Isolated and Classic Cutis Marmorata Telangiectatica Congenita
Synonym: Van Lohuizen Syndrome
Joan Tamburro, DO, Elias I Traboulsi, MD, MEd, and Millan S Patel, MD, MSc.
Author Information and AffiliationsInitial Posting: June 9, 2022.
Estimated reading time: 27 minutes
Summary
Clinical characteristics.
Isolated and classic cutis marmorata telangiectatica congenita (CMTC) are characterized by congenital skin changes including erythematous-to-violaceous, reticulated, net-like or marbled-appearing patches of skin that do not mostly or completely resolve with warming or any other acute intervention. Individuals with isolated CMTC have no other syndromic features, and skin lesions tend to fade or resolve. Those with classic CMTC may have accompanying hemihypoplasia with body asymmetry, skin atrophy or ulceration, other vascular malformations, and occasional ocular issues (early-onset glaucoma and/or peripheral retinal vascular attenuation) but do not have other malformations, dysmorphic features, or cognitive impairment. The most common location for the CMTC lesions is on the legs. An affected limb may also display weakness or be unusually susceptible to cold compared to an unaffected limb. In more than half of affected individuals, skin lesions will generally fade across a wide range in age (6 weeks to 26 years), most commonly in the first year of life, but may not resolve completely.
Diagnosis/testing.
A molecular diagnosis can be established in a proband with suggestive cutaneous findings if a mosaic heterozygous pathogenic variant in GNA11 is identified by molecular genetic testing.
Management.
Treatment of medical manifestations: Most cutaneous changes improve or resolve with time and do not require intervention. Serial exams with photography are helpful. Persistent CMTC vascular lesions may be addressed with frequency-doubled Nd:YAG, Q-switched alexandrite, and pulsed dye laser therapy, although outcomes are mixed depending on the severity and depth of the lesions. Skin ulceration is usually treated by a qualified ulcer team with advanced knowledge in pain control. Intense pulsed light therapy may be considered to aid in ulcer improvement and faster healing. Lumbar sympathetic blockade may be considered for those with chronic pain and temperature dysregulation. Shoe lifts or orthotics may be considered in those with mild leg length discrepancies, and epiphysiodesis or limb lengthening may be considered in severe cases. Weakness is typically addressed through physical therapy. Standard treatment per ophthalmologist for glaucoma and peripheral retinal vascular abnormalities is recommended.
Treatment of psychosocial issues: Parents should be counseled on how to deal with child abuse accusations that may occur when individuals (including care providers and strangers) who are not familiar with CMTC happen to see their child's skin lesions. Self-esteem issues can be a major problem for affected individuals and may be addressed proactively through resiliency training and bibliotherapy. People unfamiliar with the condition are often worried that the condition may be contagious, so providing this information up front can defuse unwanted curiosity.
Surveillance: Close monitoring of the skin for early signs of impending ulceration as determined at initial evaluation; assessment for pain, weakness, and temperature dysregulation at each visit; annual monitoring of limb lengths and girth until skeletal maturity; ophthalmologic evaluation to include measurement of intraocular pressure and consideration of peripheral retinal vascular imaging every six months for the first four years of life, then annually (throughout lifetime) or any time there is ocular pain or visible corneal clouding; annual monitoring of coping skills related to visible physical differences starting at school age.
Agents/circumstances to avoid: Blood draws or IV placement in an affected limb; cold exposure.
Genetic counseling.
Isolated and classic CMTC are typically not inherited. Most affected individuals represent simplex cases.
Vertical transmission of a GNA11 pathogenic variant has not been reported to date. The risk to sibs of a proband with somatic mosaicism for a pathogenic variant in GNA11 would be expected to be the same as in the general population.
Rarely, autosomal dominant inheritance has been reported in families with a clinical diagnosis of isolated or classic CMTC (i.e., families in which a GNA11 pathogenic variant has not been identified). Sib recurrence in families with a clinical diagnosis of isolated or classic CMTC has been described but is very rare.
Because vertical transmission of a mosaic GNA11 pathogenic variant has not been reported to date and clinically diagnosed isolated and classic CMTC is usually not inherited, risk to family members is presumed to be very low.
Diagnosis
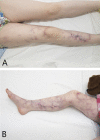
Isolated and classic cutis marmorata telangiectatica congenita (CMTC) are considered capillary malformations by the International Society for the Study of Vascular Anomalies. Clinically, CMTC is diagnosed by characteristic congenital erythematous-to-violaceous net-like or marbled areas of skin (). Several groups have attempted to develop clinical diagnostic criteria [Kienast & Hoeger 2009, Bui et al 2019], although no consensus clinical diagnostic criteria for isolated and classic CMTC have been universally accepted (see Establishing the Diagnosis). For a proposed classification system for the physical findings of CMTC, see Nomenclature.
Cutis marmorata telangiectatica congenita lesions and skin ulceration in an individual age nine years
Suggestive Findings
Isolated and classic CMTC should be suspected in individuals with the following clinical and family history findings.
Clinical findings
Persistent cutis marmorata that is more prominent or florid than physiologic cutis marmorata AND:
Vascular telangiectasia (spider veins)
Dilated, tortuous superficial veins
Body asymmetry in the form of hemihypoplasia
Family history. Because isolated and classic CMTC may be caused by a de novo, mosaic pathogenic variant, most probands represent simplex cases (i.e., a single recognized occurrence in a family).
