Summary
Clinical characteristics.
Fibrodysplasia ossificans progressiva (FOP) is characterized by congenital bilateral hallux valgus malformations and early-onset heterotopic ossification, which may be spontaneous or precipitated by trauma including intramuscular vaccinations. Painful, recurrent soft-tissue swellings (flare-ups) may precede localized heterotopic ossification. Heterotopic ossification can occur at any location, but typically affects regions in close proximity to the axial skeleton in the early/mild stages, before progressing to the appendicular skeleton. This can lead to restriction of movement as a result of ossification impacting joint mobility. Problems with swallowing and speaking can occur with ossification affecting the jaw, head, and neck, and restriction of the airway and breathing may lead to thoracic insufficiency syndrome.
Management.
Treatment of manifestations: Avoid intramuscular injections and arterial punctures. Fall prevention using household safety measures and ambulatory devices; use of protective headgear to reduce sequelae of falls; prompt medical attention after a fall with consideration of prophylactic corticosteroid use; management by a dietician for those with feeding difficulties; preventative dental care with precautions to avoid injury; orthodontic treatment with a practitioner with experience in FOP; consultation with an expert anesthetist with experience in FOP prior to elective anesthesia; use of singing, swimming, incentive spirometry; positive pressure ventilation when indicated for mechanical respiratory difficulties including thoracic insufficiency syndrome; anti-inflammatory medications for flare-ups; consider corticosteroids for flare-ups of the submandibular region or jaw, major joints, after significant soft-tissue trauma, and for prophylaxis prior to dental and surgical procedures. Conservative management for scoliosis. Consider bisphosphonates for corticosteroid-induced osteopenia; fractures should be managed by an expert in FOP; hearing aids and appliances for conductive hearing impairment; encourage hydration and avoidance of high protein and high salt intake to prevent renal stones; occupational therapy; warm water hydrotherapy for mobility difficulties; lower extremity elevation, DVT prophylaxis, and supportive stockings while avoiding traumatic compression for lymphedema. Psychological support.
Surveillance: Annual clinical evaluation including evaluation for scoliosis with orthopedist or geneticist familiar with FOP; annual nutrition evaluation and examination for jaw ankylosis; baseline pulmonary function assessment, sleep assessment, and echocardiogram before age ten years followed by annual clinical evaluation of respiratory status; annual evaluation for fracture risk; audiology assessment every 12 to 24 months; annual assessment for signs and symptoms of nephrocalcinosis, gastrointestinal complications, and skin integrity; dental examinations every six months; Doppler ultrasound if DVT is suspected.
Agents/circumstances to avoid: Avoid procedures that predispose to soft-tissue injury, including intramuscular injections such as vaccinations, arterial punctures, dental procedures, procedures related to anesthesia, biopsies, removal of heterotopic bone, and all nonemergent surgical procedures. Avoid contact sports, overstretching of soft tissues, muscle fatigue, and passive range of motion. Avoid falls. In individuals with thoracic insufficiency syndrome, avoid supplemental oxygen, which can suppress respiratory drive.
Genetic counseling.
FOP is inherited in an autosomal dominant manner. The majority of affected individuals represent simplex cases (i.e., a single occurrence in a family) resulting from a de novo
ACVR1 pathogenic variant. Rarely, an individual diagnosed with FOP has an affected parent. If a parent of the proband is affected and/or is known to have the pathogenic variant identified in the proband, the risk to sibs is 50%. Once the ACVR1 pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
Diagnosis
There are no formal diagnostic criteria for fibrodysplasia ossificans progressiva (FOP).
Suggestive Findings
FOP should be suspected in individuals with the following clinical and radiographic findings.
Clinical findings
Congenital hallux valgus deformity that is most often bilateral
Progressive heterotopic ossification (extraosseous bone formation) that may manifest as a palpable mass. Ossification is either spontaneous or in response to soft-tissue trauma, including iatrogenic trauma from vaccinations or surgical procedures.
