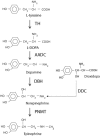
Clinical Description
Dopamine beta-hydroxylase (DBH) deficiency is characterized by a lack of sympathetic noradrenergic function but normal parasympathetic and sympathetic cholinergic function. Affected individuals exhibit profound deficits in autonomic regulation of cardiovascular function, but apparently only subtle signs of central nervous system dysfunction [Robertson et al 1986, Man in 't Veld et al 1987, Timmers et al 2004, Jepma et al 2011].
Onset. Although DBH deficiency appears to be present from birth, the diagnosis is not generally recognized until late childhood, when orthostatic hypotension becomes more severe.
Features by age. The full clinical spectrum of DBH deficiency is not known because of the limited number of cases reported. Clinical features reported in 21 affected individuals (13 female, 8 male) are included in Table 3.
Clinical features of DBH deficiency are included in Table 3.
Table 3.
Clinical Features of DBH Deficiency
View in own window
| Feature | # of Individuals 1 |
|---|
| Severe orthostatic hypotension | 21/21 (100%) |
| Anemia | 9/15 (60%) |
| Ptosis of eyelids | 12/14 (86%) |
| Hyperflexible or hypermobile joints | 6/10 (60%) |
| EKG abnormalities 2 | 2/12 (17%) |
| Epileptiform symptoms | 4/12 (33%) |
| Nasal stuffiness | 10/10 (100%) |
| Hypoglycemia | 4/12 (33%) |
| Sluggish deep-tendon reflexes | 3/9 (33%) |
| Increased plasma creatinine | 6/11 (54%) |
| Polyuria/nocturia | 3/9 (33%) |
| High palate | 9/10 (90%) |
| Increased BUN | 6/9 (67%) |
| Muscle hypotonia | 3/9 (33%) |
| Postprandial hypotension | 3/7 (43%) |
| Sleep irregularities | 5/7 (71%) |
| Impaired ejaculation | 4/4 (100%) |
- 1.
Number of individuals with the finding/total number evaluated for the finding
- 2.
Presyncopal symptoms include dizziness, blurred vision, dyspnea, nuchal discomfort, and occasionally chest pain. Symptoms may worsen in hot environments or after heavy meals or alcohol ingestion. Occasional bouts of unexplained diarrhea occur.
Renal function. Elevated blood urea nitrogen has been noted in six affected individuals in the USA [Garland et al 2005a, Garland et al 2009]. This may be evidence of a loss of renal function. A nephrologist who evaluated an individual age 16 years with a disproportionately high BUN (32 mg/dL) and slightly elevated creatinine (1.09 mg/dL) proposed that renal perfusion was reduced and that a BUN/Cr ratio <25 should be targeted. Although droxidopa acutely improved the ratio, the BUN/Cr ratio was further increased after a year of droxidopa treatment. The estimated GFR of an affected female age 57 years was reduced to 18 mL/min/1.73 m2 [Emily Garland, personal observation]. Another patient with unexpectedly low eGFR and elevated creatinine was found, by electron microscopy, to have abnormal, fused mitochondria in the proximal, but not the distal, tubules. Associated problems with the glomerular-tubular balance can be at least partially reversed by treatment with droxidopa [Wassenberg et al 2017].
Cognitive function. Despite the lack of norepinephrine, persons with DBH deficiency apparently have relatively normal mental status. Five affected individuals and ten matched healthy unaffected participants underwent a comprehensive battery of neurocognitive testing in addition to brain MRI, pupillometry, and EEG. Performance of the affected individuals, whether on or off droxidopa treatment, was similar to that of the unaffected individuals in most respects, suggesting that other systems compensate for absent norepinephrine in affected individuals. Brain MRI studies revealed a smaller total brain volume in the affected individuals compared to unaffected individuals, although relative proportions of white and gray matter and cerebrospinal fluid were similar in the two groups. In addition, affected individuals had a temporal-attention deficit when they were not on treatment. During an attentional-blink task, participants were asked to identify two digits, separated by a variable number of letters. Attentional blink refers to the deficit in processing the second digit when it is presented within 200-400 msec of the first. Accuracy in identifying the second digit was impaired in affected individuals not on treatment but performance improved with droxidopa treatment [Jepma et al 2011].
Ptosis. Ptosis of the eyelids, defined as a reduction in the margin reflex distance, is common in individuals with DBH deficiency and can be noted at an early age. It was reported in the first descriptions of individuals with this disorder in the late 1980s and in more than 85% of all cases in the published literature. Levator function is intact. Some individuals undergo levator advancement surgery [Phillips et al 2013], which may mask this aspect of the phenotype.
Olfactory function is relatively unaffected in individuals with DBH deficiency, who have intact noradrenergic neurons, in contrast to the marked deficit in individuals with pure autonomic failure, who have peripheral neuronal degeneration [Garland et al 2011].
Hypoglycemia. Because so few individuals have been diagnosed with DBH deficiency, there has not been a clear explanation for the occurrence of hypoglycemic episodes in some of the individuals. It is not known if this is related to the absence of norepinephrine and epinephrine, or the elevated levels of dopamine. Investigators have speculated that it may result from loss of the counterregulatory actions of epinephrine that protect against hypoglycemia [Man in 't Veld et al 1987]. In contrast to the report of hypoglycemia during the perinatal period, a girl age 15 years studied with a hyperglycemic clamp had a normal fasting glucose level but insulin resistance [Arnold et al 2017]. Her hyperinsulinemia persisted after a year of droxidopa treatment, despite improved orthostatic tolerance and restoration of plasma norepinephrine [Arnold et al 2017].
High palate. Physicians who inspect the palate often report that patients with DBH deficiency have a high, arched palate [Man in 't Veld et al 1988, Cheshire et al 2006; Emily Garland, unpublished findings]. This, however, is generally a subjective determination; it is not known how frequently it is either not assessed or reported incorrectly.
Life span. Four persons with DBH deficiency are known to have died. A woman age 57 years had chronic kidney disease and was undergoing treatment for breast cancer. Her listed cause of death at an assisted living facility was cardiac arrhythmia. Autopsy of a male age 28 years reported "scattered pyknotic cerebral neurons, isolated microfoci of cortical gliosis, cardiac arteriolar smooth muscle hypertrophy, scattered fibrosis in the cardiac conduction system, and sclerotic renal glomeruli." Cardiac dysrhythmia, possibly related to fibrosis in the cardiac conduction system, may have contributed to the patient's sudden demise [Cheshire et al 2006]. One individual died at age 20 years, possibly by suicide. A person age 63 died of unknown causes. Other affected individuals are likely to be deceased, but there are no published reports of causes of death or of effects of the disorder on life span.
One individual was not diagnosed with DBH deficiency until age 73 years despite having long-lasting orthostatic hypotension [Despas et al 2010], suggesting that DBH deficiency may not necessarily shorten the life span.