Establishing the Diagnosis
Clinical Diagnosis
While there are no universally accepted clinical diagnostic criteria, the authors suggest that an affected individual should have all three of the following:
Congenital, net-like pattern of red-to-purplish cutaneous erythema that is obvious at rest
Erythema that does not mostly or completely resolve with warming or any other acute intervention
Note: These proposed clinical diagnostic criteria have not been validated.
Molecular Diagnosis
A molecular diagnosis of isolated and classic CMTC can be established in a proband with suggestive cutaneous findings if a mosaic heterozygous pathogenic (or likely pathogenic) variant in GNA11 is identified by molecular genetic testing (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include likely pathogenic variants. (2) Identification of a heterozygous GNA11 variant of uncertain significance does not establish or rule out the diagnosis.
Because the few reported individuals with a molecular diagnosis have a GNA11 pathogenic variant that was postzygotic (and thus mosaic), more than one tissue may need to be tested [Jordan et al 2020, Polubothu et al 2020].
Experience suggests that sequence analysis of DNA derived from affected skin or subcutaneous tissue has a higher detection rate than that of peripheral blood-derived DNA, where the variant is often absent.
Because CMTC is a focal disorder, pathogenic variants may only be detectable in affected tissues.
Failure to detect a
GNA11 pathogenic variant does not exclude a clinical diagnosis of CMTC in individuals with suggestive features, given that low-level mosaicism may be observed [
Jordan et al 2020]. Furthermore, pathogenic variants in other as-yet-unidentified genes may lead to CMTC.
Sensitivity to detect low-level mosaicism of a GNA11 pathogenic variant is theoretically greatest using massively parallel sequencing (also known as next-generation sequencing) in tissues other than blood.
Table 1.
Molecular Genetic Testing Used in Isolated and Classic Cutis Marmorata Telangiectatica Congenita
View in own window
| Gene 1 | Method | Proportion of Probands with a Pathogenic Variant 2 Detectable by Method |
|---|
|
GNA11
| Sequence analysis 3 | Dependent on tissue analyzed & method 4 |
| Unknown 5 | NA | |
- 1.
- 2.
- 3.
Sequence analysis detects variants that are benign, likely benign, of uncertain significance, likely pathogenic, or pathogenic. Variants may include small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected. For issues to consider in interpretation of sequence analysis results, click here.
- 4.
Most identified GNA11 pathogenic variants are missense gain-of-function pathogenic variants. Loss-of-function pathogenic variants may lead to other conditions (see Genetically Related Disorders).
- 5.
Next-generation sequencing of affected tissues has failed to detect GNA11 variants in most people with isolated and classic CMTC, so genetic heterogeneity is very likely present.
Clinical Characteristics
Clinical Description
To date, nearly 500 individuals with all clinical subtypes of cutis marmorata telangiectatica congenita (CMTC), including isolated and classic CMTC, have been published (reviewed in Bui et al [2019]). CMTC should be distinguished from cutis marmorata (CM), which is a normal physiologic finding that is apparent at rest in some newborns because of immaturity of the vascular system. CM consists of a homogeneous, fine, lacy, or reticular capillary change that worsens (or becomes apparent) with cold or emotion, completely (or nearly completely) resolves with warming, and usually fades by age four to six months. No ulcerations or skin atrophy are present. In contrast, while CMTC lesions often show changes with warming or strong emotion, the lesions do not completely disappear acutely (regardless of the intervention) as expected. If complete or near-complete resolution occurs acutely with any intervention, the individual has CM, not CMTC.
While individuals with the physical finding of CMTC may have a spectrum of features (see Nomenclature), this chapter focuses on the clinical subtypes of isolated and classic CMTC. Syndromes in which CMTC is one feature (syndromic CMTC) or situations where CMTC is one of several features (CMTC plus) are outside the scope of this chapter (see Differential Diagnosis).
Isolated CMTC. The skin changes consist of irregular, marbled or net-like, reddish-to-violaceous, flat skin lesions that may occur on a background of CM (see ). Presumably depending on the thickness of the dermal layer, CMTC lesions may have mildly diffuse or sharp borders. This group comprises 57.5% of reported individuals who have CMTC as a feature.
Classic CMTC is defined as CMTC with accompanying hemihypoplasia. Ocular problems, especially glaucoma, may be seen in a subset of affected individuals. Apart from these, no significant dysmorphism or other congenital anomalies are present.
Note: Care should be taken by the clinician to distinguish whether there is hemihypoplasia on one side versus hemihyperplasia on the other side, as the presence of hemihyperplasia indicates that a different diagnosis should be sought.
Skin/dermatologic findings. In more than half of affected individuals, skin lesions will generally fade across a wide range in age (6 weeks to 26 years), most commonly in the first year of life. For the remaining half, the skin lesions are lifelong.
Temperature dysregulation. An affected limb may be cold all the time. Acute pain may occur in an affected limb with cold exposure. Parents report that affected children may be hot and sweat profusely even in cool ambient temperature or may be unaware of how hot or cold they are.
Body asymmetry is reported in 37.7% of affected individuals and classically affects a leg, with a reduction in girth compared to the contralateral limb. The arms or torso may also be affected; the face is rarely affected. Hemihypoplasia usually (not always) occurs in the regions with the most visible lesions and is congenital most of the time. Leg length discrepancy has been reported in 13.6% of 485 affected individuals.
About 13% (9 of 69) of children with classic CMTC were found to have a clinically significant leg length discrepancy of 2 cm or greater [
Memarzadeh et al 2014].