Painful, recurrent soft-tissue swellings (flare-ups) that may precede localized heterotopic ossification. This may occur in the form of scalp nodules in infancy, which may be an early or presenting feature.
Limb reduction defects that may affect the fingers in atypical or nonclassic FOP and may be mistaken for a brachydactyly syndrome in individuals who have not yet developed heterotopic ossifications
Imaging findings (See .)
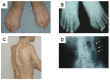
Characteristic features of FOP. A photograph (A) and radiograph (B) of the feet in an affected boy age 15 years with classic FOP show short, malformed halluces with a single, dysplastic phalanx in each great toe. A photograph of his back (C) and a radiograph (more...)
Prenatal ultrasound may identify a hallux valgus deformity as early as 23 weeks' gestation [
Maftei et al 2015].
Radiographs of the halluces demonstrate short, malformed first metatarsals and a single dysplastic phalanx.
Radiographs of affected areas demonstrate heterotopic ossification (extraosseous bone formation).
Note: Individuals with suspected FOP should avoid biopsy, elective surgery, and immunizations until diagnosis is confirmed [Kaplan et al 2019].
Establishing the Diagnosis
The diagnosis of FOP is established in a proband with hallux valgus malformations, heterotopic ossification, and/or a heterozygous pathogenic (or likely pathogenic) variant in ACVR1 identified by molecular genetic testing (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) Identification of a heterozygous ACVR1 variant of uncertain significance does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive
genomic testing (exome sequencing, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Individuals with the distinctive features described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas individuals in whom the diagnosis of FOP has not been considered are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
When the phenotypic and radiographic findings suggest the diagnosis of FOP, molecular genetic testing approaches can include single-gene testing or use of a multigene panel:
Single-gene testing. Sequence analysis of
ACVR1 to detect the most common
pathogenic variant (
c.617G>A; p.Arg206His) and other
missense variants associated with FOP. Note: Since FOP occurs through a
gain-of-function mechanism and large intragenic deletions or duplications have not been reported, testing for intragenic deletions or
duplication is not indicated in individuals in whom a diagnosis of FOP is strongly suspected. Pathogenic loss of function variants in
ACVR1 such as
nonsense, frameshift, and splice-site variants have not been described.
A skeletal dysplasia multigene panel that includes
ACVR1 and other genes of interest (see
Differential Diagnosis) may be considered to identify the genetic cause of the condition, compared to comprehensive
genomic testing, while limiting identification of variants of
uncertain significance and pathogenic variants in genes that do not explain the underlying
phenotype. Note: (1) The genes included in the panel and the diagnostic
sensitivity of the testing used for each
gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this
GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused
exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include
sequence analysis,
deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click
here. More detailed information for clinicians ordering genetic tests can be found
here.
Option 2
When the diagnosis of FOP has not been considered, including in individuals with atypical phenotypic features and/or the absence of congenital hallux malformation, comprehensive genomic testing (which does not require the clinician to determine which gene[s] are likely involved) may be the best option. Exome sequencing is most commonly used; genome sequencing is an increasingly used alternative.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Fibrodysplasia Ossificans Progressiva
View in own window
| Gene 1 | Method | Proportion of Probands with a Pathogenic Variant 2 Detectable by Method |
|---|
|
ACVR1
| Sequence analysis 3 | 100% 4 |
| Gene-targeted deletion/duplication analysis 5 | None reported |
- 1.
- 2.
- 3.
- 4.
- 5.
Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods used may include a range of techniques such as quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications.
Clinical Characteristics
Clinical Description
Fibrodysplasia ossificans progressiva (FOP) is characterized by congenital bilateral hallux valgus malformations and early-onset heterotopic ossification, which may be spontaneous or precipitated by trauma, including intramuscular vaccinations [Pignolo et al 2016].
To date, more than 800 individuals with over 20 pathogenic variants in ACVR1 have been reported (overview in Kaplan et al [2019]). The following description of the phenotypic features associated with this condition is based on these reports and reports of classic FOP phenotype in individuals who did not have molecular genetic testing.
