Clinical Description
FAM111A-related skeletal dysplasias include the milder phenotype of Kenny-Caffey syndrome (KCS) and a more severe lethal phenotype, osteocraniostenosis (OCS). To date, at least 35 individuals have been identified with a FAM111A-related skeletal dysplasia [Unger et al 2013, Guo et al 2014, Isojima et al 2014, Nikkel et al 2014, Kim et al 2015, Abraham et al 2017, Wang et al 2019, Cavole et al 2020, Deconte et al 2020, Pemberton et al 2020, Quaio et al 2020, Cheng et al 2021, Dempsey et al 2021, Lang et al 2021, Müller et al 2021, Stranneheim et al 2021, Yerawar et al 2021, Bowling et al 2022, Rosato et al 2022, Eren et al 2023]. The following description of the phenotypic features associated with these conditions are based on these reports.
Kenny-Caffey Syndrome (KCS)
Growth deficiency. The majority of individuals with KCS were born at term; intrauterine growth deficiency is not common. Postnatal short stature (height ≥2 standard deviations [SD] below the mean) is present in all affected individuals. The reported heights range from 2.6 SD below the mean to 8.2 SD below the mean [Unger et al 2013, Guo et al 2014, Isojima et al 2014, Nikkel et al 2014, Abraham et al 2017, Cavole et al 2020, Deconte et al 2020, Quaio et al 2020, Cheng et al 2021, Lang et al 2021, Yerawar et al 2021].
Relative macrocephaly is common due to the significant reduction in height with relatively preserved head circumference [Cheng et al 2021, Yerawar et al 2021].
Craniofacial features. Large anterior fontanelle and delayed anterior fontanelle closure are common in individuals with KCS. Craniosynostosis has been reported; it is predominantly basal type and leads to a V-shaped orbital roof [Unger et al 2013, Cheng et al 2021]. Characteristic facial features include frontal bossing or prominent forehead with relative macrocephaly, triangular face, short palpebral fissures, deeply set eyes, midface retrusion, short nose, narrow nasal ridge, and micrognathia or microretrognathia (see ).
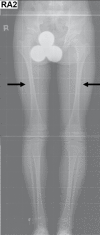
Skeletal features. Some individuals with KCS have decreased skull ossification. Long bones have cortical thickening and stenosis of the medullary cavity. In some individuals the long bones are slender. However, fractures have not been reported. Thin ribs have been reported in one individual [Kim et al 2015].
Primary hypoparathyroidism and hypocalcemia were reported in most individuals. Most presented before age two months (usually during the neonatal period) with hypocalcemic seizures [Unger et al 2013, Isojima et al 2014, Nikkel et al 2014, Kim et al 2015, Abraham et al 2017, Wang et al 2019, Cavole et al 2020, Deconte et al 2020, Quaio et al 2020, Cheng et al 2021, Yerawar et al 2021, Eren et al 2023]. Severity of hypocalcemia is variable, and most affected individuals require lifelong vitamin D and calcium supplements.
Ectopic calcification. Primary hypoparathyroidism results in hyperphosphatemia; elevated serum phosphorous can cause ectopic soft tissue calcifications.
Other ophthalmologic features include myopia, hypermetropia, astigmatism, and pseudopapilledema. Refractive errors were present in more than 80% of individuals with KCS, requiring corrective lenses without affecting daily function. Microphthalmia was reported in one individual with KCS [Lang et al 2021].
Dental anomalies include oligodontia or hypodontia, thin enamel or enamel hypoplasia, increased dental caries (4 individuals), retention of primary dentition, and delayed eruption of secondary dentition or loss of secondary dentition prior to age 40 years (3 individuals). Dental manifestations were found in more than 60% of individuals with KCS, and many individuals required dental prostheses [Guo et al 2014, Nikkel et al 2014, Wang et al 2019, Cavole et al 2020, Cheng et al 2021].
Genitourinary anomalies. Small testes were reported in three males with KCS [Cavole et al 2020, Cheng et al 2021]. Micropenis was reported in one individual [Cavole et al 2020].
High-pitched voice was noted in four in individuals with KCS [Unger et al 2013, Cheng et al 2021].