Some leg length discrepancies resolve or stabilize in the first two years of life, and growth trajectories are not linear, requiring ongoing monitoring; differences in the range of 1 cm to 6.8 cm have been reported.
An affected limb may also display weakness or be unusually susceptible to cold compared to an unaffected limb.
Eye findings. Although uncommon (specific incidence is not well defined), infantile or juvenile glaucoma may occur. Elevation of intraocular pressure can lead to optic nerve damage and loss of vision. Peripheral retinal vascular attenuation has also been reported in a number of affected individuals and can lead to retinal ischemia. Dilated fundus examination, and if vascular changes are suspected, retinal imaging, can help detect the presence of retinal ischemia (see Management).
Social. Parents should be counseled on how to deal with child abuse accusations that may occur when individuals (including care providers and strangers) who are not familiar with CMTC notice their child's skin lesions. Parents should also be provided with lay language suitable for discussing the condition with friends, family, and even strangers. People unfamiliar with the condition are often worried that the condition may be contagious; providing this information up front can defuse unwanted curiosity.
Self-esteem issues can be a major problem for affected individuals and may be addressed proactively through resiliency training and bibliotherapy with books such as Rare Is Everywhere.
Giving children age-appropriate language to describe their condition can help with curious peers.
Children should be coached on strategies like smiling broadly until they get a smile in return and age-appropriate one-liners to manage staring.
Genotype-Phenotype Correlations
No genotype-phenotype correlations for isolated and classic CMTC have been identified.
Nomenclature
Clinically, the authors classify the physical finding of CMTC into the following groups:
Isolated CMTC. The skin changes consist of irregular, marbled or net-like, reddish-to-violaceous, flat skin lesions that may occur on a background of CM in the absence of significant dysmorphic features, body asymmetry, or major congenital anomalies.
Classic CMTC refers to CMTC with body asymmetry, usually hemihypoplasia, and occasional ocular abnormalities (early-onset glaucoma and/or peripheral retinal vascular attenuation) without dysmorphic features or other congenital anomalies.
Syndromic CMTC refers to CMTC or CMTC-like lesions associated with a known syndrome (see
Differential Diagnosis).
CMTC plus is a term the authors use to characterize CMTC that is associated with congenital anomalies and/or dysmorphic features in a pattern that does not fit with a known syndrome. This group likely comprises multiple conditions.
Previous terms used to refer to isolated and classic CMTC:
Congenital phlebectasia or congenital generalized phlebectasia
Naevus vascularis reticularis
Congenital livedo reticularis
Prevalence
Isolated and classic CMTC are rare conditions, with fewer than 500 individuals reported in the literature. Unfortunately, individuals with any type of CMTC (see Nomenclature) are frequently referred to in the literature as having "CMTC," without further delineation of CMTC subtype, and the older literature often includes more recently recognized diagnoses (e.g., diffuse capillary malformation with overgrowth, phakomatosis pigmentovascularis type V) under the blanket term of "CMTC." CMTC is found in nearly equal proportions between the sexes, with a slight predominance in females (1.2:1 female:male ratio).
Differential Diagnosis
Isolated and classic cutis marmorata telangiectatica congenita (CMTC) are congenital conditions; similar skin lesions occurring later in life are referred to as livedo reticularis or livedo racemosa (see A CMTC-like skin appearance seen later in life at the end of this section).
The primary differential diagnosis for congenital isolated CMTC is physiologic cutis marmorata (CM). CM is a normal physiologic finding that is apparent at rest in a minority of newborns. CM consists of a homogeneous, fine, lacy, or reticular capillary change that worsens (or becomes apparent) with cold or emotion, completely (or nearly completely) resolves with warming, and usually fades by age four to six months. No ulcerations or skin atrophy are present. In contrast, while CMTC lesions often show changes with warming or strong emotion, the lesions do not rapidly resolve, regardless of the intervention. If complete or near-complete resolution occurs acutely with any intervention, the individual has physiologic CM, not CMTC.
Syndromic CMTC refers to CMTC or CMTC-like lesions associated with a known syndrome (see Table 3). Some of these diagnoses have early-onset clinical and laboratory findings that rule out isolated and classic CMTC. For others, adhering to the diagnostic criteria proposed by Kienast & Hoeger [2009] minus the "absence of venectasia" criterion or using the diagnostic criteria described in Establishing the Diagnosis should lead to correct identification of isolated or classic CMTC.
Table 3.