Table 2.
Select Features of Fibrodysplasia Ossificans Progressiva
View in own window
| Feature | % of Persons
w/Feature 1 | Comment |
|---|
|
Bilateral congenital hallux valgus malformations
| >97% | Hallux valgus, malformed 1st metatarsal, &/or monophalangism Often 1st clinical feature Present in 100% of persons w/common c.617G>A (p.Arg206His) variant & classic phenotype
|
|
Heterotopic ossification
| ~100% |
|
|
Inflammatory soft-tissue swellings
| ~100% | May be spontaneous or triggered by trauma |
|
Scalp nodules
| ~40% |
|
Other
skeletal
manifestations
| Osteochondromas | ~90% | Proximal medial tibia is most common site. |
| Cervical spine fusions | ~80% | Affecting C2 to C7 |
| Short broad femoral neck | ~70% | |
| Scoliosis | ~65% | May be rapidly progressive |
| Thumb malformations | ~50% | |
| Distal limb reduction defects | <3% | May be misdiagnosed as a brachydactyly syndrome |
Hallux valgus malformation. Hallux valgus malformations are present from birth and may be identifiable on prenatal imaging [Maftei et al 2015]. The first metatarsal is short with a hallux valgus malformation and/or monophalangism with a single dysplastic phalanx (see ) [Towler et al 2020]. Additional hallux malformations can include a delta-shaped, dysplastic proximal phalanx. Hallux valgus malformations are most often bilateral but can be unilateral or absent in a minority of individuals with atypical FOP.
Heterotopic ossification
Extraosseous bone formation (abnormal bone formation in soft connective tissues outside of the normal skeleton) may manifest as a palpable hard lump or mass. Onset of ossification in individuals with the most common
pathogenic variant (
c.617G>A [p.Arg206His]) is age one to ten years, while onset of heterotopic ossification may be later in some individuals with atypical FOP.
Heterotopic ossification can be spontaneous or in response to soft-tissue trauma, including iatrogenic trauma from intramuscular vaccinations, falls, and surgical procedures. Painful, recurrent soft-tissue swelling may precede localized heterotopic ossification.
Heterotopic ossification can occur at any location, typically affecting regions in close proximity to the axial skeleton in the early/mild stages, before progressing to the appendicular skeleton. This can lead to restriction of movement as a result of ossification affecting joint mobility. Ossification of the jaw, head, and neck can affect swallowing and speaking.
Heterotopic ossification occurring in the thoracic region, submandibular region, throat, or other locations near the airway may impact the airway or respiratory function. In addition, costovertebral involvement, ossification of intercostal muscles, paravertebral muscles, and aponeuroses, as well as progressive spinal deformity with kyphoscoliosis may lead to thoracic insufficiency syndrome, the predominant cause of mortality. Pneumonia, hypoxemia, hypercarbia, pulmonary hypertension, and right-sided heart failure may occur in individuals with thoracic insufficiency syndrome.
Soft-tissue swellings
Soft-tissue swellings (flare-ups) may be spontaneous or follow an injury. They are characterized by painful swellings in soft connective tissue including skeletal muscles, tendons, ligaments, fascia, and aponeuroses. They may precede the development of localized heterotopic ossification.
Scalp nodules occurring in neonates and infants have been described in 40% of individuals from a national disease registry [
Piram et al 2011]. The nodules were large, firm, immobile, and tender, with rapid growth when they first appear. They generally regress spontaneously without treatment. The overlying skin was normal. Scalp nodules may be a localized manifestation of the soft-tissue swellings.
Additional skeletal malformations and manifestations variably seen:
Variable thumb
malformations may be present in some individuals, including hypoplasia and dysplastic phalanges.
Limb reduction defects affecting the fingers may be seen in atypical FOP.
Cervical spine fusions between C2 and C7 may be noted on cervical spine radiographs and may contribute to limitations in mobility as heterotopic ossification progresses. This occurs from intra-articular ankylosis of facet joints and early degenerative changes of the cervical spine.
