
Prognosis. Data are lacking as to whether life span in females with RPS6KA3-ID is impacted. Hunter [2002] noted that one affected female had died at age 48.