Syndromes of Known Genetic Cause in Which CMTC or CMTC-Like Lesions Are Present
View in own window
Involved Genes /
Chromosomes | Syndrome | Genetic Mechanism (MOI) | Key Features / Comment |
|---|
ARHGAP3
DLL4
DOCK6
EOGT
NOTCH1
RBPJ
| Adams-Oliver syndrome (OMIM PS100300) | Germline pathogenic variant(s) (AD, AR) | Should be suspected in any persons w/CMTC in combination w/aplasia cutis congenita of scalp, terminal transverse limb reduction defects, complex congenital heart disease, or dilated, tortuous vein(s) on scalp |
BRD4
HDAC8
NIPBL
RAD21
SMC1A
SMC3
|
Cornelia de Lange syndrome
| Germline pathogenic variant (AD, XL) 1 | Cutis marmorata that is much more prominent than expected & is persistent (60% of affected persons); also assoc w/synophrys, growth failure, feeding difficulties, limb deformity, ID |
CBS
MTHFR
| Homocystinuria (See Homocystinuria Caused by Cystathionine Beta-Synthase Deficiency.) | Germline pathogenic variants (AR) | Cutis marmorata that is much more prominent than expected & persistent; also assoc w/tall stature, ectopia lentis, DD |
|
RASA1
| RASA1-related Parkes Weber syndrome (See Capillary Malformation-Arteriovenous Malformation Syndrome.) | Germline pathogenic variant (AD) | Capillary malformations (sometimes reticulate or lace-like) ± overgrowth |
|
POLE
| IMAGE-I syndrome (OMIM 618336) / FILS syndrome (OMIM 615139) | Germline pathogenic variants (AR) | CMTC-like lesions (often present at birth), IUGR, short stature, metaphyseal dysplasia, immunodeficiency, congenital adrenal hypoplasia |
|
GNA11
| Diffuse capillary malformation w/overgrowth 2 | Postzygotic (mosaic) | Capillary malformations (sometimes reticulate or lace-like) ± overgrowth |
|
GNA11
| Phakomatosis pigmentovascularis type V (phakomatosis cesiomarmorata) | Postzygotic (mosaic) | Should be suspected in any person w/CMTC & extensive or unusual & permanent common dermal melanocytosis (previously called "Mongolian spots") or other forms of uncommon dermal melanocytosis. 3 This is important to identify, due to the risk of melanoma in this condition. Multiple café au lait spots, often atypical, may develop over time. Hemihypoplasia & GNA11 mosaic activating variants may also be seen. 4 |
|
GNAQ
| Sturge-Weber syndrome (OMIM 185300) | Postzygotic (mosaic) | Capillary malformations (sometimes reticulate or lace-like) ± overgrowth; also assoc w/overgrowth of affected tissue |
|
PIK3CA
|
PIK3CA-related overgrowth spectrum
| Postzygotic (mosaic) | Capillary malformations (sometimes reticulate or lace-like) ± overgrowth; also assoc w/overgrowth of affected tissue |
| Chromosome 13 | Patau syndrome | Trisomy | Cutis marmorata that is much more prominent than expected & persistent; also assoc w/cardiac, ocular, renal & brain malformations, & polydactyly |
| Chromosome 18 | Edward syndrome | Cutis marmorata that is much more prominent than expected & persistent; also assoc w/IUGR & neural tube & abdominal wall defects. |
| Chromosome 21 | Down syndrome | Cutis marmorata that is much more prominent than expected & persistent; also assoc w/hypotonia, specific facial features, & complex congenital heart disease |
AD = autosomal dominant; AR = autosomal recessive; DD = developmental delay; ID = intellectual disability; IUGR = intrauterine growth restriction, MOI = mode of inheritance; XL = X-linked
- 1.
NIPBL-related Cornelia de Lange syndrome (CdLS), RAD21-related CdLS, SMC3-related CdLS, and BRD4-related CdLS are inherited in an autosomal dominant manner; HDAC8-related CdLS and SMC1A-related CdLS are inherited in an X-linked manner.
- 2.
- 3.
- 4.
Other types of syndromic CMTC
Cutis marmorata that is much more prominent than expected and is persistent may be seen in neonatal lupus (or any primary antiphospholipid antibody syndrome).
Hypoplasia of an affected limb may be seen in "inverse Klippel-Trenaunay" in association with geographic capillary malformations, and in diffuse capillary malformation with undergrowth [
Cubiró et al 2020] with patchy or reticulated, poorly demarcated, pink-red or light red-to-purple capillary stains.
Genuine diffuse phlebectasia (Bockenheimer disease)
CMTC plus. CMTC may occur in association with congenital anomalies or dysmorphism that precludes a diagnosis of isolated or classic CMTC and does not fit with a known syndrome. Additional findings reported in such individuals include the ocular findings described below, which may also occur in other types of CMTC.
Ocular manifestations have been reported in 3.7% to 10% of affected individuals with any type of CMTC [
Kienast & Hoeger 2009,
Bui et al 2019] and were identified before age one year in more than 70% of those affected. However, unlike classic CMTC, individuals with CMTC plus with ocular manifestations also have other manifestations (see
Other manifestations below).
Neurologic manifestations
Developmental delays and seizures were found to be the most commonly reported neurologic manifestations in CMTC of any type, along with a wide variety of other findings including skull shape changes, brain arteriovenous malformation, intellectual disability, hydrocephalus, corpus callosum agenesis, brain ischemia, microcephaly, and hearing impairment – some of which may be incidental associations.
Transient ischemic attacks and cerebral vascular malformations were described in association with a homozygous truncating variant in
ARL6IP6 in a child with CMTC plus [
Abumansour et al 2015]. Marked dysmorphism, brain atrophy, focal seizures, global developmental delay, hepatomegaly, and various dermal melanocytic lesions were also described in this child, raising the possibility that the child may also have had phakomatosis pigmentovascularis type V. Transient ischemic attacks (with or without seizures) and developmental delay have been reported in at least two other individuals with CMTC plus [
Hinek et al 2008,
Van Schaik et al 2015].
Other manifestations include cardiac defects (reported at a rate of 5.2%), genitourinary defects (4.6%), and abdominal defects (2.3%).
A CMTC-like skin appearance seen later in life is referred to either as livedo reticularis or livedo racemosa [Hartig et al 2020].