Scoliosis affects up to 65% of individuals, may be rapidly progressive as a result of paravertebral lesions, and may contribute to thoracic insufficiency syndrome.
Pelvic radiographs may identify
congenital short broad femoral necks, which rarely affect function.
Developmental hip dysplasia is present in 60% of individuals with acute hip pain.
Osteochondromas are reported in up to 90% of individuals, with the proximal medial tibia the most common location.
Enchondromas, a benign tumor originating in cartilaginous tissue, have been described in several individuals [
Tabas et al 1993,
Rafati et al 2016]; the prevalence is unknown.
Fractures. Individuals with FOP are at increased risk for fractures of both normotropic and heterotopic bone because of the increased risk for falls, immobility, and corticosteroid-related osteopenia. Fractures in individuals with FOP usually heal with minimal heterotopic bone formation. Open reduction and internal fixation can lead to rapid onset of heterotopic ossification and is not recommended.
Hearing loss. Conductive hearing loss is present in 50% of individuals with FOP and may be slowly progressive. Onset is usually in childhood and may result from middle ear ossification. In some individuals, a sensorineural component may be present. Acute hearing loss is not usually associated with FOP and should prompt evaluation for other causes.
Renal stones. Individuals with FOP have a threefold increased risk of renal stones, which may be due to a combination of immobilization coupled with increased bone turnover. There has been no comprehensive study of stone composition in individuals with FOP.
Lymphedema may occur with flare-ups affecting the limbs. This may be acute, subacute, or chronic. In some individuals, underlying deep vein thrombosis may be present.
Genotype-Phenotype Correlations
The c.617G>A (p.Arg206His) pathogenic variant is associated with the classic FOP phenotype, including bilateral hallux valgus malformations and early-onset heterotopic ossification [Kaplan et al 2009].
Specific gain-of-function variants at amino acid residue 328 (p.Gly328Arg [c.982G>A and c.982G>C], p.Gly328Trp [c.982G>T], and p.Gly328Glu [c.983G>A]) have been associated with a characteristic phenotype that includes limb reduction defects, which may be misdiagnosed as an amniotic band defect or a brachydactyly syndrome, most commonly brachydactyly type B [Kaplan et al 2009].
Nomenclature
In the 2023 revision of the Nosology of Genetic Skeletal Disorders [Unger et al 2023], FOP is referred to as ACVR1-related fibrodysplasia ossificans progressiva and is included in the disorganized development of skeletal components group.
Prevalence
Based on studies in French [Baujat et al 2017] and British [Connor & Evans 1982] populations, the prevalence of FOP is estimated at one in one million (0.6-1.36:1,000,000). Individuals with FOP have been reported in diverse populations, and no racial, ethnic, sex, or geographic predisposition has been identified.
Differential Diagnosis
The diagnosis of fibrodysplasia ossificans progressiva (FOP) is often missed, due in part to the rarity of the condition. Nearly 90% of individuals with FOP initially receive a misdiagnosis, with two thirds undergoing unnecessary and potentially dangerous procedures that lead to permanent harm and lifelong disability in as many as 50% [Kitterman et al 2005].
Disorders that may present with clinical features similar to those of FOP are summarized in Table 3.
Table 3.
Genes of Interest in the Differential Diagnosis of Fibrodysplasia Ossificans Progressiva
View in own window
| Gene(s) | Disorder | MOI | Clinical Features of Differential Diagnosis Disorder |
|---|
| Overlapping w/FOP | Distinguishing from FOP |
|---|
EXT1
EXT2
|
Hereditary multiple osteochondromas
| AD | Multiple osteochondromas arising from growth plate in juxtaphyseal region of long bones or from surface of flat bones |
|
|
GNAS
| Progressive osseous heteroplasia
(See Disorders of GNAS Inactivation.) | AD 1 | Extensive bone formation (episodic & cumulative) w/in soft connective tissues | No hallux malformations or inflammatory soft-tissue swellings Individuals w/POH typically develop ossification w/in superficial dermal layer of the skin (which is unaffected in FOP. Predominance of membranous rather than endochondral bone formation
|
|
PTPN11
| Metachondromatosis
(OMIM 156250) | AD | Osteochondromas & enchondromas |
|
|
ROR2
| Brachydactyly type B1
(OMIM 113000) | AD | Distal limb (terminal) reduction-type defects w/brachydactyly | No heterotopic ossification |
Disorders to consider in individuals presenting with an isolated clinical feature characteristic of FOP [Kaplan et al 2008]:
Hallux malformations may represent
isolated congenital malformations (e.g., isolated brachydactyly) or juvenile bunions.