Other
Developmental delay is rarely reported. The severity of developmental delay was reported as mild in one individual [
Deconte et al 2020]. One individual with
FAM111A-related skeletal dysplasia presented with microcephaly and intellectual disability. The clinical
phenotype was described as intermediate between KCS and Sanjad-Sakati syndrome (OMIM
211410) [
Cavole et al 2020].
Growth hormone deficiency was reported in three individuals with KCS [
Isojima et al 2014,
Kim et al 2015]; however, further investigation is required to establish a causal relationship.
Prognosis. Based on current evidence, individuals with KCS have a normal life span.
Osteocraniostenosis (OCS)
Growth deficiency. Intrauterine growth deficiency is almost universal. Postnatal short stature (height ≥2 SD below the mean) was present in all affected individuals.
Microcephaly (head circumference ≥2 SD below the mean) is common in individuals with OCS. Information regarding detailed structural brain imaging is limited in this group.
Craniofacial features. Cloverleaf-shaped skull with large anterior fontanel was reported in all individuals/fetuses with OCS [Unger et al 2013, Pemberton et al 2020, Rosato et al 2022, Eren et al 2023]. One fetus with OCS and microcephaly had craniosynostosis [Pemberton et al 2020]. Characteristic facial features included frontal bossing, triangular face, short palpebral fissures, midface retrusion, ear anomalies, low-set ears, short nose, narrow mouth, micrognathia, and retrognathia. Two fetuses with OCS had microretrognathia detected at 20 weeks' gestation by ultrasound [Müller et al 2021, Rosato et al 2022].
Skeletal features. Decreased skull ossification was reported in ten individuals with OCS and could be detected as early as 20 weeks' gestation [Unger et al 2013, Müller et al 2021, Rosato et al 2022. Eren et al 2023]. Hypomineralized skull is a distinguishing skeletal feature in OCS. The long bones are slender with cortical thickening, stenosis of the medullary cavity, and flared metaphyses in all individuals with OCS. Fractures of the long bones (e.g., femur, radius, ulna) and rib fractures have been reported in the antenatal and perinatal period in three individuals [Rosato et al 2022]. Additional skeletal features (e.g., camptodactyly, platyspondyly) were also noted [Rosato et al 2022].
Thin ribs and thoracic hypoplasia can be detected early in the antenatal period. Pulmonary hypoplasia often leads to respiratory distress in newborns and is the main cause of early mortality [Rosato et al 2022, Eren et al 2023]. Surviving neonates with OCS typically require aggressive respiratory support. One child with OCS survived until age 21 months [Unger et al 2013].
Primary hypoparathyroidism and hypocalcemia. Two of six individuals had documented hypocalcemic seizures that occurred early in the neonatal period [Unger et al 2013, Eren et al 2023]. Affected individuals required vitamin D and calcium supplementation.
Ophthalmologic features. Microphthalmia is reported in half of individuals. Other ocular features (e.g., papilledema, refractive errors, early-onset cataract) were not identified due to the perinatal lethality of OCS.
Genitourinary anomalies. Micropenis was reported in six individuals with OCS. [Unger et al 2013, Rosato et al 2022, Eren et al 2023]. Small testes were reported in one individual [Eren et al 2023].
Splenic aplasia or hypoplasia was reported in four individuals with OCS [Unger et al 2013, Müller et al 2021, Rosato et al 2022].
Nomenclature
Kenny-Caffey syndrome has also been referred to as Kenny-Caffey syndrome type 2. "Type 2" is intended to distinguish Kenny-Caffey syndrome caused by heterozygous pathogenic variants in FAM111A from a clinically similar disorder caused by biallelic pathogenic variants in TBCE [Rosato et al 2022]. The TBCE-related phenotype is referred to as a "recessive variant of the Kenny Caffey" in OMIM (OMIM 244460) and designated "Kenny-Caffey syndrome type 1."
In the 2023 revision of the Nosology of Genetic Skeletal Disorders [Unger et al 2023], Kenny-Caffey syndrome caused by heterozygous FAM111A pathogenic variants is designated "Kenny-Caffey syndrome, dominant, FAM111A-related," while the autosomal recessive skeletal dysplasia associated with TBCE pathogenic variants is designated "Sanjad-Sakati syndrome, recessive, TBCE-related." While these two conditions have some phenotypic overlap, they represent distinct clinical and genotypic entities (see Table 3a).