Livedo reticularis (LR) is net-like and uniform. Some authors divide it into four subcategories: physiologic LR (synonymous with CM); primary LR (usually as a result of embolism, thrombosis, or occlusion of arterioles as seen with multiple cholesterol emboli, vasculitis, and other disorders); idiopathic LR; and amantadine-induced LR.
Livedo racemosa presents with a less uniform, broken net-like appearance and irregular mottling, usually intensifying with warming. It occurs secondary to regional arteriole occlusion, such as in thrombotic/hypercoagulable disorders; with emboli such as gas bubbles in decompression sickness, cholesterol emboli, or crystals in calciphylaxis; or with vasculitis (e.g., Sneddon syndrome, COVID-19, etc.).
Management
No clinical practice guidelines for cutis marmorata telangiectatica congenita (CMTC) have been published.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with isolated and classic CMTC, the evaluations summarized in Table 4 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 4.
Recommended Evaluations Following Initial Diagnosis in Individuals with Isolated and Classic Cutis Marmorata Telangiectatica Congenita
View in own window
| System/Concern | Evaluation | Comment |
|---|
|
Skin
| Full skin exam to identify areas of skin atrophy & ulceration | Consider referral to dermatologist. |
Musculoskeletal/
Pain
| Assessment for significant limb asymmetry, abnormal joints, or weakness in affected limbs | Consider referral to orthopedist if gait is affected or leg length discrepancy is >1.5 cm. |
| Assessment for chronic pain & temperature dysregulation | Typically seen in affected limbs |
|
Eyes
| Dilated eye exam w/intraocular pressure measurement & peripheral vascular imaging (as soon as feasible) | To screen for glaucoma & peripheral retinal vascular abnormalities |
Genetic
counseling
| By genetics professionals 1 | To inform affected persons & their families re nature, MOI (not typically inherited), & implications of CMTC to facilitate medical & personal decision making |
Family support
& resources
| Assess need for:
| Consider referral to psychologist for those experiencing social & self-esteem issues due to their diagnosis. |
MOI = mode of inheritance
- 1.
Medical geneticist, certified genetic counselor, certified advanced genetic nurse
Treatment of Manifestations
Table 5.
Treatment of Manifestations in Individuals with Isolated and Classic Cutis Marmorata Telangiectatica Congenita
View in own window
Manifestation/
Concern | Treatment | Considerations/Other |
|---|
|
Skin ulceration
| Ulcer care may need to be less aggressive than other wound treatments. | Since skin w/reticulate erythema does not heal like uninvolved skin, standard ulcer care is not typically sufficient. |
| A qualified ulcer team w/advanced knowledge in pain control is often required. | Assoc pain may be severe. |
| Ongoing cleaning of ulcer bed is needed. | Since these ulcers occur on already compromised skin, the length of time for ulcer healing may be longer than for other causes of ulcers. |
| Consideration of intense pulsed light therapy, which may → ulcer improvement & faster ulcer healing. 1 | |
Persistent CMTC
vascular lesions
| Frequency-doubled Nd:YAG, Q-switched alexandrite, & pulsed dye laser therapy may be tried but has mixed results depending on severity & depth of lesions. 2 | Anecdotally, some children worsen w/this therapy. Laser therapy is typically not needed; usually done for cosmetic reasons.
|
Chronic pain /
Temperature
dysregulation
| Lumbar sympathetic blockade may be considered. | Used successfully to treat chronic pain & marked temperature difference in an affected foot in 1 child w/classic CMTC 3 |
Leg length
discrepancy
| Mild leg length discrepancy: shoe lifts or orthotics may be considered. | |
| Severe leg length discrepancy: epiphysiodesis or limb lengthening surgery may be considered. | Surgery should be performed before skeleton is mature (age ~14 yrs in girls; ~16 yrs in boys). |
Weakness in
hypoplastic limb
| Physical therapy | |
Glaucoma &
peripheral retinal
vascular
abnormalities
| Standard treatment per ophthalmologist | |
Issues related
to physical
appearance
| Treatment may incl:
Bibliotherapy to normalize differences; Resilience training; Training in strategies for dealing w/staring or unwanted attention; Preparing parents to deal w/potential child abuse allegations.
| Consider referral to psychologist. |
Surveillance
Table 6.
Recommended Surveillance for Individuals with Isolated and Classic Cutis Marmorata Telangiectatica Congenita
View in own window
| System/Concern | Evaluation | Frequency |
|---|
|
Skin
| Close monitoring for early signs of impending ulceration of skin | As determined at initial eval |
|
Musculoskeletal
| Monitoring of limb length & girth | Annually until skeletal maturity |
| Assessment for pain, weakness, & temperature dysregulation | At each visit |
|
Eyes
| Ophthalmologic eval to incl measurement of intraocular pressure & consideration of peripheral retinal vascular imaging | Every 6 mos until age 4 yrs, then annually (throughout lifetime) 1 or when there is ocular pain or visible corneal clouding |
|
Self-esteem
| Monitoring of coping skills related to visible differences | Annually starting at school age |
Agents/Circumstances to Avoid
Avoid blood draws or IV placement in affected limbs because of potential vein fragility.
Cold exposure of affected limbs may be painful for some people with CMTC.
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
Isolated and classic cutis marmorata telangiectatica congenita (CMTC) are typically not inherited. Most affected individuals represent simplex cases (i.e., a single affected family member).