Tumor-like swellings may be associated with sarcoma, desmoid tumor, aggressive juvenile fibromatosis, or lymphedema.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with fibrodysplasia ossificans progressiva (FOP), the evaluations summarized in Table 4 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 4.
Recommended Evaluations Following Initial Diagnosis in Individuals with Fibrodysplasia Ossificans Progressiva
View in own window
| System/Concern | Evaluation | Comment |
|---|
|
Overall
| Clinical staging based on published criteria (See Table 5.) | |
Hallux
malformations
| Functional assessment w/orthopedist &/or physiotherapist | Rarely requires intervention |
Heterotopic
ossification
| Clinical assessment of extent of heterotopic ossification & impact on function | Avoid elective medical, surgical, & dental procedures. |
Thoracic
insufficiency
syndrome
| Consider pulmonary & sleep studies | If clinical concern for mechanical respiratory insufficiency |
Painful, recurrent
soft-tissue swelling
| History & physical exam for areas of soft-tissue swelling | |
|
Other
| Consultation w/clinical geneticist &/or genetic counselor | |
Note: Further evaluation may be indicated for participation in clinical trials.
Table 5.
Clinical Staging of Fibrodysplasia Ossificans Progressiva
View in own window
| Feature | Clinical Stage |
|---|
| Early/Mild | Moderate | Late/Severe | Profound | End Stage |
|---|
|
History of flare-ups
| None; or if present, limited to scalp, neck, or back | Mostly limited to axial regions & upper limbs | In any location | In any location | In any location |
|
Body regions affected
| Neck, back, upper limbs | Neck, back, chest, upper & lower limbs | Neck, back, chest, upper & lower limbs, jaw | Neck, back, chest, upper & lower limbs, jaw & distal limbs (wrists & ankles) | Ankyloses of most or all joints |
|
Thoracic insufficiency (TI)
| | Limited chest expansion | Rigid chest wall, no chest expansion, diaphragmatic breathing | Symptomatic TI syndrome (pulmonary hypertension & right-sided heart failure) | SymptomaticTI syndrome (pulmonary hypertension & right-sided heart failure) |
|
Other complications
| | | | Pneumonia, pressure ulcers | Recurrent respiratory infections |
|
ADL
| No or minimal assistance required due to mild joint limitations or physical delay in developmental milestones | Some assistance required | Assistance needed for most activities | Dependent for all ADL | Dependent for all ADL |
|
Ambulation
| Unaffected or cannot evaluate due to very young age | Walks; may use wheelchair in extenuating circumstances (e.g., long distances) | Walks w/assistive device &/or uses wheelchair | Wheelchair bound | Mostly bed bound |
|
CAJIS
| ≤4 | 5-18 | 19-24 | ≥24 | ≥28 |
ADL = activities of daily living; CAJIS = cumulative analog joint involvement scale (for FOP)
- 1.
Treatment of Manifestations
There is currently no definitive medical treatment for FOP and management is supportive. Clinical trials investigating experimental treatments are in progress (see Therapies Under Investigation). Guidelines for the management of individuals with FOP have been developed by a multidisciplinary team of experts [Kaplan et al 2019].
A hallmark of FOP management is prevention of soft-tissue injury and muscle damage to prevent inflammatory soft-tissue swellings and heterotopic ossification [Kaplan et al 2019].
Table 6.
Treatment of Manifestations in Individuals with Fibrodysplasia Ossificans Progressiva
View in own window
| Manifestation/Concern | Treatment | Considerations/Other |
|---|
|
Prevention of soft-tissue & muscle injury
| Avoid intramuscular injections. | Routine DTP vaccinations are particularly harmful. |
| Avoid arterial punctures. | Routine venipuncture poses minimal risk. |
| Biopsies of lesions are never indicated. | Biopsies are likely to cause heterotopic ossification. |
| Precautions during dental care |
|
|
Fall-related injuries
| Fall prevention | Modification of activity Improved household safety (e.g., install handrails, secure loose carpeting, remove objects from walkways, eliminate uneven flooring.) Use of ambulatory devices
|
| Reduce sequelae of falls | Use of protective headgear |
| Treatment following a fall |
|
|
Feeding difficulties due to jaw ankylosis when heterotopic ossification affects jaw region
| Referral to dietician to consider supplemental intake or modified food consistency | |
|
Dental care
|
|
|
|
Orthodontic concerns
| Orthodontic treatment by practitioner w/experience w/FOP | |
|
Requirement for anesthesia
| Consult w/expert anesthetist w/experience w/FOP prior to elective anesthesia. | If general anesthesia is required, an awake intubation by nasotracheal fiber-optic technique should be performed because of neck malformations, jaw ankylosis, sensitive airway, & risk of an obstructing neck flare-up. Highly skilled FOP-aware anesthesiologists should be present for all elective intubations.
|
|
Mechanical respiratory difficulties incl thoracic insufficiency syndrome
| Singing, swimming, incentive spirometry Positive pressure ventilation when indicated
| Avoid respiratory infections. Consider subcutaneous vaccination for influenza & pneumococcus in the proband. 1Recommend pertussis & influenza vaccination in family members. 2 Avoid supplemental oxygen, which can suppress respiratory drive.
|
|
Painful, recurrent soft-tissue swelling (flare-ups)
| NSAIDs or COX-2 inhibitors (oral or topical) Other anti-inflammatory medications including mast cell stabilizers, leukotriene inhibitors Consider corticosteroids, particularly for flare-ups affecting the submandibular region or jaw, major joints, & after significant soft-tissue trauma. 1 Consider oral corticosteroids for prophylaxis prior to dental & surgical procedures. 1
| Consider prophylactic treatment to prevent gastrointestinal complications due to NSAIDs or COX-2 inhibitors. Avoid narcotic analgesia if possible. No definitive evidence for use of bisphosphonates or imatinib
|
|
Scalp nodules
| No treatment required | Usually spontaneously regress |
|
Scoliosis
| Conservative management | Avoid traditional operative approaches. |
|
Corticosteroid-induced osteopenia
| Consider bisphosphonates according to standard treatment protocols | Bisphosphonates may play role in managing soft-tissue swellings. 1 |
|
Fractures
| Consult w/FOP expert | Fractures usually heal w/minimal heterotopic bone formation. Avoid open reduction & internal fixation, which can precipitate heterotopic ossification.
|
|
Conductive hearing impairment
| Hearing aids & appliances | |
|
Renal stones
|
| |
|
Problems w/ADL
| OT | |
|
Mobility issues
| Warm water hydrotherapy | Avoid passive joint movement. |
|
Lymphedema
|
| |
|
Depression
| Psychological support | |
ADL = activities of daily living; DVT = deep vein thrombosis; NSAID = nonsteroidal anti-inflammatory drug; OT = occupational therapy
- 1.
- 2.
Anti-influenza medication (oseltamivir) at first sign of influenza-like illness, while contacting medical practitioner
Note: Treatments for which no definitive evidence supports their use in FOP include chemotherapy, radiotherapy, bone marrow transplantation, and the chronic use of antiangiogenic agents, calcium binders, colchicine, fluoroquinolone antibiotics, propranolol, mineralization inhibitors, PPAR-gamma antagonists, and TNF-α inhibitors [Kaplan et al 2019].