Vertical transmission of a GNA11 pathogenic variant has not been reported to date. Rarely, autosomal dominant inheritance has been reported in families with a clinical diagnosis of isolated or classic CMTC (i.e., families in which a GNA11 pathogenic variant has not been identified).
Risk to Family Members
Parents of a proband
Sibs of a proband
The risk to sibs of a proband with somatic mosaicism for a pathogenic variant in GNA11 would be expected to be the same as in the general population.
Note: This is a relatively new area for clinical genetics as only a small (albeit growing) number of disorders are known to be caused by mosaic pathogenic variants. Counseling for sib recurrence risk in CMTC should emphasize that, while no pregnancy is at zero risk, all evidence suggests that the risk for recurrence is very low (<1%).
Sib recurrence in families with a clinical diagnosis of isolated or classic CMTC has been described but is very rare. Isolated CMTC was reported in two sisters, one of whom also had scleroderma [
Andreev & Pramatarov 1979].
Offspring of a proband
All individuals with a molecular diagnosis of isolated or classic CMTC have had somatic mosaicism for a pathogenic variant in GNA11, suggesting that mutation occurred post fertilization in one cell of the multicellular embryo. Therefore, the risk for transmission to offspring is expected to be less than 50%.
The risk to offspring of a proband with a clinical diagnosis of isolated or classic CMTC is presumed to be low but slightly greater than that of the general population (vertical transmission has been observed in only four families to date).
Other family members. The risk to other family members of a proband with a mosaic GNA11 pathogenic variant is expected to be similar to that of the general population.
Prenatal Testing and Preimplantation Genetic Testing
Because vertical transmission of a mosaic GNA11 pathogenic variant has not been reported to date and clinically diagnosed isolated and classic CMTC is usually not inherited, the risk to family members is presumed to be very low. While prenatal diagnosis and preimplantation genetic testing (PGT) are possible in families with a known GNA11 pathogenic variant, prenatal testing and PGT are usually not indicated for family members unless they show features of the condition.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella
support organizations and/or registries for the benefit of individuals with this disorder
and their families. GeneReviews is not responsible for the information provided by other
organizations. For information on selection criteria, click here.
CMTC Alliance
CMTC alliance is a world-wide non profit with a mission to help patient and families that have been diagnosed with CMTC and to aid other patients with vascular anomalies to achieve diagnosis and treatment
3715 Wesley Chapel Road
Zanesville OH 43701
CMTC-OVM
CMTC-OVM is a worldwide non-profit patient organization that aims to improve the quality of life of people suffering from vascular abnormalities (blood vessel abnormalities), such as CMTC (‘Van Lohuizen syndrome’), and stimulate scientific research into these disorders.
Kapelweg 154B
Netherlands
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Isolated and Classic Cutis Marmorata Telangiectatica Congenita: Genes and Databases
View in own window
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Table B.
OMIM Entries for Isolated and Classic Cutis Marmorata Telangiectatica Congenita (View All in OMIM)
View in own window
|
139313 | GUANINE NUCLEOTIDE-BINDING PROTEIN, ALPHA-11; GNA11 |
|
219250 | CUTIS MARMORATA TELANGIECTATICA CONGENITA; CMTC |
Molecular Pathogenesis
Isolated and classic cutis marmorata telangiectatica congenita (CMTC) affects capillaries and venules. Pathologically, vessels are abundant and show tortuous dilatation. Capillary lakes, venous dilatation, and stenoses are also seen, and vessels may have an irregular intima and elastic lamina [Way et al 1974, Mazereeuw-Hautier et al 2002, Sahin et al 2006]. Skin biopsies sometimes show no findings [Petrozzi et al 1970].
Mosaic activating pathogenic variants in GNA11, such as Arg183Cys and Gln209Leu, are thought to cause disease by activating downstream growth signaling pathways: the p38 MAPK signaling pathway and the JNK and ERK pathways, respectively [Thomas et al 2016].
Research studies to elucidate the genetic architecture of CMTC currently are being conducted by Dr Beth Drolet at the University of Wisconsin, Dr Pierre Vabres at the Université de Bourgogne, and Dr Miikka Vikkula at the Institut de Duve, Université catholique de Louvain.
Mechanism of disease causation.
GNA11 pathogenic variants appear to cause disease through a gain-of-function mechanism.
Note: No prenatal exposure or teratogen has been identified to cause isolated or classic CMTC. When four affected infants (3 with classic CMTC and 1 with isolated CMTC) who all lived less than 20 km from each other were diagnosed over an 18-month period in Australia, no teratogens meeting modified Bradford Hill epidemiologic criteria for causal association could be identified [Rogers & Poyzer 1982].
Table 7.
Notable GNA11 Pathogenic Variants
View in own window
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
Cancer and Benign Tumors
Pathogenic activating variants in GNA11, specifically the p.Gln209Pro pathogenic variant, is seen in close to 50% of primary uveal melanomas [Van Raamsdonk et al 2010].
Chapter Notes
Author Notes
Dr Tamburro is a dermatologist who has attended CMTC patient conferences for years to provide expertise to the CMTC community.
Dr Traboulsi is a pediatric ophthalmologist and clinical geneticist who cares for multiple patients with CMTC and is a team member with Dr Tamburro in the Cleveland Clinic Vascular Anomalies Clinic.
Dr Patel is a medical geneticist who has attended several CMTC patient conferences to provide expertise. He researches Adams-Oliver syndrome and sees patients with CMTC.