Surveillance
Table 7.
Recommended Surveillance for Individuals with Fibrodysplasia Ossificans Progressiva
View in own window
| System/Concern | Evaluation | Frequency |
|---|
|
Musculoskeletal
| Clinical eval w/orthopedist &/or clinical geneticist w/experience in managing FOP Careful eval for scoliosis, which may be progressive
| At least annually; more frequently when clinically indicated |
Feeding
issues
| Anthropometric assessment Nutrition eval to monitor weight & caloric intake Clinical history & physical exam for jaw ankylosis
| Annually |
|
Mechanical respiratory difficulties, incl thoracic insufficiency syndrome
|
| Baseline pulmonary function assessment, sleep assessments, & echocardiogram before age 10 yrs (earlier if indicated) Annual clinical eval w/investigations as clinically indicated to guide specific respiratory therapies, incl positive pressure ventilation
|
|
Fracture risk
|
| Annually & as clinically indicated |
|
Hearing loss
| Audiology assessment | Every 12-24 mos |
|
Nephrocalcinosis
| Clinical assessment for signs/symptoms of nephrocalcinosis | Annually w/additional investigations as clinically indicated according to signs/symptoms |
|
Gastrointestinal
| Clinical assessment for signs/symptoms of gastric complications due to NSAID & corticosteroid management | Annually & as clinically indicated |
|
Pressure sores
| Eval of skin integrity | At each clinical visit |
|
Oral health
| Age-appropriate dental exam | Every 6 mos |
|
Lymphedema
| Doppler ultrasound if underlying DVT suspected | As clinically indicated |
DVT = deep vein thrombosis; NSAID = nonsteroidal anti-inflammatory drug
Agents/Circumstances to Avoid
It is imperative that iatrogenic harm is limited by avoiding procedures that predispose to soft-tissue injury, including intramuscular injections such as vaccinations, dental procedures, procedures related to anesthesia, biopsies, removal of heterotopic bone, and all nonemergent surgical procedures [Kaplan et al 2019].
Other activities to avoid include soft-tissue injuries, contact sports, overstretching of soft tissues, muscle fatigue, and passive range of motion (caution is required during treatment with physical therapists) [Kaplan et al 2019].
Falls should be actively avoided. Protective headwear should be considered for children who have upper limb involvement to prevent fall-induced head injury. Mobility aids may be effective in reducing falls in all age groups [Kaplan et al 2019].
In individuals with thoracic insufficiency syndrome, avoid supplemental oxygen, which can suppress respiratory drive.
Administration of vaccinations must be carefully managed in individuals with FOP. Detailed guidelines are available [Kaplan et al 2019] (see pdf). In brief, intramuscular vaccinations and all diphtheria-tetanus-pertussis (DTP) type vaccinations should be avoided. When the benefit outweighs the risk, subcutaneous vaccinations may be given at least six to eight weeks following recovery from soft-tissue flare-ups. Vaccinations to prevent respiratory disease (influenza, pneumococcal) are particularly important, and family members of individuals with FOP should receive influenza and pertussis vaccinations.
Evaluation of Relatives at Risk
It may be appropriate to clarify the genetic status of apparently asymptomatic young sibs of an affected individual in order to identify individuals at risk of iatrogenic harm (e.g., intramuscular injections) and other sources of trauma that may precipitate heterotopic ossification.
Note: For adult at-risk family members of a proband with classic FOP, molecular genetic testing in the absence of supportive physical examination findings (i.e., hallux deformity and signs of heterotopic ossification) is not usually required. However, for the evaluation of adult family members of a proband with atypical FOP, molecular genetic testing is recommended because manifestations of FOP may not be clinically apparent on physical examination.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
It is not known whether women with FOP have impaired fertility. Pregnancy in women with FOP is uncommon, as the disease manifestations at reproductive age limit reproductive potential. FOP poses major life-threatening risks to mother and fetus because of potential mechanical restrictions secondary to heterotopic ossification affecting the pelvis and surrounding regions, as well as breathing difficulties in later pregnancy secondary to restrictive chest wall disease. There is an increased risk of thromboembolism exacerbated by immobility. Ideally, pregnancy in a woman with FOP should be provided at a high-risk pregnancy center and follow established guidelines for the management of pregnancy in women with FOP (see Kaplan et al [2019]).