Acknowledgments
We thank the many patients and parents who have taught us over the years and the CMTC Alliance and CMTC-OVM organizations for their dedication to people with this condition.
References
Literature Cited
Abumansour
IS, Hijazi
H, Alazmi
A, Alzahrani
F, Bashiri
FA, Hassan
H, Alhaddab
M, Alkuraya
FS. ARL6IP6, a susceptibility locus for ischemic stroke, is mutated in a patient with syndromic cutis marmorata telangiectatica congenita.
Hum Genet.
2015;134:815-22.
[
PubMed: 25957586]
Amitai
DB, Fichman
S, Merlob
P, Morad
Y, Lapidoth
M, Metzker
A. Cutis marmorata telangiectatica congenita: clinical findings in 85 patients.
Pediatr Dermatol.
2000;17:100-4.
[
PubMed: 10792796]
Andreev
VC, Pramatarov
K. Cutis mamorata telangiectatica congenita in two sisters.
Br J Dermatol.
1979;101:345-50.
[
PubMed: 508599]
Ayturk
UM, Couto
JA, Hann
S, Mulliken
JB, Williams
KL, Huang
AY, Fishman
SJ, Boyd
TK, Kozakewich
HP, Bischoff
J, Greene
AK, Warman
ML. Somatic activating mutations in GNAQ and GNA11 are associated with congenital hemangioma.
Am J Hum Genet.
2016;98:789-95.
[
PMC free article: PMC4833432] [
PubMed: 27058448]
Couto
JA, Ayturk
UM, Konczyk
DJ, Goss
JA, Huang
AY, Hann
S, Reeve
JL, Liang
MG, Bischoff
J, Warman
ML, Greene
AK. A somatic GNA11 mutation is associated with extremity capillary malformation and overgrowth.
Angiogenesis.
2017;20:303-6.
[
PMC free article: PMC5511772] [
PubMed: 28120216]
Cubiró
X, Rozas-Muñoz
E, Castel
P, Roé Crespo
E, Garcia-Melendo
C, Puig
L, Baselga
E.
Clinical and genetic evaluation of six children with diffuse capillary malformation and undergrowth.
Pediatr Dermatol.
2020;37:833-8.
[
PubMed: 32608066]
Dedania
VS, Moinuddin
O, Lagrou
LM, Sathrasala
S, Cord Medina
FM, Del Monte
MA, Chang
EY, Bohnsack
BL, Besirli
CG. Ocular manifestations of cutis marmorata telangiectatica congenita.
Ophthalmol Retina.
2019;3:791-801.
[
PubMed: 31147303]
del Boz González
J, Serrano Martín
MM, Vera Casaño
A. Cutis marmorata telangiectatica congenita. Review of 33 cases.
An Pediatr (Barc). 2008;69:557-64.
[
PubMed: 19128769]
Deshpande
AJ. Cutis marmorata telangiectatica congenital successfully treated with intense pulsed light therapy: a case report.
J Cosmet Laser Ther.
2018;20:145-7.
[
PubMed: 29020473]
Elitt
MS, Tamburro
JE, Moran
RT, Traboulsi
E. Cutis marmorata telangiectatica congenita: a focus on its diagnosis, ophthalmic anomalies, and possible etiologic factors.
Ophthalmic Genet.
2020;41:101-7.
[
PubMed: 32233697]
Garzon
MC, Huang
JT, Enjolras
O, Frieden
IJ. Vascular malformations. Part II: associated syndromes.
J Am Acad Dermatol.
2007;56:541-64.
[
PubMed: 17367610]
Haidari W, Light JG, Castellanos B, Jorizzo JL. Cutis marmorata telangiectasia congenita with painful ulcerations. Dermatol Online J. 2020;15:26:13030. [
PubMed: 32815697]
Happle
R.
Phacomatosis pigmentovascularis revisited and reclassified.
Arch Dermatol.
2005;141:385-8.
[
PubMed: 15781681]
Hartig
F, Reider
N, Sojer
M, Hammer
A, Ploner
T, Muth
CM, Tilg
H, Köhler
A. Livedo racemosa - the pathophysiology of decompression-associated cutis marmorata and right/left shunt.
Front Physiol.
2020;11:994.
[
PMC free article: PMC7497564] [
PubMed: 33013436]
Hinek
A, Jain
S, Taylor
G, Nykanen
D, Chitayat
D. High copper levels and increased elastolysis in a patient with cutis marmorata teleangiectasia congenita.
Am J Med Genet A.
2008;146A:2520-7.
[
PubMed: 18792971]
Jia
D, Rajadurai
VS, Chandran
S. Cutis marmorata telangiectatica congenita with skin ulceration: a rare benign skin vascular malformation.
BMJ Case Rep.
2018;2018:bcr2018226763. [
PMC free article: PMC6194448] [
PubMed: 30297497]
Jordan
M, Carmignac
V, Sorlin
A, Kuentz
P, Albuisson
J, Borradori
L, Bourrat
E, Boute
O, Bukvic
N, Bursztejn
AC, Chiaverini
C, Delobel
B, Fournet
M, Martel
J, Goldenberg
A, Hadj-Rabia
S, Mahé
A, Maruani
A, Mazereeuw
J, Mignot
C, Morice-Picard
F, Moutard
ML, Petit
F, Pasteur
J, Phan
A, Whalen
S, Willems
M, Philippe
C, Vabres
P. Reverse phenotyping in patients with skin capillary malformations and mosaic GNAQ or GNA11 mutations defines a clinical spectrum with genotype-phenotype correlation.