See MotherToBaby for further information on medication use during pregnancy.
Therapies Under Investigation
Research to develop treatments for FOP has focused on targeted inhibition of the ACVR1 receptor, ACVR1 ligands, BMP pathway signaling, pre-osseous chondrogenic heterotopic ossification, and inflammatory triggers of disease activity.
Palovarotene is a RAR-gamma agonist that reduces BMP signaling and may reduce the volume of heterotopic ossification.
REGN2477 is an antibody that binds to activin A and blocks its activity. By binding and blocking activin A, REN2477 may prevent the formation and stop the growth of heterotopic ossification in individuals with FOP. REGN2477 is currently in Phase II clinical trials in individuals older than age 18 years.
Sirolimus is an mTOR inhibitor that may reduce heterotopic ossification. Sirolimus is currently in Phase II clinical trials in individuals older than age six years.
Several other agents are currently undergoing safety and tolerability assessment in Phase I clinical trials. Further information on therapies under investigation is available in Kaplan et al [2019].
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for further information on clinical studies for FOP.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
Fibrodysplasia ossificans progressiva (FOP) is inherited in an autosomal dominant manner.
Risk to Family Members
Parents of a proband
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's parents:
If a parent of the
proband is affected and/or is known to have the
pathogenic variant identified in the proband, the risk to the sibs is 50%. The
penetrance of FOP is estimated to be near complete with intrafamilial clinical variability.
If the parents have not been tested for the
ACVR1 pathogenic variant but are clinically unaffected, the risk to the sibs of a
proband appears to be low. However, sibs of a proband with clinically unaffected parents are still presumed to be at increased risk for FOP because of the possibility of parental
germline mosaicism or, if the proband has atypical FOP, the absence of clinical manifestations in a
heterozygous parent.
Offspring of a proband. Each child of an individual with FOP has a 50% chance of inheriting the ACVR1 pathogenic variant; as the penetrance is estimated to be near complete, a child who has inherited a gain-of-function ACVR1 variant is expected to develop features of FOP.
Other family members. The risk to other family members depends on the genetic status of the proband's parents: if a parent has the ACVR1 pathogenic variant, the parent's family members are at risk.
Prenatal Testing and Preimplantation Genetic Testing
Once the ACVR1 pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Fibrodysplasia Ossificans Progressiva: Genes and Databases
View in own window
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Molecular Pathogenesis
ACVR1 encodes ACVR1, a transmembrane serine/threonine kinase. All individuals with FOP have overactive BMP signaling mediated by ACVR1. Disease-causing variants cause a conformational change of the receptor that alters its sensitivity and activity, resulting in induced BMP signaling in a BMP-independent and BMP-responsive manner to activate downstream signaling. In contrast to mouse models of BMP-pathway overactivation, which are embryonic-lethal, disease-causing variants in ACVR1 are more mildly activating, which may explain its compatibility with life [Shore & Kaplan 2010].
Mechanism of disease causation. Gain of function. To date, only missense variants in the GS-rich and protein kinase functional domains have been described in individuals with FOP, suggesting a mechanism of action involving altered protein function and BMP pathway activation, rather than a loss-of-function mechanism [Kaplan et al 2009, Shore & Kaplan 2010].
Table 8.
Notable ACVR1 Pathogenic Variants
View in own window
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
- 1.
Cancer and Benign Tumors
Somatic gain-of-function variants in ACVR1 have been identified in 20%-25% of diffuse intrinsic pontine gliomas (DIPG), with a wider variant spectrum than that seen in FOP. There is no reported increased incidence of DIPG in individuals with FOP [Han et al 2018].