J invest Dermatol.
2020;140:1106-1110.e2.
[
PubMed: 31726051]
Kienast
AK, Hoeger
PH. Cutis marmorata telangiectatica congenita: a prospective study of 27 cases and review of the literature with proposal of diagnostic criteria.
Clin Exp Dermatol.
2009;34:319-23.
[
PubMed: 19196300]
Kurczynski
TW. Hereditary cutis marmorata telangiectatica congenita.
Pediatrics.
1982;70:52-3.
[
PubMed: 7088633]
MacGibeny
MA, John
AM, Milgraum
DM, Wassef
C, Milgraum
SS. Early cutis marmorata telangiectatica congenita masquerading as ulcerated retiform purpura: a diagnostic trap.
Pediatr Dermatol.
2020;37:979-80.
[
PubMed: 32749028]
Mazereeuw-Hautier
J, Carel-Caneppele
S, Bonafé
JL. Cutis marmorata telangiectatica congenita: report of two persistent cases.
Pediatr Dermatol.
2002;19:506-9.
[
PubMed: 12437551]
Memarzadeh
A, Pengas
I, Syed
S, Eastwood
DM. Limb length discrepancy in cutis marmorata telangiectatica congenita: an audit of assessment and management in a multidisciplinary setting.
Br J Dermatol.
2014;170:681-6.
[
PubMed: 24641785]
Petrozzi
JW, Rahn
EK, Mofenson
H, Greensher
J. Cutis marmorata telangiectatica congenita.
Arch Dermatol.
1970;101:74-7.
[
PubMed: 5416798]
Polubothu
S, Al-Olabi
L, Carmen Del Boente
M, Chacko
A, Eleftheriou
G, Glover
M, Jiménez-Gallo
D, Jones
EA, Lomas
D, Fölster-Holst
R, Syed
S, Tasani
M, Thomas
A, Tisdall
M, Torrelo
A, Aylett
S, Kinsler
VA. GNA11 mutation as a cause of Sturge-Weber syndrome: expansion of the phenotypic spectrum of Gα/11 mosaicism and the associated clinical diagnoses.
J Invest Dermatol.
2020;140:1110-13.
[
PMC free article: PMC7187890] [
PubMed: 31838126]
Richards
S, Aziz
N, Bale
S, Bick
D, Das
S, Gastier-Foster
J, Grody
WW, Hegde
M, Lyon
E, Spector
E, Voelkerding
K, Rehm
HL, et al.
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
Genet Med.
2015;17:405-24.
[
PMC free article: PMC4544753] [
PubMed: 25741868]
Rogers
M, Poyzer
KG. Cutis marmorata telangiectatica congenita.
Arch Dermatol.
1982;118:895-9.
[
PubMed: 7138045]
Sahin
A, Celebi
N, Doğan
R, Canbay
O, Uzümcügil
F, Aypar
U. Lumbar sympathetic blockade in a patient with cutis marmorata telangiectatica congenita.
Paediatr Anaesth.
2006;16:1292-3.
[
PubMed: 17121565]
Sassalos
TM, Fields
TS, Levine
R, Gao
H. Retinal neovascularization from a patient with cutis marmorata telangiectatica congenita.
Retin Cases Brief Rep.
2021;15:77-80.
[
PubMed: 29543621]
Thomas
AC, Zeng
Z, Rivière
JB, O'Shaughnessy
R, Al-Olabi
L, St-Onge
J, Atherton
DJ, Aubert
H, Bagazgoitia
L, Barbarot
S, Bourrat
E, Chiaverini
C, Chong
WK, Duffourd
Y, Glover
M, Groesser
L, Hadj-Rabia
S, Hamm
H, Happle
R, Mushtaq
I, Lacour
JP, Waelchli
R, Wobser
M, Vabres
P, Patton
EE, Kinsler
VA. Mosaic activating mutations in GNA11 and GNAQ are associated with phakomatosis pigmentovascularis and extensive dermal melanocytosis.
J Invest Dermatol.
2016;136:770-8.
[
PMC free article: PMC4803466] [
PubMed: 26778290]
Tracey
EH, Eversman
A, Knabel
D, Irfan
M. Cutis marmorata telangiectatica congenita successfully treated with intense pulse light and pulsed dyed laser therapy: a case report.
J Cosmet Laser Ther.
2020;22:177-9.
[
PubMed: 33586582]
Van Raamsdonk
CD, Griewank
KG, Crosby
MB, Garrido
MC, Vemula
S, Wiesner
T, Obenauf
AC, Wackernagel
W, Green
G, Bouvier
N, Sozen
MM, Baimukanova
G, Roy
R, Heguy
A, Dolgalev
I, Khanin
R, Busam
K, Speicher
MR, O'Brien
J, Bastian
BC. Mutations in GNA11 in uveal melanoma.
N Engl J Med.
2010;363:2191-9.
[
PMC free article: PMC3107972] [
PubMed: 21083380]
Van Schaik
SM, Reneman
L, Engelen
M, Roos
YB, Poll-The
BT. Stroke-like episodes and cutis marmorata telangiectatica congenita.
J Child Neurol
2015;30:129-32.
[
PubMed: 24525998]
Way
BH, Herrmann
J, Gilbert
EF, Johnson
SA, Opitz
JM. Cutis marmorata telangiectatica congenita.
J Cutan Pathol.
1974;1:10-25.
[
PubMed: 4220056]